
HDL in Immune-Inflammatory Responses: Implications beyond Cardiovascular Diseases




Abstract
:1. Introduction
2. HDL in Immune-Inflammatory Diseases: What Are HDL-C Levels Telling Us?
HDL Composition in Auto-Immune Diseases
3. The Role of HDL on Immune Cell Function
3.1. Anti-Oxidative, Anti-Inflammatory, and Vascular-Protective Effects of HDL
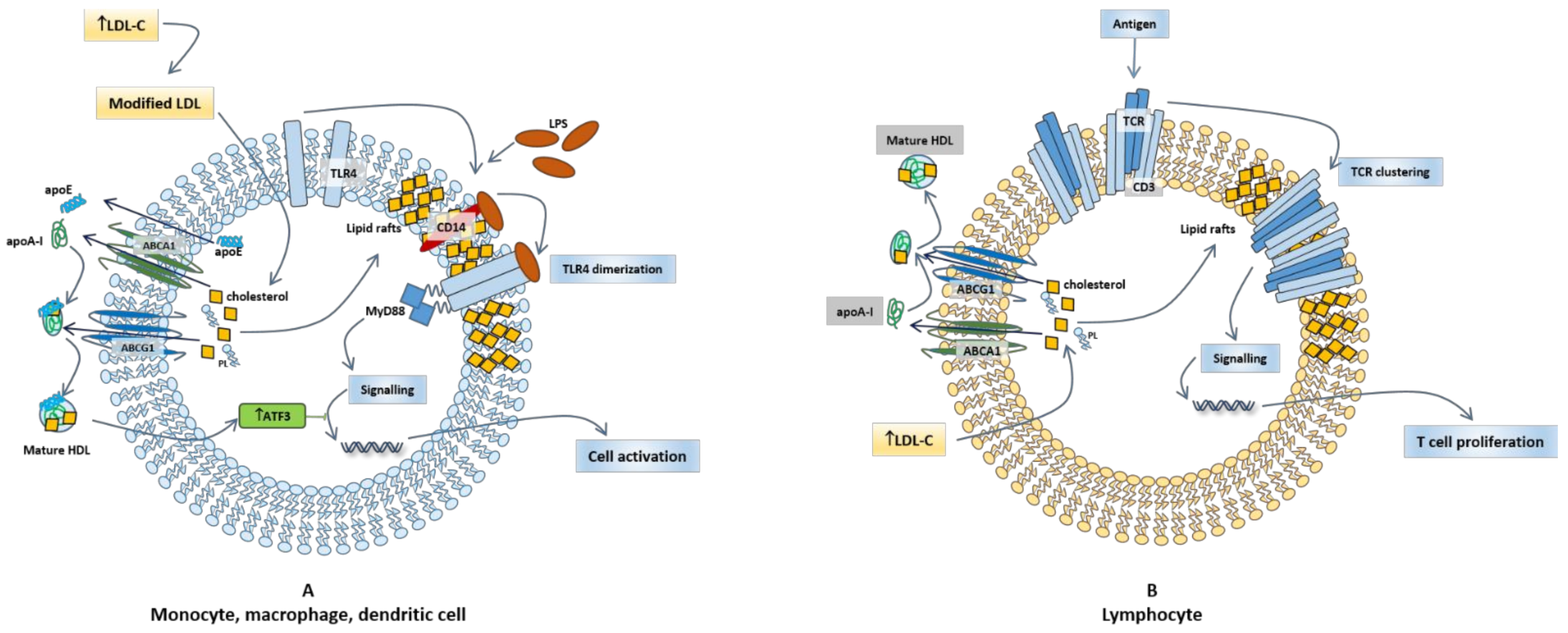
3.2. HDL, Lipid Rafts, and Immunomodulatory Function
3.3. HDL, Lipid Rafts in Innate Immune Cells
3.4. HDL, Cholesterol, and Lymphocyte Activation and Proliferation
4. Genetics as a Proxy to Study the Impact of HDL Cholesterol and Functions in Inflammation and Immune Disorders
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bonacina, F.; Da Dalt, L.; Catapano, A.L.; Norata, G. Metabolic adaptations of cells at the vascular-immune interface during atherosclerosis. Mol. Asp. Med. 2020, 100918. [Google Scholar]
- Wilson, P.W.; Abbott, R.D.; Castelli, W.P. High density lipoprotein cholesterol and mortality. The Framingham Heart Study. Arteriosclerosis 1988, 8, 737–741. [Google Scholar] [CrossRef] [Green Version]
- Emerging Risk Factors Collaboration; Di Angelantonio, E.; Sarwar, N.; Perry, P.; Kaptoge, S.; Ray, K.K.; Thompson, A.; Wood, A.M.; Lewington, S.; Sattar, N.; et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009, 302, 1993–2000. [Google Scholar] [CrossRef] [Green Version]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: Two prospective cohort studies. Eur. Heart J. 2017, 38, 2478–2486. [Google Scholar] [CrossRef] [Green Version]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Low HDL Cholesterol and High Risk of Autoimmune Disease: Two Population-Based Cohort Studies Including 117341 Individuals. Clin. Chem. 2019, 65, 644–652. [Google Scholar] [CrossRef]
- Madsen, C.M.; Varbo, A.; Tybjaerg-Hansen, A.; Frikke-Schmidt, R.; Nordestgaard, B.G. U-shaped relationship of HDL and risk of infectious disease: Two prospective population-based cohort studies. Eur. Heart J. 2018, 39, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.P.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.-C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Yu, M.; Morin, E.E.; Kang, J.; Kaplan, M.J.; Schwendeman, A. High-Density Lipoprotein in Lupus: Disease Biomarkers and Potential Therapeutic Strategy. Arthritis Rheumatol. 2020, 72, 20–30. [Google Scholar] [CrossRef]
- Trakaki, A.; Marsche, G. High-Density Lipoprotein (HDL) in Allergy and Skin Diseases: Focus on Immunomodulating Functions. Biomedicines 2020, 8, 558. [Google Scholar] [CrossRef]
- Romanato, G.; Scarpa, M.; Angriman, I.; Faggian, D.; Ruffolo, C.; Marin, R.; Zambon, S.; Basato, S.; Zanoni, S.; Filosa, T.; et al. Plasma lipids and inflammation in active inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2009, 29, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Matyus, S.P.; Braun, P.J.; Wolak-Dinsmore, J.; Saenger, A.K.; Shalaurova, I.; Warner, S.M.; Fischer, T.J.; Connelly, M.A. HDL particle number measured on the Vantera(R), the first clinical NMR analyzer. Clin. Biochem. 2015, 48, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.P.; Oeser, A.; Raggi, P.; Sokka, T.; Pincus, T.; Solus, J.F.; Linton, M.F.; Fazio, S.; Stein, C.M. Lipoprotein subclasses determined by nuclear magnetic resonance spectroscopy and coronary atherosclerosis in patients with rheumatoid arthritis. J. Rheumatol. 2010, 37, 1633–1638. [Google Scholar] [CrossRef] [Green Version]
- Lilleby, V.; Haugen, M.; Mørkrid, L.; Frøslie, F.K.; Holven, K.B.; Førre, Ø. Body composition, lipid and lipoprotein levels in childhood-onset systemic lupus erythematosus. Scand. J. Rheumatol. 2007, 36, 40–47. [Google Scholar] [CrossRef]
- O’Neill, F.; Riwanto, M.; Charakida, M.; Colin, S.; Manz, J.; McLoughlin, E.; Khan, T.; Klein, N.; Kay, C.W.; Patel, K.; et al. Structural and functional changes in HDL with low grade and chronic inflammation. Int. J. Cardiol. 2015, 188, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.; Speer, T.; Colin, S.; Charakida, M.; Zewinger, S.; Staels, B.; Chinetti-Gbaguidi, G.; Hettrich, I.; Rohrer, L.; O’Neill, F.; et al. HDL in children with CKD promotes endothelial dysfunction and an abnormal vascular phenotype. J. Am. Soc. Nephrol. 2014, 25, 2658–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiesa, S.T.; Charakida, M.; McLoughlin, E.; Nguyen, H.C.; Georgiopoulos, G.; Motran, L.; Elia, Y.; Marcovecchio, M.L.; Dunger, D.B.; Dalton, R.N.; et al. Elevated high-density lipoprotein in adolescents with Type 1 diabetes is associated with endothelial dysfunction in the presence of systemic inflammation. Eur. Heart J. 2019, 40, 3559–3566. [Google Scholar] [CrossRef] [Green Version]
- Rysz, J.; Gluba-Brzózka, A.; Rysz-Górzyńska, M.; Franczyk, B. The Role and Function of HDL in Patients with Chronic Kidney Disease and the Risk of Cardiovascular. Dis. Int. J. Mol. Sci. 2020, 21, 601. [Google Scholar] [CrossRef] [Green Version]
- Rosenson, R.S.; Jr, H.B.B.; Ansell, B.J.; Barter, P.J.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease, Nature reviews. Cardiology 2016, 13, 48–60. [Google Scholar]
- Srivastava, R.A.K. Dysfunctional HDL in diabetes mellitus and its role in the pathogenesis of cardiovascular disease. Mol. Cell. Biochem. 2018, 440, 167–187. [Google Scholar] [CrossRef]
- Pirillo, A.; Catapano, A.L.; Norata, G.D. HDL in infectious diseases and sepsis. Handb. Exp. Pharmacol. 2015, 224, 483–508. [Google Scholar] [PubMed] [Green Version]
- Tietge, U.J.F. The impact of myeloperoxidase on HDL function in myocardial infarction. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 137–142. [Google Scholar] [CrossRef]
- Chiesa, S.T.; Charakida, M. High-Density Lipoprotein Function and Dysfunction in Health and Disease. Cardiovasc. Drugs 2019, 33, 207–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, M.; Grossman, J.; Fitzgerald, J.; Dahlin-Lee, E.; Wallace, D.J.; Thong, B.Y.; Badsha, H.; Kalunian, K.; Charles, C.; Navab, M.; et al. Proinflammatory high-density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 2006, 54, 2541–2549. [Google Scholar] [CrossRef]
