
Crosstalk between Interleukin-1β and Type I Interferons Signaling in Autoinflammatory Diseases


Abstract
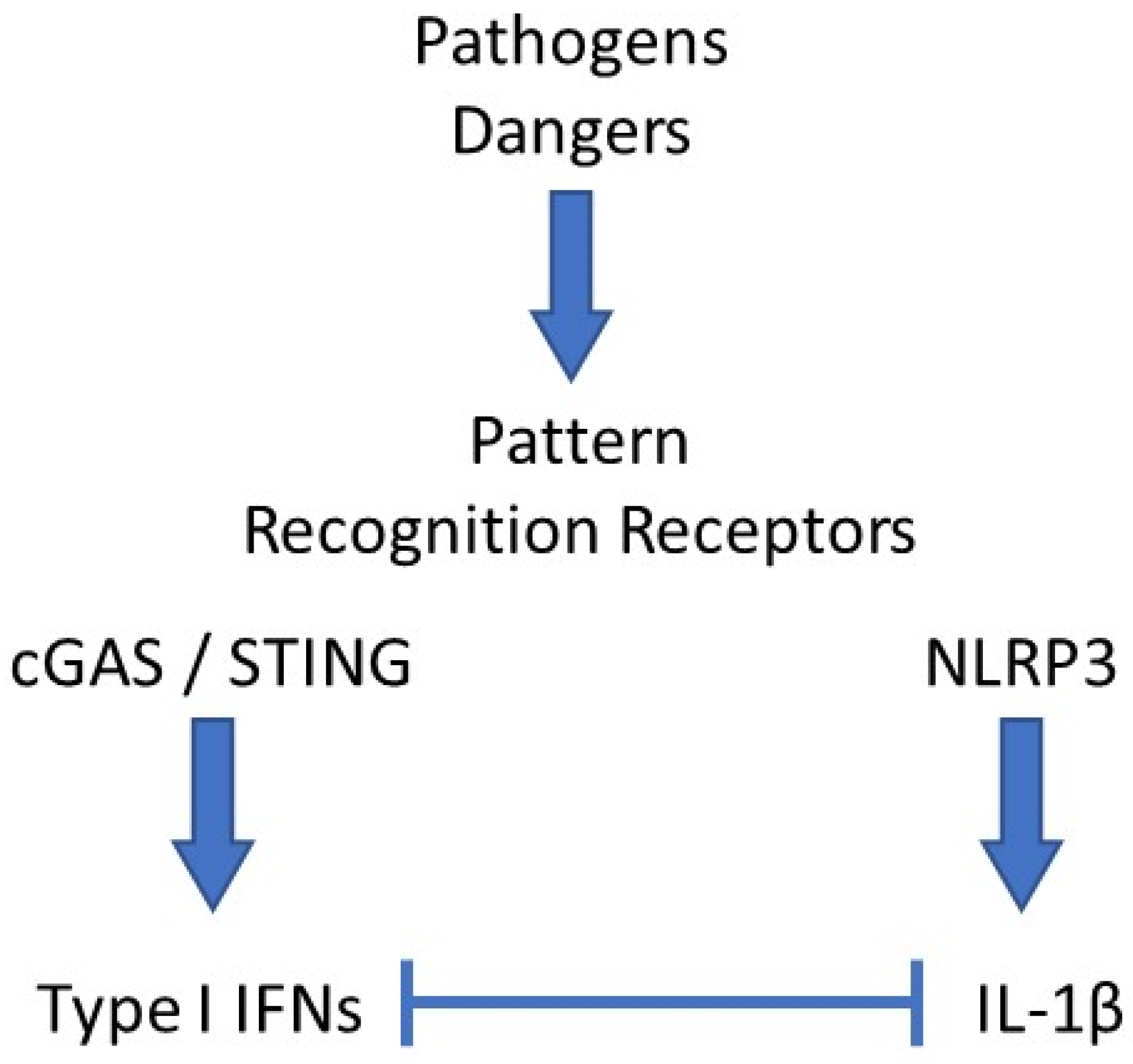
:1. Introduction
2. Type I IFNs in Autoinflammation
3. IL-1β in Autoinflammation
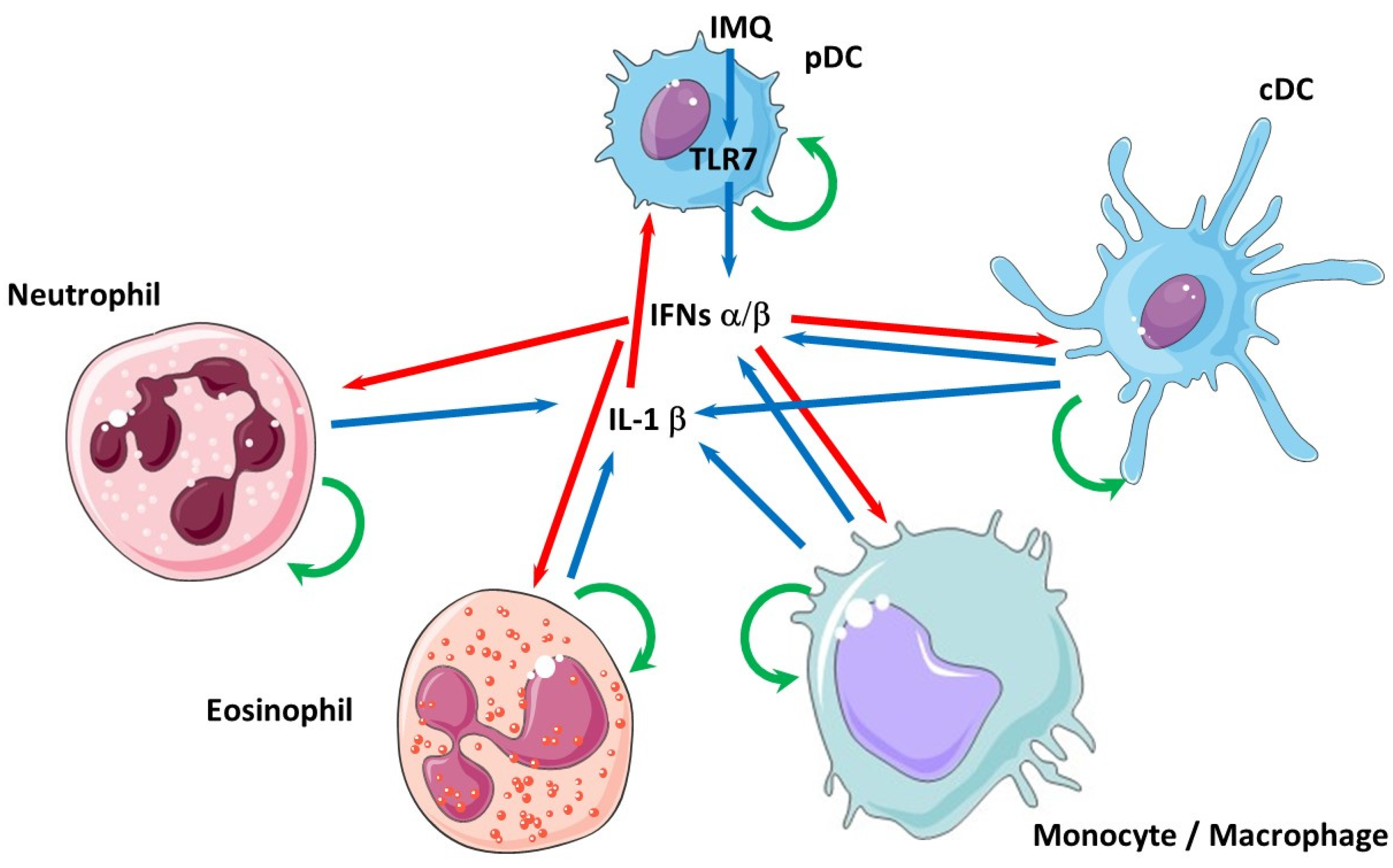
4. Interplay between Type I IFNs and IL-1β in Inflammatory/Autoimmune Diseases
5. Therapeutic Consequences
5.1. Targeting Type I IFNs in Il-1β-Dependent Diseases
5.2. Targeting IL-1β in Interferonopathies
6. Perspectives
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ma, J.; Zhao, S.; Gao, X.; Wang, R.; Liu, J.; Zhou, X.; Zhou, Y. The Roles of Inflammasomes in Host Defense against Mycobacterium tuberculosis. Pathogens 2021, 10, 120. [Google Scholar] [CrossRef]
- Schoggins, J.W. Recent advances in antiviral interferon-stimulated gene biology. F1000Research 2018, 7, 309. [Google Scholar] [CrossRef] [Green Version]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Li, S.-F.; Gong, M.-J.; Zhao, F.-R.; Shao, J.-J.; Xie, Y.-L.; Zhang, Y.-G.; Chang, H.-Y. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell. Physiol. Biochem. 2018, 51, 2377–2396. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Smith, A.; Jurado, K.A.; Iwasaki, A.; Garlick, J.A.; Kagan, J.C. An Antiviral Branch of the IL-1 Signaling Pathway Restricts Immune-Evasive Virus Replication. Mol. Cell 2018, 71, 825–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarreberg, L.D.; Wilkins, C.; Ramos, H.J.; Green, R.; Davis, M.A.; Chow, K.; Gale, M. Interleukin-1beta Signaling in Dendritic Cells Induces Antiviral Interferon Responses. mBio 2018, 9, 2. [Google Scholar]
- Jamilloux, Y.; Henry, T.; Belot, A.; Viel, S.; Fauter, M.; El Jammal, T.; Walzer, T.; François, B.; Sève, P. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun. Rev. 2020, 19, 102567. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [Green Version]
- Land, W.G. The Role of Damage-Associated Molecular Patterns in Human Diseases: Part I—Promoting inflammation and immunity. Sultan Qaboos Univ. Med. J. 2015, 15, e9–e21. [Google Scholar]