- Holzer, M.; Wolf, P.; Curcic, S.; Birner-Gruenberger, R.; Weger, W.; Inzinger, M.; El-Gamal, D.; Wadsack, C.; Heinemann, A.; Marsche, G. Psoriasis alters HDL composition and cholesterol efflux capacity. J. Lipid Res. 2012, 53, 1618–1624. [Google Scholar] [CrossRef] [Green Version]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the complexities of the HDL lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef] [Green Version]
- Giraud, C.; Tournadre, A.; Pereira, B.; Dutheil, F.; Soubrier, M.; Lhomme, M.; Kontush, A.; Sébédio, J.-L.; Capel, F. Alterations of HDL particle phospholipid composition and role of inflammation in rheumatoid arthritis. J. Physiol. Biochem. 2019, 75, 453–462. [Google Scholar] [CrossRef]
- Jury, E.C.; Kabouridis, P.S.; Flores-Borja, F.; Mageed, R.A.; Isenberg, D.A. Altered lipid raft-associated signaling and ganglioside expression in T lymphocytes from patients with systemic lupus erythematosus. J. Clin. Investig. 2004, 113, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Nambiar, M.P.; Warke, V.G.; Fisher, C.U.; Mitchell, J.; Delaney, N.; Tsokos, G.C. Alterations in lipid raft composition and dynamics contribute to abnormal T cell responses in systemic lupus erythematosus. J. Immunol. 2004, 172, 7821–7831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montecucco, F.; Favari, E.; Norata, G.D.; Ronda, N.; Nofer, J.R.; Vuilleumier, N. Impact of systemic inflammation and autoimmune diseases on apoA-I and HDL plasma levels and functions. Handb. Exp. Pharm. 2015, 224, 455–482. [Google Scholar]
- Greco, D.; Gualtierotti, R.; Agosti, P.; Adorni, M.P.; Ingegnoli, F.; Rota, M.; Bernini, F.; Meroni, P.L.; Ronda, N. Anti-atherogenic Modification of Serum Lipoprotein Function in Patients with Rheumatoid Arthritis after Tocilizumab Treatment, a Pilot Study. J. Clin. Med. 2020, 9, 2157. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, J.F.; Bonfa, E.; Borba, E.F. Systemic lupus erythematosus and lupus dyslipoproteinemia. Autoimmun. Rev. 2008, 7, 246–250. [Google Scholar] [CrossRef]
- Hahn, B.H.; Grossman, J.; Ansell, B.J.; Skaggs, B.J.; McMahon, M. Altered lipoprotein metabolism in chronic inflammatory states: Proinflammatory high-density lipoprotein and accelerated atherosclerosis in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, 213. [Google Scholar] [CrossRef] [Green Version]
- Toloza, S.M.A.; Uribe, A.G.; McGwin, G.; Alarcón, G.S.; Fessler, B.J.; Bastian, H.M.; Vilá, L.M.; Wu, R.; Shoenfeld, Y.; Roseman, J.M.; et al. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA). XXIII. Baseline predictors of vascular events. Arthritis Rheum. 2004, 50, 3947–3957. [Google Scholar]
- Gamal, S.M.; Fawzy, S.M.; Abdo, M.; Elgengehy, F.T.; Ghoniem, S.; Alkemry, A. Immunological profile and dyslipidemia in Egyptian Systemic Lupus Erythematosus patients. Egypt. Rheumatol. 2017, 39, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Li, L.; Wang, Z.; Song, W.; Zhang, Z. Dyslipidemia in patients with systemic lupus erythematosus: Association with disease activity and B-type natriuretic peptide levels. Biomed. Rep. 2016, 4, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Kiss, E.; Seres, I.; Tarr, T.; Kocsis, Z.; Szegedi, G.; Paragh, G. Reduced paraoxonase1 activity is a risk for atherosclerosis in patients with systemic lupus erythematosus. Ann. N. Y. Acad. Sci. 2007, 1108, 83–91. [Google Scholar] [CrossRef]
- Gaál, K.; Tarr, T.; Lőrincz, H.; Borbás, V.; Seres, I.; Harangi, M.; Fülöp, P.; Paragh, G. High-density lipopoprotein antioxidant capacity, subpopulation distribution and paraoxonase-1 activity in patients with systemic lupus erythematosus. Lipids Health Dis. 2016, 15, 60. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Skaggs, B.J.; Sahakian, L.; Grossman, J.; Fitzgerald, J.; Ragavendra, N.; Charles-Schoeman, C.; Chernishof, M.; Gorn, A.; Witztum, J.L.; et al. High plasma leptin levels confer increased risk of atherosclerosis in women with systemic lupus erythematosus, and are associated with inflammatory oxidised lipids. Ann. Rheum. Dis. 2011, 70, 1619–1624. [Google Scholar] [CrossRef] [PubMed]
- G., H.B.; Rao, V.S.; Kakkar, V.V. Friend Turns Foe: Transformation of Anti-Inflammatory HDL to Proinflammatory HDL during Acute-Phase Response. Cholesterol 2011, 2011, 274629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Lenten, B.J.; Wagner, A.C.; Nayak, D.P.; Hama, S.; Navab, M.; Fogelman, A.M. High-density lipoprotein loses its anti-inflammatory properties during acute influenza a infection. Circulation 2001, 103, 2283–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.K.; Vivekanandan-Giri, A.; Tang, C.; Knight, J.S.; Mathew, A.; Padilla, R.L.; Gillespie, B.W.; Carmona-Rivera, C.; Liu, X.; Subramanian, V.; et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: An additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. 2014, 66, 2532–2544. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.P.; Oeser, A.; Raggi, P.; Solus, J.F.; Avalos, I.; Linton, M.F.; Fazio, S.; Stein, C.M. Lipoprotein subclasses and particle size determined by nuclear magnetic resonance spectroscopy in systemic lupus erythematosus. Clin. Rheumatol. 2008, 27, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Su, J.; Svenungsson, E.; Hurt-Camejo, E.; Jensen-Urstad, K.; Angelin, B.; Båvenholm, P.; Frostegård, J. Dyslipidaemia and lipoprotein pattern in systemic lupus erythematosus (SLE) and SLE-related cardiovascular disease. Scand. J. Rheumatol. 2009, 38, 184–189. [Google Scholar] [CrossRef]
- Juárez-Rojas, J.; Medina-Urrutia, A.; Posadas-Sánchez, R.; Jorge-Galarza, E.; Mendoza-Pérez, E.; Caracas-Portilla, N.; Cardoso-Saldaña, G.; Muñoz-Gallegos, G.; Posadas-Romero, C. High-density lipoproteins are abnormal in young women with uncomplicated systemic lupus erythematosus. Lupus 2008, 17, 981–987. [Google Scholar] [CrossRef]