- Land, W.G. The Role of Damage-Associated Molecular Patterns (DAMPs) in Human Diseases: Part II: DAMPs as diagnostics, prognostics and therapeutics in clinical medicine. Sultan Qaboos Univ. Med. J. 2015, 15, e157–e170. [Google Scholar] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Moghaddas, F.; Masters, S.L. Monogenic autoinflammatory diseases: Cytokinopathies. Cytokine 2015, 74, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Szymczak, F.; Colli, M.L.; Mamula, M.J.; Evans-Molina, C.; Eizirik, D.L. Gene expression signatures of target tissues in type 1 diabetes, lupus erythematosus, multiple sclerosis, and rheumatoid arthritis. Sci. Adv. 2021, 7, eabd7600. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.F.; Rody, T.; Forny-Germano, L.; Cerdeiro, S.; Bellio, M.; Ferreira, S.T.; Munoz, D.P.; De Felice, F.G. Interleukin-1beta mediates alterations in mitochondrial fusion/fission proteins and memory impairment induced by amyloid-beta oligomers. J. Neuroinflam. 2021, 18, 54. [Google Scholar] [CrossRef]
- Mariotte, A.; De Cauwer, A.; Po, C.; Abou-Faycal, C.; Pichot, A.; Paul, N.; Aouadi, I.; Carapito, R.; Frisch, B.; Macquin, C.; et al. A mouse model of MSU-induced acute inflammation in vivo suggests imiquimod-dependent targeting of Il-1beta as relevant therapy for gout patients. Theranostics 2020, 10, 2158–2171. [Google Scholar] [CrossRef]
- Nehmar, R.; Alsaleh, G.; Voisin, B.; Flacher, V.; Mariotte, A.; Saferding, V.; Puchner, A.; Niederreiter, B.; Vandamme, T.; Schabbauer, G.; et al. Therapeutic Modulation of Plasmacytoid Dendritic Cells in Experimental Arthritis. Arthritis Rheumatol. 2017, 69, 2124–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. Ser. B Boil. Sci. 1957, 147, 258–267. [Google Scholar]
- Duncan, C.J.; Randall, R.E.; Hambleton, S. Genetic Lesions of Type I Interferon Signalling in Human Antiviral Immunity. Trends Genet. 2021, 37, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Reich, N.C. Too much of a good thing: Detrimental effects of interferon. Semin. Immunol. 2019, 43, 101282. [Google Scholar] [CrossRef] [PubMed]
- Melki, I.; Rose, Y.; Uggenti, C.; Van Eyck, L.; Frémond, M.-L.; Kitabayashi, N.; Rice, G.I.; Jenkinson, E.M.; Boulai, A.; Jeremiah, N.; et al. Disease-associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J. Allergy Clin. Immunol. 2017, 140, 543–552.e5. [Google Scholar] [CrossRef] [Green Version]
- Volpi, S.; Insalaco, A.; Caorsi, R.; Santori, E.; Messia, V.; Sacco, O.; Terheggen-Lagro, S.; Cardinale, F.; Scarselli, A.; Pastorino, C.; et al. Efficacy and Adverse Events During Janus Kinase Inhibitor Treatment of SAVI Syndrome. J. Clin. Immunol. 2019, 39, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Kretschmer, S.; Lee-Kirsch, M.A. Type I interferon-mediated autoinflammation and autoimmunity. Curr. Opin. Immunol. 2017, 49, 96–102. [Google Scholar] [CrossRef]
- Savic, S.; Caseley, E.A.; McDermott, M.F. Moving towards a systems-based classification of innate immune-mediated diseases. Nat. Rev. Rheumatol. 2020, 16, 222–237. [Google Scholar] [CrossRef]
- Gupta, S.; Kaplan, M.J. Bite of the wolf: Innate immune responses propagate autoimmunity in lupus. J. Clin. Investig. 2021, 131, e144918. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and Granulopoiesis Signatures in Systemic Lupus Erythematosus Blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Mai, L.; Asaduzzaman, A.; Noamani, B.; Fortin, P.R.; Gladman, D.D.; Touma, Z.; Urowitz, M.B.; Wither, J. The baseline interferon signature predicts disease severity over the subsequent 5 years in systemic lupus erythematosus. Arthritis Res. 2021, 23, 29. [Google Scholar] [CrossRef]
- Nehar-Belaid, D.; Hong, S.; Marches, R.; Chen, G.; Bolisetty, M.; Baisch, J.; Walters, L.; Punaro, M.; Rossi, R.J.; Chung, C.-H.; et al. Mapping systemic lupus erythematosus heterogeneity at the single-cell level. Nat. Immunol. 2020, 21, 1094–1106. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.-C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Manca, M.L.; Angelotti, F.; Melillo, D.; Pratesi, F.; Puxeddu, I.; Boraschi, D.; Migliorini, P. IL-1 family cytokines and soluble receptors in systemic lupus erythematosus. Arthritis Res. 2018, 20, 27. [Google Scholar] [CrossRef] [Green Version]
- Cafarelli, F.; Coladonato, L.; Lopalco, G.; Cacciapaglia, F.; Cantarini, L.; Iannone, F. Successful treatment with anakinra of refractory pericarditis in systemic lupus erythematosus. Clin. Exp. Rheumatol. 2020, 39, 227. [Google Scholar]
- Kubler, L.; Bittmann, I.; Kuipers, J.G. Macrophage activation syndrome triggered by active systemic lupus erythematosus: Successful treatment by interleukin-1 inhibition (anakinra). Z. Rheumatol. 2020, 79, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Kanneganti, T.-D. Inflammasome activation and assembly at a glance. J. Cell Sci. 2017, 130, 3955–3963. [Google Scholar] [CrossRef] [Green Version]