- Skaggs, B.J.; Hahn, B.H.; Sahakian, L.; Grossman, J.; McMahon, M. Dysfunctional, pro-inflammatory HDL directly upregulates monocyte PDGFRbeta, chemotaxis and TNFalpha production. Clin. Immunol. 2010, 137, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.K.; Seto, N.L.; Vivekanandan-Giri, A.; Yuan, W.; Playford, M.P.; Manna, Z.; Hasni, S.A.; Kuai, R.; Mehta, N.N.; Schwendeman, A.; et al. Lupus high-density lipoprotein induces proinflammatory responses in macrophages by binding lectin-like oxidised low-density lipoprotein receptor 1 and failing to promote activating transcription factor 3 activity. Ann. Rheum. Dis. 2017, 76, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-R.; Lee, E.-Y.; Park, J.K.; Song, Y.W.; Cho, K.-H. Patients with Rheumatoid Arthritis Show Altered Lipoprotein Profiles with Dysfunctional High-Density Lipoproteins that Can Exacerbate Inflammatory and Atherogenic Process. PLoS ONE 2016, 11, e0164564. [Google Scholar] [CrossRef] [PubMed]
- Myasoedova, E.; Crowson, C.S.; Kremers, H.M.; Roger, V.L.; Fitz-Gibbon, P.D.; Therneau, T.M.; Gabriel, S.E. Lipid paradox in rheumatoid arthritis: The impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Ann. Rheum. Dis. 2011, 70, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.P.; Oeser, A.; Raggi, P.; Gebretsadik, T.; Shintani, A.K.; Sokka, T.; Pincus, T.; Avalos, I.; Stein, C.M. Increased coronary-artery atherosclerosis in rheumatoid arthritis: Relationship to disease duration and cardiovascular risk factors. Arthritis Rheum. 2005, 52, 3045–3053. [Google Scholar] [CrossRef]
- Liao, K.P.; Cai, T.; Gainer, V.S.; Cagan, A.; Murphy, S.N.; Liu, C.; Churchill, S.; Shaw, S.Y.; Kohane, I.; Solomon, D.H.; et al. Lipid and lipoprotein levels and trend in rheumatoid arthritis compared to the general population. Arthritis Care Res. 2013, 65, 2046–2050. [Google Scholar] [CrossRef]
- Charles-Schoeman, C.; Watanabe, J.; Lee, Y.Y.; Furst, D.E.; Amjadi, S.; Elashoff, D.; Park, G.; McMahon, M.; Paulus, H.E.; Fogelman, A.M.; et al. Abnormal function of high-density lipoprotein is associated with poor disease control and an altered protein cargo in rheumatoid arthritis. Arthritis Rheum. 2009, 60, 2870–2879. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, J.; Charles-Schoeman, C.; Miao, Y.; Elashoff, D.; Lee, Y.Y.; Katselis, G.S.; Lee, T.D.; Reddy, S.T. Proteomic profiling following immunoaffinity capture of high-density lipoprotein: Association of acute-phase proteins and complement factors with proinflammatory high-density lipoprotein in rheumatoid arthritis. Arthritis Rheum. 2012, 64, 1828–1837. [Google Scholar] [CrossRef] [Green Version]
- Vivekanandan-Giri, A.; Slocum, J.L.; Byun, J.; Tang, C.; Sands, R.L.; Gillespie, B.W.; Heinecke, J.W.; Saran, R.; Kaplan, M.J.; Pennathur, S. High density lipoprotein is targeted for oxidation by myeloperoxidase in rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 1725–1731. [Google Scholar] [CrossRef] [PubMed]
- Rosso, L.G.; Lhomme, M.; Meroño, T.; Sorroche, P.; Catoggio, L.; Soriano, E.; Saucedo, C.; Malah, V.; Dauteuille, C.; Boero, L.; et al. Altered lipidome and antioxidative activity of small, dense HDL in normolipidemic rheumatoid arthritis: Relevance of inflammation. Atherosclerosis 2014, 237, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Tejera-Segura, B.; Macía-Díaz, M.; Machado, J.D.; De Vera-González, A.; García-Dopico, J.A.; Olmos, J.M.; Hernández, J.L.; Díaz-González, F.; González-Gay, M.A.; Ferraz-Amaro, I. HDL cholesterol efflux capacity in rheumatoid arthritis patients: Contributing factors and relationship with subclinical atherosclerosis. Arthritis Res. Ther. 2017, 19, 113. [Google Scholar] [CrossRef] [Green Version]
- Charles-Schoeman, C.; Lee, Y.Y.; Grijalva, V.; Amjadi, S.; Fitzgerald, J.; Ranganath, V.K.; Taylor, M.; McMahon, M.; E Paulus, H.; Reddy, S.T. Cholesterol efflux by high density lipoproteins is impaired in patients with active rheumatoid arthritis. Ann. Rheum. Dis. 2012, 71, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Ronda, N.; Favari, E.; Borghi, M.O.; Ingegnoli, F.; Gerosa, M.; Chighizola, C.; Zimetti, F.; Adorni, M.P.; Bernini, F.; Meroni, P.L. Impaired serum cholesterol efflux capacity in rheumatoid arthritis and systemic lupus erythematosus. Ann. Rheum. Dis. 2014, 73, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Tam, L.-S.; Tomlinson, B.; Chu, T.T.-W.; Li, M.; Leung, Y.-Y.; Kwok, L.-W.; Li, T.K.; Yu, T.; Zhu, Y.-E.; Wong, K.-C.; et al. Cardiovascular risk profile of patients with psoriatic arthritis compared to controls--the role of inflammation. Rheumatology 2008, 47, 718–723. [Google Scholar] [CrossRef] [Green Version]
- Mallbris, L.; Granath, F.; Hamsten, A.; Ståhle, M. Psoriasis is associated with lipid abnormalities at the onset of skin disease. J. Am. Acad. Derm. 2006, 54, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Borska, L.; Kremlacek, J.; Andrys, C.; Krejsek, J.; Hamakova, K.; Borsky, P.; Palicka, V.; Rehacek, V.; Malkova, A.; Fiala, Z. Systemic Inflammation, Oxidative Damage to Nucleic Acids, and Metabolic Syndrome in the Pathogenesis of Psoriasis. Int. J. Mol. Sci. 2017, 18, 2238. [Google Scholar] [CrossRef] [Green Version]
- Nakhwa, Y.C.; Rashmi, R.; Basavaraj, K.H. Dyslipidemia in Psoriasis: A Case Controlled Study. Int. Sch. Res. Not. 2014, 2014, 729157. [Google Scholar] [CrossRef] [Green Version]
- Pietrzak, A.; Chabros, P.; Grywalska, E.; Kicinski, P.; Pietrzak-Franciszkiewicz, K.; Krasowka, D.; Kandzierski, G. Serum lipid metabolism in psoriasis and psoriatic arthritis—An update. Arch. Med. Sci. Ams. 2019, 15, 369–375. [Google Scholar] [CrossRef]
- Pietrzak, A.; Grywalska, E.; Walankiewicz, M.; Lotti, T.; Roliński, J.; Myśliński, W.; Chabros, P.; Piekarska-Myślińska, D.; Reich, K. Psoriasis and metabolic syndrome in children: Current data. Clin. Exp. Derm. 2017, 42, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Veetil, B.M.A.; Matteson, E.L.; Maradit-Kremers, H.; McEvoy, M.T.; Crowson, C.S. Trends in lipid profiles in patients with psoriasis: A population-based analysis. BMC Derm. 2012, 12, 20. [Google Scholar]
- Uyanik, B.S.; Ari, Z.; Onur, E.; Gündüz, K.; Tanülkü, S.; Durkan, K. Serum lipids and apolipoproteins in patients with psoriasis. Clin. Chem. Lab. Med. 2002, 40, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.; Asha, K.; Sharma, S.B.; Arora, V.K.; Aggarwal, A. Dyslipidaemia & oxidative stress in patients of psoriasis: Emerging cardiovascular risk factors. Indian J. Med. Res. 2017, 146, 708–713. [Google Scholar]
- Miller, I.; Skaaby, T.; Ellervik, C.; Jemec, G. Quantifying cardiovascular disease risk factors in patients with psoriasis: A meta-analysis. Br. J. Derm. 2013, 169, 1180–1187. [Google Scholar] [CrossRef]
- Yu, Y.; Sheth, N.; Krishnamoorthy, P.; Saboury, B.; Raper, A.; Baer, A.; Ochotony, R.; Doveikis, J.; DerOhannessian, S.; Van Voorhees, A.S.; et al. Aortic vascular inflammation in psoriasis is associated with HDL particle size and concentration: A pilot study. Am. J. Cardiovasc. Dis. 2012, 2, 285–292. [Google Scholar] [PubMed]
- Tom, W.L.; Playford, M.P.; Admani, S.; Natarajan, B.; Joshi, A.A.; Eichenfield, L.F.; Mehta, N.N. Characterization of Lipoprotein Composition and Function in Pediatric Psoriasis Reveals a More Atherogenic Profile. J. Investig. Dermatol. 2016, 136, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Wolk, R.; Armstrong, E.J.; Hansen, P.R.; Thiers, B.; Lan, S.; Tallman, A.M.; Kaur, M.; Tatulych, S. Effect of tofacitinib on lipid levels and lipid-related parameters in patients with moderate to severe psoriasis. J. Clin. Lipidol. 2017, 11, 1243–1256. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Qin, S.; Dang, L.; Song, G.; Yao, S.; Yang, N.; Li, Y. Psoriasis decreases the anti-oxidation and anti-inflammation properties of high-density lipoprotein. Biochim. Biophys. Acta 2014, 1841, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Staniak, H.L.; Bittencourt, M.S.; Santos, I.D.S.; Sharovsky, R.; Sabbag, C.; Goulart, A.C.; Lotufo, P.A.; Benseñor, I.M. Association between psoriasis and coronary calcium score. Atherosclerosis 2014, 237, 847–852. [Google Scholar] [CrossRef] [Green Version]
- Holzer, M.; Wolf, P.; Inzinger, M.; Trieb, M.; Curcic, S.; Pasterk, L.; Weger, W.; Heinemann, A.; Marsche, G. Anti-psoriatic therapy recovers high-density lipoprotein composition and function. J. Investig. Dermatol. 2014, 134, 635–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, N.N.; Li, R.; Krishnamoorthy, P.; Yu, Y.; Farver, W.; Rodrigues, A.; Raper, A.; Wilcox, M.; Baer, A.; DerOhannesian, S.; et al. Abnormal lipoprotein particles and cholesterol efflux capacity in patients with psoriasis. Atherosclerosis 2012, 224, 218–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Huang, Y. Meta-analysis of the association between asthma and serum levels of high-density lipoprotein cholesterol and low-density lipoprotein cholesterol. Ann. Allergy Asthma Immunol. 2017, 118, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Barochia, A.V.; Gordon, E.M.; Kaler, M.; Cuento, R.A.; Theard, P.; Figueroa, D.M.; Yao, X.; Weir, N.A.; Sampson, M.L.; Stylianou, M.; et al. High density lipoproteins and type 2 inflammatory biomarkers are negatively correlated in atopic asthmatics. J. Lipid Res. 2017, 58, 1713–1721. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, D.; Fraser, S.; Oh, J.; Huber, A.M.; Schulman, Y.; Bhagtani, R.H.; Khan, Z.S.; Tesfa, L.; Hall, C.B.; Macian, F. Inflammation, metabolic dysregulation, and pulmonary function among obese urban adolescents with asthma. Am. J. Respir. Crit. Care Med. 2015, 191, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirillo, D.J.; Agrawal, Y.; Cassano, P.A. Lipids and pulmonary function in the Third National Health and Nutrition Examination Survey. Am. J. Epidemiol. 2002, 155, 842–848. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-W.; Lee, E.H.; Kim, H.J.; Bae, D.-J.; Han, S.; Kim, D.; Jang, A.S.; Uh, S.-T.; Kim, Y.H.; Erle, D.J.; et al. Apolipoprotein A1 potentiates lipoxin A4 synthesis and recovery of allergen-induced disrupted tight junctions in the airway epithelium. Clin. Exp. Allergy 2013, 43, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, T.; Ruhdorfer, S.; Weigl, L.; Wessner, D.; Heinrich, J.; Döring, A.; Wichmann, H.-E.; Ring, J. Intake of unsaturated fatty acids and HDL cholesterol levels are associated with manifestations of atopy in adults. Clin. Exp. Allergy 2003, 33, 1360–1367. [Google Scholar] [CrossRef]
- Agón-Banzo, P.J.; Sanmartin, R.; García-Malinis, A.J.; Hernández-Martín, Á.; Puzo, J.; Doste, D.; Pardos, C.; Gilaberte, Y. Body mass index and serum lipid profile: Association with atopic dermatitis in a paediatric population. Australas. J. Derm. 2020, 61, e60–e64. [Google Scholar] [CrossRef]
- Trieb, M.; Wolf, P.; Knuplez, E.; Weger, W.; Schuster, C.; Peinhaupt, M.; Holzer, M.; Trakaki, A.; Eichmann, T.; Lass, A.; et al. Abnormal composition and function of high-density lipoproteins in atopic dermatitis patients. Allergy 2019, 74, 398–402. [Google Scholar] [CrossRef] [Green Version]
- Norata, G.D.; Catapano, A.L. Molecular mechanisms responsible for the antiinflammatory and protective effect of HDL on the endothelium. Vasc. Health Risk Manag. 2005, 1, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norata, G.D.; Pirillo, A.; Catapano, A.L. HDLs, immunity, and atherosclerosis. Curr. Opin. Lipidol. 2011, 22, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Catapano, A.L. HDL and adaptive immunity: A tale of lipid rafts. Atherosclerosis 2012, 225, 34–35. [Google Scholar] [CrossRef] [PubMed]
- Bonacina, F.; Pirillo, A.; Catapano, A.L.; Norata, G.D. Cholesterol membrane content has a ubiquitous evolutionary function in immune cell activation: The role of HDL. Curr. Opin. Lipidol. 2019, 30, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Sala, F.; Catapano, A.L.; Norata, G.D. High density lipoproteins and atherosclerosis: Emerging aspects. J. Geriatr. Cardiol. 2012, 9, 401–407. [Google Scholar]
- Pirillo, A.; Catapano, A.L.; Norata, G.D. Biological Consequences of Dysfunctional HDL. Curr. Med. Chem. 2019, 26, 1644–1664. [Google Scholar] [CrossRef]
- Marsillach, J.; Becker, J.O.; Vaisar, T.; Hahn, B.H.; Brunzell, J.D.; Furlong, C.E.; De Boer, I.H.; McMahon, M.A.; Hoofnagle, A.N. DCCT/EDIC Research Group Paraoxonase-3 is depleted from the high-density lipoproteins of autoimmune disease patients with subclinical atherosclerosis. J. Proteome Res. 2015, 14, 2046–2054. [Google Scholar] [CrossRef] [Green Version]