- Sönmez, H.E.; Özen, S. A clinical update on inflammasomopathies. Int. Immunol. 2017, 29, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Malcova, H.; Strizova, Z.; Milota, T.; Striz, I.; Sediva, A.; Cebecauerova, D.; Horvath, R. IL-1 Inhibitors in the Treatment of Monogenic Periodic Fever Syndromes: From the Past to the Future Perspectives. Front. Immunol. 2021, 11, 619257. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, M.; Crupi, R.; Di Paola, R.; Ontario, M.L.; Bella, R.; Calabrese, E.J.; Crea, R.; Cuzzocrea, S.; Calabrese, V. Inflammasomes, hormesis, and antioxidants in neuroinflammation: Role of NRLP3 in Alzheimer disease. J. Neurosci. Res. 2017, 95, 1360–1372. [Google Scholar] [CrossRef]
- Camilli, G.; Bohm, M.; Piffer, A.C.; Lavenir, R.; Williams, D.L.; Neven, B.; Grateau, G.; Georgin-Lavialle, S.; Quintin, J. beta-Glucan-induced reprogramming of human macrophages inhibits NLRP3 inflammasome activation in cryopyrinopathies. J. Clin. Investig. 2020, 130, 4561–4573. [Google Scholar] [CrossRef]
- Dilger, R.N.; Johnson, R.W. Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J. Leukoc. Biol. 2008, 84, 932–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musella, A.; Fresegna, D.; Rizzo, F.R.; Gentile, A.; De Vito, F.; Caioli, S.; Guadalupi, L.; Bruno, A.; Dolcetti, E.; Buttari, F.; et al. ’Prototypical’ proinflammatory cytokine (IL-1) in multiple sclerosis: Role in pathogenesis and therapeutic targeting. Expert Opin. Targets 2020, 24, 37–46. [Google Scholar] [CrossRef]
- McGinley, P.M.; Goldschmidt, C.H.; Rae-Grant, A.D. Diagnosis and Treatment of Multiple Sclerosis: A Review. JAMA 2021, 325, 765–779. [Google Scholar] [CrossRef]
- Chisari, C.G.; Sgarlata, E.; Arena, S.; Toscano, S.; Luca, M.; Patti, F. Rituximab for the treatment of multiple sclerosis: A review. J. Neurol. 2021, 1–25. [Google Scholar]
- Masanneck, L.; Eichler, S.; Vogelsang, A.; Korsen, M.; Wiendl, H.; Budde, T.; Meuth, S.G. The STING-IFN-beta-Dependent Axis Is Markedly Low in Patients with Relapsing-Remitting Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 9249. [Google Scholar] [CrossRef] [PubMed]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Förster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I Interferon Inhibits Interleukin-1 Production and Inflammasome Activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer-Barber, K.D.; Yan, B. Clash of the Cytokine Titans: Counter-regulation of interleukin-1 and type I interferon-mediated inflammatory responses. Cell. Mol. Immunol. 2017, 14, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludigs, K.; Parfenov, V.; Du Pasquier, R.A.; Guarda, G. Type I IFN-mediated regulation of IL-1 production in inflammatory disorders. Cell. Mol. Life Sci. 2012, 69, 3395–3418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kempen, T.S.; Wenink, M.H.; Leijten, E.F.; Radstake, T.R.; Boes, M. Perception of self: Distinguishing autoimmunity from autoinflammation. Nat. Rev. Rheumatol. 2015, 11, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Costa, C.; Eixarch, H.; Keller, C.W.; Amman, L.; Martínez-Banaclocha, H.; Midaglia, L.; Sarró, E.; Machín-Díaz, I.; Villar, L.M.; et al. NLRP3 inflammasome as prognostic factor and therapeutic target in primary progressive multiple sclerosis patients. Brain 2020, 143, 1414–1430. [Google Scholar] [CrossRef]
- Piancone, F.; Saresella, M.; Marventano, I.; La Rosa, F.; Santangelo, M.A.; Caputo, D.; Mendozzi, L.; Rovaris, M.; Clerici, M. Monosodium Urate Crystals Activate the Inflammasome in Primary Progressive Multiple Sclerosis. Front. Immunol. 2018, 9, 983. [Google Scholar] [CrossRef]
- Nehmar, R.; Mariotte, A.; De Cauwer, A.; Sibilia, J.; Bahram, S.; Georgel, P. Therapeutic Perspectives for Interferons and Plasmacytoid Dendritic Cells in Rheumatoid Arthritis. Trends Mol. Med. 2018, 24, 338–347. [Google Scholar] [CrossRef]
- Kohase, M.; Zhang, Y.; Lin, J.X.; Yamazaki, S.; Sehgal, P.B.; Vilček, J. Interleukin-1 can inhibit interferon-beta synthesis and its antiviral action: Comparison with tumor necrosis factor. J. Interferon. Res. 1988, 8, 559–570. [Google Scholar] [CrossRef]
- SSlim, J.; Afridi, M.S. Managing Adverse Effects of Interferon-Alfa and Ribavirin in Combination Therapy for HCV. Infect. Dis. Clin. N. Am. 2012, 26, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, J.A.; Perry, C.M. Topical imiquimod: A review of its use in the management of anogenital warts, actinic keratoses, basal cell carcinoma and other skin lesions. Drugs 2007, 67, 2187–2210. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tummala, R. Anifrolumab, a monoclonal antibody to the type I interferon receptor subunit 1, for the treatment of systemic lupus erythematosus: An overview from clinical trials. Mod. Rheumatol. 2021, 31, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Clere-Jehl, R.; Mariotte, A.; Meziani, F.; Bahram, S.; Georgel, P.; Helms, J. JAK–STAT Targeting Offers Novel Therapeutic Opportunities in Sepsis. Trends Mol. Med. 2020, 26, 987–1002. [Google Scholar] [CrossRef]
- Melki, I.; Frémond, M.-L. Type I Interferonopathies: From a Novel Concept to Targeted Therapeutics. Curr. Rheumatol. Rep. 2020, 22, 32. [Google Scholar] [CrossRef] [PubMed]
- Mailhot, B.; Christin, M.; Tessandier, N.; Sotoudeh, C.; Bretheau, F.; Turmel, R.; Pellerin, È.; Wang, F.; Bories, C.; Joly-Beauparlant, C.; et al. Neuronal interleukin-1 receptors mediate pain in chronic inflammatory diseases. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Conaghan, P.G.; Alten, R.; Deodhar, A.; Sullivan, E.; Blackburn, S.; Tian, H.; Gandhi, K.; Jugl, S.M.; Strand, V. Relationship of pain and fatigue with health-related quality of life and work in patients with psoriatic arthritis on TNFi: Results of a multi-national real-world study. RMD Open 2020, 6, e001240. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Edelson, B.T. New Insights into the Role of IL-1 beta in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. J. Immunol. 2017, 198, 4553–4560. [Google Scholar] [CrossRef] [Green Version]
- Ozdogan, H.; Ugurlu, S.; Uygunoglu, U.; Tutuncu, M.; Gul, A.; Akman, G.; Siva, A. The efficacy of anti- IL-1 treatment in three patients with coexisting familial Mediterranean fever and multiple sclerosis. Mult. Scler. Relat. Disord. 2020, 45, 102332. [Google Scholar] [CrossRef]
- Iula, L.; Keitelman, I.A.; Sabbione, F.; Fuentes, F.; Guzman, M.; Galletti, J.G.; Gerber, P.P.; Ostrowski, M.; Geffner, J.R.; Jancic, C.C.; et al. Autophagy Mediates Interleukin-1β Secretion in Human Neutrophils. Front. Immunol. 2018, 9, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esnault, S.; Kelly, E.A.; Nettenstrom, L.M.; Cook, E.B.; Seroogy, C.M.; Jarjour, N.N. Human eosinophils release IL-1ß and increase expression of IL-17A in activated CD4+T lymphocytes. Clin. Exp. Allergy 2012, 42, 1756–1764. [Google Scholar] [CrossRef] [Green Version]