- Barter, P.J.; Nicholls, S.; Rye, K.-A.; Anantharamaiah, G.M.; Navab, M.; Fogelman, A.M. Antiinflammatory properties of HDL. Circ. Res. 2004, 95, 764–772. [Google Scholar] [CrossRef]
- Riwanto, M.; Landmesser, U. High density lipoproteins and endothelial functions: Mechanistic insights and alterations in cardiovascular disease. J. Lipid. Res. 2013, 54, 3227–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norata, G.D.; Pirillo, A.; Catapano, A.L. Modified HDL: Biological and physiopathological consequences. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 371–386. [Google Scholar] [CrossRef]
- Callegari, E.; Norata, G.D.; Inoue, H.; Catapano, A.L. Oxidized-HDL3 modulates the expression of Cox-2 in human endothelial cells. Int. J. Mol. Med. 2006, 18, 209–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norata, G.D.; Banfi, C.; Pirillo, A.; Tremoli, E.; Hamsten, A.; Catapano, A.L.; Eriksson, P. Oxidised-HDL3 induces the expression of PAI-1 in human endothelial cells. Role of p38MAPK activation and mRNA stabilization. Br. J. Haematol. 2004, 127, 97–104. [Google Scholar] [PubMed]
- Munford, R.S.; Hall, C.L.; Lipton, J.M.; Dietschy, J.M. Biological activity, lipoprotein-binding behavior, and in vivo disposition of extracted and native forms of Salmonella typhimurium lipopolysaccharides. J. Clin. Invest. 1982, 70, 877–888. [Google Scholar] [CrossRef] [Green Version]
- Grunfeld, C.; Marshall, M.; Shigenaga, J.K.; Moser, A.H.; Tobias, P.; Feingold, K.R. Lipoproteins inhibit macrophage activation by lipoteichoic acid. J. Lipid. Res. 1999, 40, 245–252. [Google Scholar] [CrossRef]
- Gautier, T.; Lagrost, L. Plasma PLTP (phospholipid-transfer protein): An emerging role in ’reverse lipopolysaccharide transport’ and innate immunity. Biochem. Soc. Trans. 2011, 39, 984–988. [Google Scholar] [CrossRef] [Green Version]
- Hailman, E.; Albers, J.J.; Wolfbauer, G.; Tu, A.-Y.; Wright, S.D. Neutralization and transfer of lipopolysaccharide by phospholipid transfer protein. J. Biol. Chem. 1996, 271, 12172–12178. [Google Scholar] [CrossRef] [Green Version]
- Vesy, C.J.; Kitchens, R.L.; Wolfbauer, G.; Albers, J.J.; Munford, R.S. Lipopolysaccharide-binding protein and phospholipid transfer protein release lipopolysaccharides from gram-negative bacterial membranes. Infect. Immun. 2000, 68, 2410–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norata, G.D.; Marchesi, P.; Pirillo, A.; Uboldi, P.; Chiesa, G.; Maina, V.; Garlanda, C.; Mantovani, A.; Catapano, A.L. Long pentraxin 3, a key component of innate immunity, is modulated by high-density lipoproteins in endothelial cells. Arter. Thromb. Vasc. Biol. 2008, 28, 925–931. [Google Scholar] [CrossRef] [Green Version]
- Porte, R.; Davoudian, S.; Asgari, F.; Parente, R.; Mantovani, A.; Garlanda, C.; Bottazzi, B. The Long Pentraxin PTX3 as a Humoral Innate Immunity Functional Player and Biomarker of Infections and Sepsis. Front. Immunol. 2019, 10, 794. [Google Scholar] [CrossRef]
- Norata, G.D.; Marchesi, P.; Venu, V.K.P.; Pasqualini, F.; Anselmo, A.; Moalli, F.; Pizzitola, I.; Garlanda, C.; Mantovani, A.; Catapano, A.L. Deficiency of the long pentraxin PTX3 promotes vascular inflammation and atherosclerosis. Circulation 2009, 120, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Doni, A.; Musso, T.; Morone, D.; Bastone, A.; Zambelli, V.; Sironi, M.; Castagnoli, C.; Cambieri, I.; Stravalaci, M.; Pasqualini, F.; et al. An acidic microenvironment sets the humoral pattern recognition molecule PTX3 in a tissue repair mode. J. Exp. Med. 2015, 212, 905–925. [Google Scholar] [CrossRef]
- Bonacina, F.; Barbieri, S.; Cutuli, L.; Amadio, P.; Doni, A.; Sironi, M.; Tartari, S.; Mantovani, A.; Bottazzi, B.; Garlanda, C.; et al. Vascular pentraxin 3 controls arterial thrombosis by targeting collagen and fibrinogen induced platelets aggregation. Biochim. Biophys. Acta 2016, 1862, 1182–1190. [Google Scholar] [CrossRef]
- Bonacina, F.; Moregola, A.; Porte, R.; Baragetti, A.; Bonavita, E.; Salatin, A.; Grigore, L.; Pellegatta, F.; Molgora, M.; Sironi, M.; et al. Pentraxin 3 deficiency protects from the metabolic inflammation associated to diet-induced obesity. Cardiovasc. Res. 2019, 115, 1861–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norata, G.D.; Pirillo, A.; Ammirati, E.; Catapano, A.L. Emerging role of high density lipoproteins as a player in the immune system. Atherosclerosis 2012, 220, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Catapano, A.L.; Pirillo, A.; Bonacina, F.; Norata, G.D. HDL in innate and adaptive immunity. Cardiovasc. Res. 2014, 103, 372–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorci-Thomas, M.G.; Thomas, M.J. Microdomains, Inflammation, and Atherosclerosis. Circ. Res. 2016, 118, 679–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonacina, F.; Coe, D.; Wang, G.; Longhi, M.P.; Baragetti, A.; Moregola, A.; Garlaschelli, K.; Uboldi, P.; Pellegatta, F.; Grigore, L.; et al. Myeloid apolipoprotein E controls dendritic cell antigen presentation and T cell activation. Nat. Commun. 2018, 9, 3083. [Google Scholar] [CrossRef]
- Baragetti, A.; Bonacina, F.; Catapano, A.L.; Norata, G.D. Effect of lipids and lipoproteins on hematopoietic cell metabolism and commitment in atherosclerosis. Immunometabolism 2021, 3, e210014. [Google Scholar]
- Murphy, A.J.; Dragoljevic, D.; Tall, A.R. Cholesterol efflux pathways regulate myelopoiesis: A potential link to altered macrophage function in atherosclerosis. Front. Immunol. 2014, 5, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala, F.; Cutuli, L.; Grigore, L.; Pirillo, A.; Chiesa, G.; Catapano, A.L.; Norata, G.D. Prevalence of classical CD14++/CD16- but not of intermediate CD14++/CD16+ monocytes in hypoalphalipoproteinemia. Int. J. Cardiol. 2013, 168, 2886–2889. [Google Scholar] [CrossRef]
- Ruysschaert, J.M.; Lonez, C. Role of lipid microdomains in TLR-mediated signalling. Biochim. Biophys. Acta 2015, 1848, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Welch, C.; Pagler, T.A.; Ranalletta, M.; Lamkanfi, M.; Han, S.; Ishibashi, M.; Li, R.; Wang, N.; Tall, A.R. Increased inflammatory gene expression in ABC transporter-deficient macrophages: Free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation 2008, 118, 1837–1847. [Google Scholar] [CrossRef]