- Bassi, G.; Grimaudo, M.; Panseri, S.; Montesi, M. Advanced Multi-Dimensional Cellular Models as Emerging Reality to Reproduce In Vitro the Human Body Complexity. Int. J. Mol. Sci. 2021, 22, 1195. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Kinast, V.; Döring, M.; Lipps, C.; Duran, V.; Spanier, J.; Tegtmeyer, P.-K.; Wirth, D.; Cicin-Sain, L.; Alcamí, A.; et al. Human monocyte-derived macrophages inhibit HCMV spread independent of classical antiviral cytokines. Virulence 2018, 9, 1669–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.A.; Gale, M., Jr. Interleukin-1β Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol. Cell 2019, 74, 801–815. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Pascual, V. Type I Interferon in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Immunity 2006, 25, 383–392. [Google Scholar] [CrossRef] [Green Version]
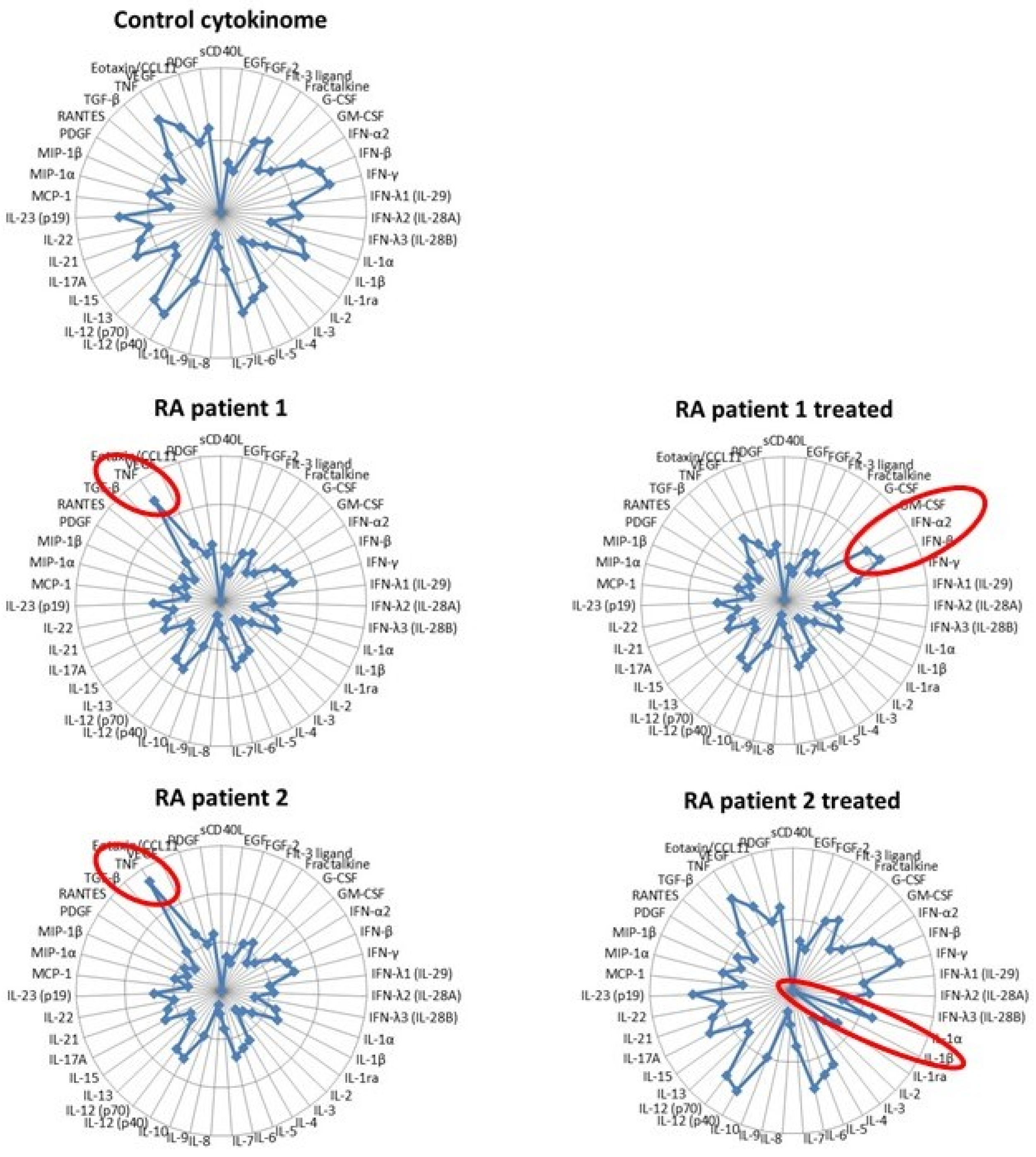
- Lliso-Ribera, G.; Humby, F.; Lewis, M.; Nerviani, A.; Mauro, D.; Rivellese, F.; Kelly, S.; Hands, R.; Bene, F.; Ramamoorthi, N.; et al. Synovial tissue signatures enhance clinical classification and prognostic/treatment response algorithms in early inflammatory arthritis and predict requirement for subsequent biological therapy: Results from the pathobiology of early arthritis cohort (PEAC). Ann. Rheum. Dis. 2019, 78, 1642–1652. [Google Scholar]
- Costantini, S.; Castello, G.; Colonna, G. Human Cytokinome: A new challenge for systems biology. Bioinformation 2010, 5, 166–167. [Google Scholar] [CrossRef] [Green Version]
- Brzustewicz, E.; Bzoma, I.; Daca, A.; Szarecka, M.; Bykowska, M.S.; Witkowski, J.M.; Bryl, E. Heterogeneity of the cytokinome in undifferentiated arthritis progressing to rheumatoid arthritis and its change in the course of therapy. Move toward personalized medicine. Cytokine 2017, 97, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Di Domizio, J.; Mylonas, A.; Belkhodja, C.; DeMaria, O.; Navarini, A.A.; Lapointe, A.-K.; French, L.E.; Vernez, M.; Gilliet, M. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat. Commun. 2018, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Brennan, F.; Jackson, A.; Chantry, D.; Maini, R.; Feldmann, M. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet 1989, 2, 244–247. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disease | Type | Genetic Defect | Cytokine Profile | Treatment |