- De Nardo, D.; Labzin, L.I.; Kono, H.; Seki, R.; Schmidt, S.V.; Beyer, M.; Xu, D.; Zimmer, S.; Lahrmann, C.; Schildberg, F.A.; et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat. Immunol. 2014, 15, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Li, X.; Chavakis, T. Immunometabolic control of hematopoiesis. Mol. Asp. Med. 2021, 77, 100923. [Google Scholar] [CrossRef]
- Murphy, A.J.; Bijl, N.; Yvan-Charvet, L.; Welch, C.B.; Bhagwat, N.; Reheman, A.; Wang, Y.; Shaw, J.A.; Levine, R.L.; Ni, H.; et al. Cholesterol efflux in megakaryocyte progenitors suppresses platelet production and thrombocytosis. Nat. Med. 2013, 19, 586–594. [Google Scholar] [CrossRef] [Green Version]
- Dragoljevic, D.; Kraakman, M.J.; Nagareddy, P.R.; Ngo, D.; Shihata, W.; Kammoun, H.L.; Whillas, A.; Lee, M.K.S.; Al-Sharea, A.; Pernes, G.; et al. Defective cholesterol metabolism in haematopoietic stem cells promotes monocyte-driven atherosclerosis in rheumatoid arthritis. Eur. Heart J. 2018, 39, 2158–2167. [Google Scholar] [CrossRef] [Green Version]
- van der Vorst, E.P.C.; Theodorou, K.; Wu, Y.; Hoeksema, M.A.; Goossens, P.; Bursill, C.A.; Aliyev, T.; Huitema, L.F.A.; Tas, S.W.; Wolfs, I.M.J.; et al. High-Density Lipoproteins Exert Pro-inflammatory Effects on Macrophages via Passive Cholesterol Depletion and PKC-NF-kappaB/STAT1-IRF1 Signaling. Cell. Metab. 2017, 25, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Smoak, K.A.; Aloor, J.J.; Madenspacher, J.; Merrick, B.A.; Collins, J.B.; Zhu, X.; Cavigiolio, G.; Oda, M.N.; Parks, J.S.; Fessler, M.B. Myeloid differentiation primary response protein 88 couples reverse cholesterol transport to inflammation. Cell. Metab. 2010, 11, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Fotakis, P.; Kothari, V.; Thomas, D.G.; Westerterp, M.; Molusky, M.M.; Altin, E.; Abramowicz, S.; Wang, N.; He, Y.; Heinecke, J.W.; et al. Anti-Inflammatory Effects of HDL (High-Density Lipoprotein) in Macrophages Predominate Over Proinflammatory Effects in Atherosclerotic Plaques. Arter. Thromb. Vasc. Biol. 2019, 39, e253–e272. [Google Scholar] [CrossRef]
- Anderson, H.A.; Roche, P.A. MHC class II association with lipid rafts on the antigen presenting cell surface. Biochim. Biophys. Acta 2015, 1853, 775–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerterp, M.; Gautier, E.L.; Ganda, A.; Molusky, M.M.; Wang, W.; Fotakis, P.; Wang, N.; Randolph, G.J.; D’Agati, V.D.; Yvan-Charvet, L.; et al. Cholesterol Accumulation in Dendritic Cells Links the Inflammasome to Acquired Immunity. Cell. Metab. 2017, 25, 1294–1304 e1296. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.H.; Yuan, S.G.; Peng, D.Q.; Zhao, S.P. HDL and ApoA-I inhibit antigen presentation-mediated T cell activation by disrupting lipid rafts in antigen presenting cells. Atherosclerosis 2012, 225, 105–114. [Google Scholar] [CrossRef]
- Perrin-Cocon, L.; Diaz, O.; Carreras, M.; Dollet, S.; Guironnet-Paquet, A.; André, P.; Lotteau, V. High-density lipoprotein phospholipids interfere with dendritic cell Th1 functional maturation. Immunobiology 2012, 217, 91–99. [Google Scholar] [CrossRef]
- Tiniakou, I.; Drakos, E.; Sinatkas, V.; Van Eck, M.; Zannis, V.I.; Boumpas, D.; Verginis, P.; Kardassis, D. High-density lipoprotein attenuates Th1 and th17 autoimmune responses by modulating dendritic cell maturation and function. J. Immunol. 2015, 194, 4676–4687. [Google Scholar] [CrossRef] [Green Version]
- Perrin-Cocon, L.; Coutant, F.; Agaugué, S.; Deforges, S.; André, P.; Lotteau, V. Oxidized low-density lipoprotein promotes mature dendritic cell transition from differentiating monocyte. J. Immunol. 2001, 167, 3785–3791. [Google Scholar] [CrossRef] [Green Version]
- Coutant, F.; Agaugué, S.; Perrin-Cocon, L.; André, P.; Lotteau, V. Sensing environmental lipids by dendritic cell modulates its function. J. Immunol. 2004, 172, 54–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutant, F.; Miossec, P. Altered dendritic cell functions in autoimmune diseases: Distinct and overlapping profiles. Nat. Rev. Rheumatol. 2016, 12, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.A.; Waddington, K.E.; Pineda-Torra, I.; Jury, E.C. Transcriptional Regulation of T-Cell Lipid Metabolism: Implications for Plasma Membrane Lipid Rafts and T-Cell Function. Front. Immunol. 2017, 8, 1636. [Google Scholar] [CrossRef] [Green Version]
- Mailer, R.K.W.; Gisterå, A.; Polyzos, K.A.; Ketelhuth, D.F.J.; Hansson, G.K. Hypercholesterolemia Enhances T Cell Receptor Signaling and Increases the Regulatory T Cell Population. Sci. Rep. 2017, 7, 15655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Ballester, M.; Herrero-Cervera, A.; Vinué, Á.; Martínez-Hervás, S.; González-Navarro, H. Impact of Cholesterol Metabolism in Immune Cell Function and Atherosclerosis. Nutrients 2020, 12, 2021. [Google Scholar]
- Jury, E.C.; Flores-Borja, F.; Kabouridis, P.S. Lipid rafts in T cell signalling and disease. Semin. Cell. Dev. Biol. 2007, 18, 608–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.J.; Ong, K.L.; Shrestha, S.; Chen, K.; Tabet, F.; Barter, P.J.; Rye, K.-A. Inhibition of arthritis in the Lewis rat by apolipoprotein A-I and reconstituted high-density lipoproteins. Arter. Thromb. Vasc. Biol. 2014, 34, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Tavori, H.; Su, Y.R.; Yancey, P.G.; Giunzioni, I.; Wilhelm, A.J.; Blakemore, J.L.; Zabalawi, M.; Linton, M.F.; Sorci-Thomas, M.G.; Fazio, S. Macrophage apoAI protects against dyslipidemia-induced dermatitis and atherosclerosis without affecting HDL. J. Lipid. Res. 2015, 56, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Ait-Oufella, H.; Salomon, B.L.; Potteaux, S.; Robertson, A.-K.L.; Gourdy, P.; Zoll, J.; Merval, R.; Esposito, B.; Cohen, J.L.; Fisson, S.; et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 2006, 12, 178–180. [Google Scholar] [CrossRef]
- Klingenberg, R.; Gerdes, N.; Badeau, R.M.; Gisterå, A.; Strodthoff, D.; Ketelhuth, D.F.J.; Lundberg, A.M.; Rudling, M.; Nilsson, S.K.; Olivecrona, G.; et al. Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J. Clin. Invest. 2013, 123, 1323–1334. [Google Scholar] [CrossRef]
- Cheng, H.-Y.; Gaddis, D.E.; Wu, R.; McSkimming, C.; Haynes, L.D.; Taylor, A.M.; McNamara, C.A.; Sorci-Thomas, M.; Hedrick, C.C. Loss of ABCG1 influences regulatory T cell differentiation and atherosclerosis. J. Clin. Invest. 2016, 126, 3236–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, A.J.; Zabalawi, M.; Owen, J.S.; Shah, D.; Grayson, J.M.; Major, A.S.; Bhat, S.; Gibbs, D.P., Jr.; Thomas, M.J.; Sorci-Thomas, M.G. Apolipoprotein A-I modulates regulatory T cells in autoimmune LDLr-/-, ApoA-I-/- mice. J. Biol. Chem. 2010, 285, 36158–36169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaddis, D.E.; Padgett, L.E.; Wu, R.; McSkimming, C.; Romines, V.; Taylor, A.M.; McNamara, C.A.; Kronenberg, M.; Crotty, S.; Thomas, M.J.; et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat. Commun. 2018, 9, 1095. [Google Scholar] [CrossRef]
- Rueda, C.M.; Rodríguez-Perea, A.L.; Moreno-Fernandez, M.; Jackson, C.M.; Melchior, J.T.; Davidson, W.S.; Chougnet, C.A. High density lipoproteins selectively promote the survival of human regulatory T cells. J. Lipid Res. 2017, 58, 1514–1523. [Google Scholar] [CrossRef] [Green Version]
- Rosenson, R.S.; Jr, H.B.B.; Barter, P.J.; Björkegren, J.L.M.; Chapman, M.J.; Gaudet, D.; Kim, D.S.; Niesor, E.; Rye, K.-A.; Sacks, F.M.; et al. HDL and atherosclerotic cardiovascular disease: Genetic insights into complex biology. Nat. Rev. Cardiol. 2018, 15, 9–19. [Google Scholar] [CrossRef]
- Bochem, A.E.; Van Der Valk, F.M.; Tolani, S.; Stroes, E.S.; Westerterp, M.; Tall, A.R. Increased systemic and plaque inflammation in ABCA1 mutation carriers with attenuation by statins. Arter. Thromb. Vasc. Biol. 2015, 35, 1663–1669. [Google Scholar] [CrossRef] [Green Version]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; Gemert, S.L.B.-V.; Wang, N.; et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef] [Green Version]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Hólm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Harslof, M.; Pedersen, K.M.; Nordestgaard, B.G.; Afzal, S. Low HDL (High-Density Lipoprotein) Cholesterol and High White Blood Cell Counts: A Mendelian Randomization Study. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Trinder, M.; Walley, K.R.; Boyd, J.H.; Brunham, L.R. Causal Inference for Genetically Determined Levels of High-Density Lipoprotein Cholesterol and Risk of Infectious Disease. Arter. Thromb. Vasc. Biol. 2020, 40, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Genga, K.R.; Trinder, M.; Kong, H.J.; Li, X.; Leung, A.K.K.; Shimada, T.; Walley, K.R.; Russell, J.A.; Francis, G.A.; Brunham, L.R.; et al. CETP genetic variant rs1800777 (allele A) is associated with abnormally low HDL-C levels and increased risk of AKI during sepsis. Sci. Rep. 2018, 8, 16764. [Google Scholar] [CrossRef] [Green Version]
- Blauw, L.L.; Wang, Y.; van Dijk, K.W.; Rensen, P.C. A novel role for CETP as immunological gatekeeper: Raising HDL to cure sepsis? Trends Endocrinol. Metab. Tem 2020, 31, 334–343. [Google Scholar] [CrossRef]
- Petropoulou, P.-I.; Berbée, J.F.; Theodoropoulos, V.; Hatziri, A.; Stamou, P.; Karavia, E.A.; Spyridonidis, A.; Karagiannides, I.; Kypreos, K.E. Lack of LCAT reduces the LPS-neutralizing capacity of HDL and enhances LPS-induced inflammation in mice. Biochim. Biophys. Acta 2015, 1852, 2106–2115. [Google Scholar] [CrossRef] [Green Version]
- Valanti, E.K.; Dalakoura-Karagkouni, K.; Sanoudou, D. Current and Emerging Reconstituted HDL-apoA-I and HDL-apoE Approaches to Treat Atherosclerosis. J. Pers. Med. 2018, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmore, S.F.; Carpenter, T.S.; Ingólfsson, H.I.; Peters, S.K.G.; Henderson, P.T.; Blanchette, C.D.; Fischer, N.O. Lipid composition dictates serum stability of reconstituted high-density lipoproteins: Implications for in vivo applications. Nanoscale 2018, 10, 7420–7430. [Google Scholar] [CrossRef]
- Van Linthout, S.; Frias, M.; Singh, N.; De Geest, B. Therapeutic potential of HDL in cardioprotection and tissue repair. Handb Exp. Pharm. 2015, 224, 527–565. [Google Scholar]
- Nieuwdorp, M.; Vergeer, M.; Bisoendial, R.J.; Roodt, J.O.‘T.; Levels, H.; Birjmohun, R.S.; Kuivenhoven, J.A.; Basser, R.; Rabelink, T.J.; Kastelein, J.J.P.; et al. Reconstituted HDL infusion restores endothelial function in patients with type 2 diabetes mellitus. Diabetologia 2008, 51, 1081–1084. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Drew, B.G.; Nakhla, S.; Duffy, S.J.; Murphy, A.J.; Barter, P.J.; Rye, K.-A.; Chin-Dusting, J.; Hoang, A.; Sviridov, D.; et al. Reconstituted high-density lipoprotein increases plasma high-density lipoprotein anti-inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J. Am. Coll. Cardiol. 2009, 53, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Shaw, J.A.; Bobik, A.; Murphy, A.; Kanellakis, P.; Blombery, P.; Mukhamedova, N.; Woollard, K.; Lyon, S.; Sviridov, D.; Dart, A.M. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ. Res. 2008, 103, 1084–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissen, S.E.; Tsunoda, T.; Tuzcu, E.M.; Schoenhagen, P.; Cooper, C.J.; Yasin, M.; Eaton, G.M.; Lauer, M.A.; Sheldon, W.S.; Grines, C.L. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: A randomized controlled trial. JAMA 2003, 290, 2292–2300. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Grégoire, J.; L’Allier, P.L.; Ibrahim, R.; Lespérance, J.; Heinonen, T.M.; Kouz, S.; Berry, C.; Basser, R.; Lavoie, M.-A.; et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: A randomized controlled trial. JAMA 2007, 297, 1675–1682. [Google Scholar] [CrossRef] [Green Version]
- Trinder, M.; Wang, Y.; Madsen, C.M.; Ponomarev, T.; Bohunek, L.; Daisely, B.A.; Kong, H.J.; Blauw, L.L.; Nordestgaard, B.G.; Tybjærg-Hansen, A.; et al. Inhibition of Cholesteryl Ester Transfer Protein Preserves High-Density Lipoprotein Cholesterol and Improves Survival in Sepsis. Circulation 2021, 143, 921–934. [Google Scholar] [CrossRef]