|---|---|---|---|---|
| STING-associated vasculopathy with onset in infancy (SAVI) | interferonopathy | STING gain-of-function | exessive type I IFN secretion | corticosteroids jakinhibs (clinical trials) |
| Systemic Lupus Erythematosus (SLE) | rheumatic autoimmune/autoinflammatory disease | multifactorial disease | IFN signature (overexpression of IFN-stimulated genes) | corticosteroids Immunosuppressants (e.g., methotrexate) biologics (e.g., antiB-cell mAb) |
| Familial Mediterranean Fever (FMF) | inflammasomopathy | mutations in MEFV (Mediterranean fever, also named PYRIN) | constitutive IL-1β secretion | colchicin biologics (IL-1β receptor antagonist, anti IL-1β mAb) |
| Alzheimer’s disease (AD) | Neurodegenerative disease | multifactorial disease | excessive IL-1β, IL-6 and TNF secretion | Cholinesterase inhibitors N-methyl D-aspartate (NMDA) antagonists anti amyloid-β mAb (clinical trials) |
| Gout | rheumatic autoinflammatory disease | multifactorial disease | excessive IL-1β secretion | colchicin biologics (IL-1β receptor antagonist, anti IL-1β mAb) |
| Rheumatoid Arthritis (RA) | rheumatic autoimmune/autoinflammatory disease | multifactorial disease | TNF overexpression IL-1β overexpression IFN signature (overexpression of IFN-stimulated genes) | corticosteroids Immunosuppressants (e.g., methotrexate) biologics (e.g., anti TNF mAb) |
| Multiple sclerosis (MS) | inflammatory, neurodegenerative disease | multifactorial disease | increased IFNγ, IL-12, IL-17 secretion/activation | IFN-β biologics (e.g., antiB-cell mAb) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgel, P. Crosstalk between Interleukin-1β and Type I Interferons Signaling in Autoinflammatory Diseases. Cells 2021, 10, 1134. https://doi.org/10.3390/cells10051134
Georgel P. Crosstalk between Interleukin-1β and Type I Interferons Signaling in Autoinflammatory Diseases. Cells. 2021; 10(5):1134. https://doi.org/10.3390/cells10051134
Chicago/Turabian StyleGeorgel, Philippe. 2021. "Crosstalk between Interleukin-1β and Type I Interferons Signaling in Autoinflammatory Diseases" Cells 10, no. 5: 1134. https://doi.org/10.3390/cells10051134
APA StyleGeorgel, P. (2021). Crosstalk between Interleukin-1β and Type I Interferons Signaling in Autoinflammatory Diseases. Cells, 10(5), 1134. https://doi.org/10.3390/cells10051134