- Trinder, M.; Genga, K.R.; Kong, H.J.; Blauw, L.L.; Lo, C.; Li, X.; Cirstea, M.; Wang, Y.; Rensen, P.C.N.; Russell, J.A.; et al. Cholesteryl Ester Transfer Protein Influences High-Density Lipoprotein Levels and Survival in Sepsis. Am. J. Respir. Crit. Care Med. 2019, 199, 854–862. [Google Scholar] [CrossRef]
- Ossoli, A.; Simonelli, S.; Varrenti, M.; Morici, N.; Oliva, F.; Stucchi, M.; Gomaraschi, M.; Strazzella, A.; Arnaboldi, L.; Thomas, M.J.; et al. Recombinant LCAT (Lecithin:Cholesterol Acyltransferase) Rescues Defective HDL (High-Density Lipoprotein)-Mediated Endothelial Protection in Acute Coronary Syndrome. Arter. Thromb. Vasc. Biol. 2019, 39, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, A.V.; Karathanasis, S.K.; Yang, Z.; Freeman, L.; Kotani, K.; Remaley, A.T. COVID-19-Associated dyslipidemia: Implications for mechanism of impaired resolution and novel therapeutic approaches. FASEB J. 2020, 34, 9843–9853. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, Q.; Zhao, X.; Dong, H.; Wu, C.; Wu, F.; Yu, B.; Lv, J.; Zhang, S.; Wu, G.; et al. Low high-density lipoprotein level is correlated with the severity of COVID-19 patients: An observational study. Lipids Health Dis. 2020, 19, 204. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chen, D.; Wu, L.; He, G.; Ye, W. Declined serum high density lipoprotein cholesterol is associated with the severity of COVID-19 infection. Clin. Chim. Acta 2020, 510, 105–110. [Google Scholar] [CrossRef]
- Wei, X.; Zeng, W.; Su, J.; Wan, H.; Yu, X.; Cao, X.; Tan, W.; Wang, H. Hypolipidemia is associated with the severity of COVID-19. J. Clin. Lipidol. 2020, 14, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Begue, F.; Tanaka, S.; Mouktadi, Z.; Rondeau, P.; Veeren, B.; Diotel, N.; Tran-Dinh, A.; Robert, T.; Vélia, E.; Mavingui, P.; et al. Altered high-density lipoprotein composition and functions during severe COVID-19. Sci. Rep. 2021, 11, 2291. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Autoimmune Disease | HDL Alterations (Level/Function) | Reference |
|---|---|---|
| Systemic lupus erythematosus | Reduced levels of HDL-C | de Carvalho JF et al., 2008 [31]; Hahn BH et al., 2008 [32]; Toloza SMA et al., 2004 [33]; Gamal SM et al., 2017 [34] |
| No difference in HDL-C levels | ||
| “Lupus dyslipoproteinemia” | Yuan J et al., 2016 [35] | |
| Reduced PON1 activity | Kiss E et al., 2007 [36]; Gaal K et al., 2011 [37] | |
| Pro-inflammatory HDL (piHDL) with increased SAA content, decreased apoA-I levels | McMahon M et al., 2011 [38]; G HB et al., 2011 [39]; Van Lenten BJ et al., 2001 [40] | |
| Oxidized HDL | Smith CK et al., 2014 [41] | |
| Reduced HDL particles/size distribution | Chung CP et al., 2008 [42]; Hua X et al., 2009 [43]; Juarez-Rojas J et al., 2008 [44] | |
| Reduced CEC | Smith CK et al., 2014 [41]; Skaggs BJ et al., 2010 [45] | |
| Reduced anti-inflammatory potential | Smith CK et al., 2016 [46] | |
| Rheumatoid arthritis | Reduced levels of HDL-C | Jae-Yong Kim et al., 2016 [47]; Myasoedova E et al., 2011 [48] |
| No difference in HDL-C | Chung CP et al., 2005 [49]; Liao KP et al., 2013 [50] | |
| Pro-inflammatory HDL (piHDL) | Hahn BH et al., 2008 [32]; Charles-Schoeman C et al., 2009 [51]; Watanabe J et al., 2012 [52] | |
| Oxidized HDL | Vivekanandan-Giri A et al., 2013 [53] | |
| Reduced anti-oxidative activity | Gomez Rosso L et al., 2014 (in patients with active disease) [54] | |
| Reduced CEC | Beatriz Tejera-Segura et al., 2017 (in patients with low or moderate disease activity compared to patients in remission) [55]; Charles-Schoeman C et al., 2012 [56]; Ronda N et al., 2014 [57] | |
| Psoriasis | Increased HDL-C levels | Tam LS et al., 2008 [58]; Mallbris L et al., 2006 [59]; Borska L et al., 2017 [60]; Nakhawa YC et al., 2014 [61] |
| Decreased HDL-C levels | Pietrzak A et al., 2019 [62]; Pietrzak A et al., 2017 [63]; Akkara Veetil BM et al., 2012 [64] | |
| Unchanged HDL-C levels | Uyanik BS et al., 2002 [65]; Asha K et al., 2017 [66]; Miller IM et al., 2013 [67] | |
| Change in HDL particle size | Yu Y et al., 2012 [68]; Tom WL et al., 2016 [69]; Wolk R et al., 2017 [70] | |
| Pro-inflammatory HDL (piHDL) | He L et al., 2014 [71]; Staniak HL et al., 2014 [72] | |
| Reduced CEC | Holzer M et al., 2012 [24]; Tom WL et al., 2016 [69]; Holzer M et al., 2014 [73]; Mehta NN et al., 2012 [74] | |
| Chron’s disease | Decreased HDL-C levels and biochemical changes in HDL particles | Romanato G et al., 2009 [10] |
| Allergic asthma | Lower HDL levels (only children) | Peng J et al., 2017 [75] |
| Inverse correlation between HDL-C levels and circulating eosinophils and monocytes | Barochia AV et al., 2017 [76]; Rastogi D et al., 2015 [77] | |
| Positive correlation of serum apoA-I levels with less severe airflow obstruction in asthmatic individuals | Cirillo DJ et al., 2002 [78] | |
| ApoA-I levels decreased in bronchoalveolar lavage fluid of patients with moderate to severe asthma | Park SW et al., 2013 [79] | |
| Atopic dermatitis | Increased HDL-C levels in patients | Schäfer T et al., 2003 [80] |
| No difference in a paediatric population | Agón-Banzo PJ et al., 2020 [81] | |
| HDL enrichment in apoA-II, SAA, and phosphatidylinositol and significant reduction in the content of apoC-III, apoE, cholesteryl ester, free cholesterol, lysophosphatidylcholine, and phosphatidylethanolamine | Trieb et al., 2019 [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonacina, F.; Pirillo, A.; Catapano, A.L.; Norata, G.D. HDL in Immune-Inflammatory Responses: Implications beyond Cardiovascular Diseases. Cells 2021, 10, 1061. https://doi.org/10.3390/cells10051061
Bonacina F, Pirillo A, Catapano AL, Norata GD. HDL in Immune-Inflammatory Responses: Implications beyond Cardiovascular Diseases. Cells. 2021; 10(5):1061. https://doi.org/10.3390/cells10051061
Chicago/Turabian StyleBonacina, Fabrizia, Angela Pirillo, Alberico L. Catapano, and Giuseppe D. Norata. 2021. "HDL in Immune-Inflammatory Responses: Implications beyond Cardiovascular Diseases" Cells 10, no. 5: 1061. https://doi.org/10.3390/cells10051061
APA StyleBonacina, F., Pirillo, A., Catapano, A. L., & Norata, G. D. (2021). HDL in Immune-Inflammatory Responses: Implications beyond Cardiovascular Diseases. Cells, 10(5), 1061. https://doi.org/10.3390/cells10051061