
Insulin Resistance and Diabetes Mellitus in Alzheimer’s Disease

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Insulin Resistance
2.1. MAPK and Insulin Resistance
2.1.1. Extracellular Signal-Regulated Kinases 1 and 2 (ERK1/2)
2.1.2. c-Jun N-Terminal Kinase (JNK)
2.1.3. p38
2.1.4. ERK5
2.2. PI3K/Akt and Insulin Resistance
2.3. mTOR and Insulin Resistance
3. Autophagy
3.1. Types of Autophagy
3.2. Autophagy in Pancreatic β Cells: A Double-Edged Sword
4. Endoplasmic Reticulum (ER) Stress
4.1. The ER Stress Response
4.1.1. PERK Pathway
4.1.2. IRE1 Pathway
4.1.3. ATF6 Pathway
4.1.4. Apoptosis-Inducing Pathways
4.2. ER Stress in Type 2 Diabetes Mellitus
5. Neurotransmitters
5.1. GABA Alterations in DM2
5.2. Effects of Dopamine on Insulin Secretion
6. Human Amylin Misfolding and T2DM
6.1. hIAPP and ER Stress in Pancreatic β-Cells
Autophagy as Defense Against hIAPP Aggregates
6.2. hIAPP Damage on Mitochondria
6.3. The Inflammatory Response to hIAPP Aggregates in T2DM
7. Molecular Pathophysiology of Alzheimer’s Disease
AD Pathologic Characteristics: The Role of Amyloid β and tau
8. The Relation Between T2DM and AD: A Molecular Approach
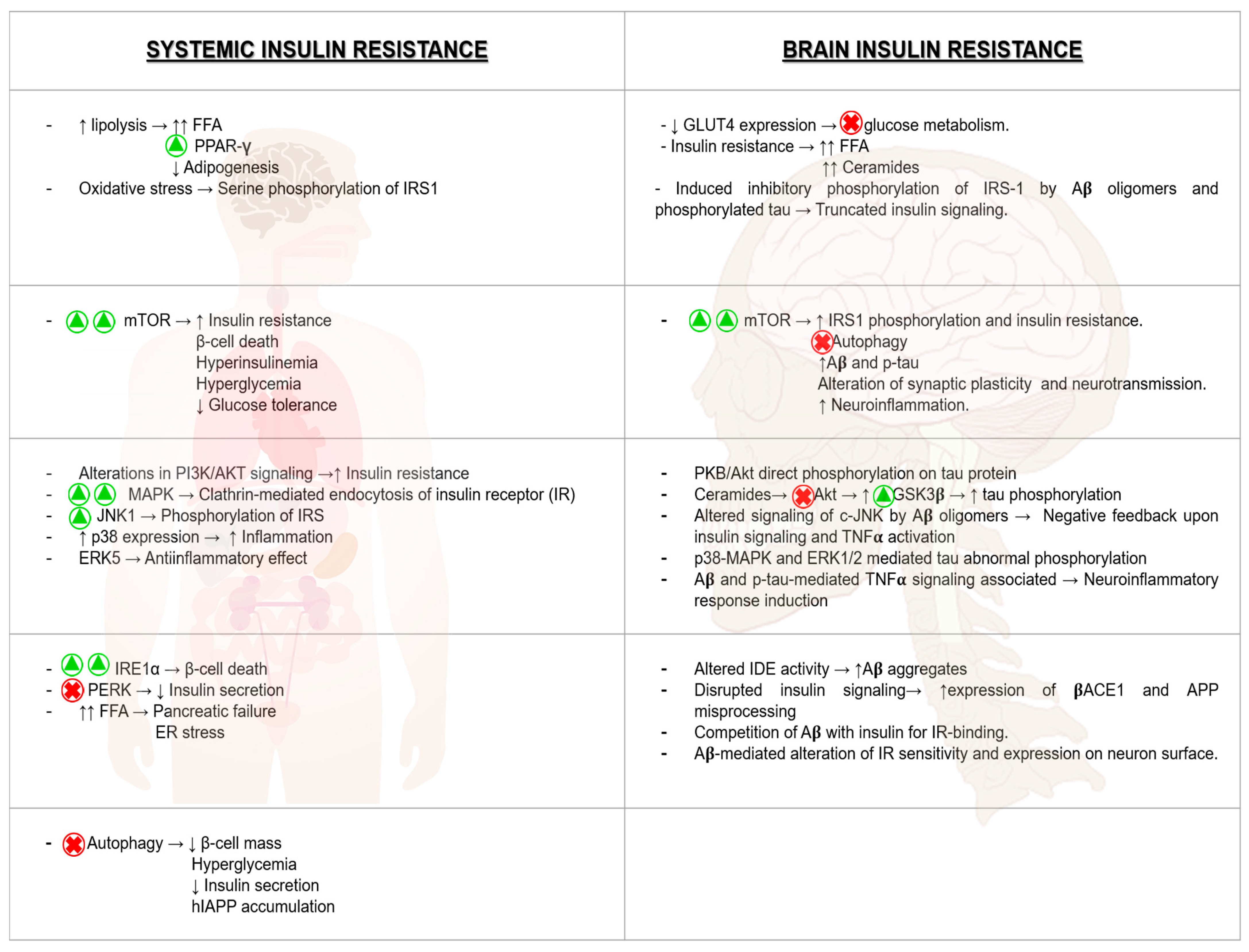
8.1. Brain Insulin Resistance and Its Impact on AD
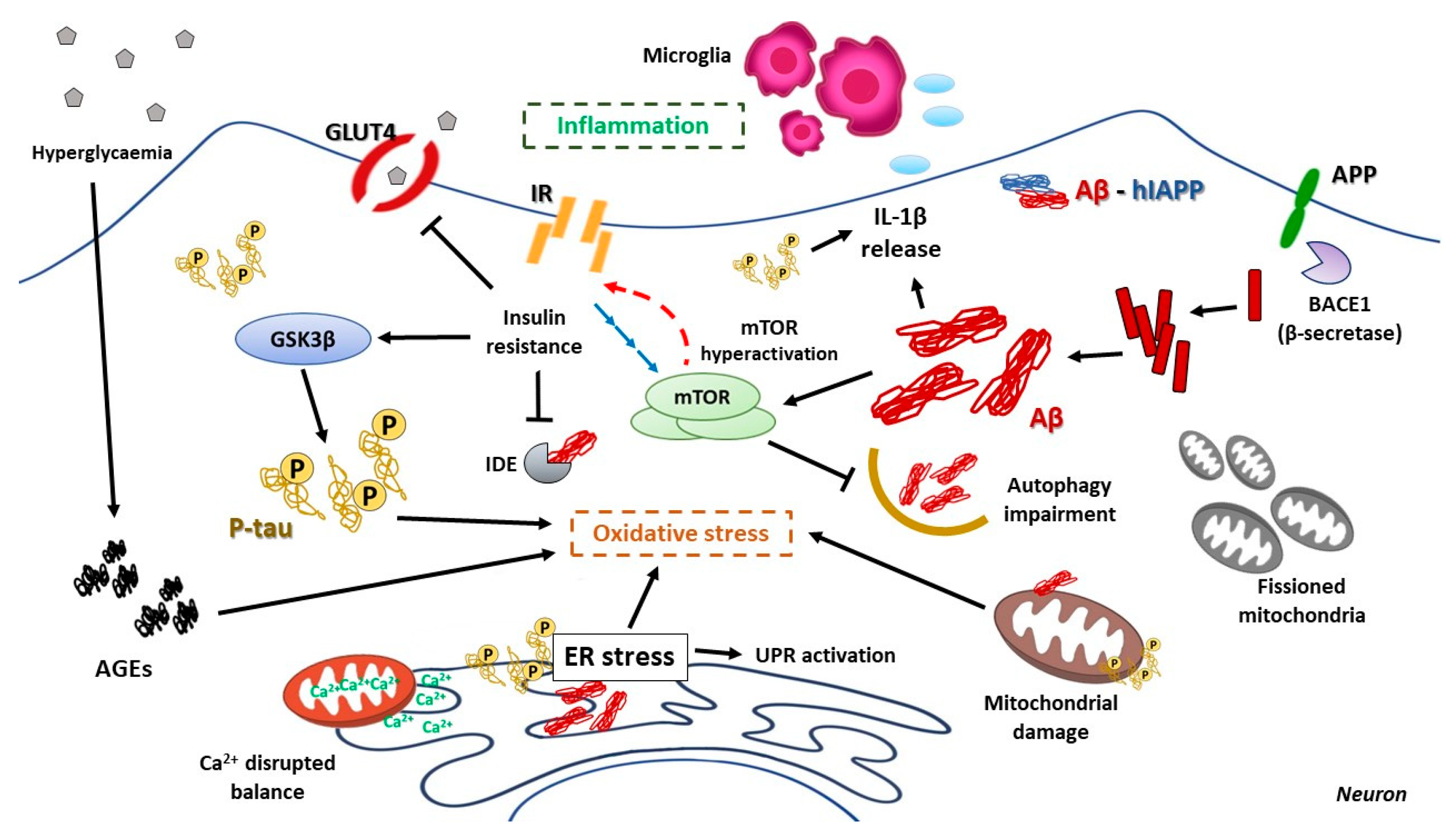
8.2. Hyperglycemia and Its Consequences on AD Pathologic Development
8.3. The Impact of ER Stress in AD
8.4. The Relevant Role of Mitochondria on AD Progression
8.5. mTOR Hyperactivation and AD
8.6. Relevance of Autophagy in AD Neuronal Homeostasis
8.7. Inflammation as a Harmful Fuel in AD
9. The Crosstalk Between T2DM and AD: The “Type 3 Diabetes Mellitus”
10. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Classification and diagnosis of diabetes: Standards of medical care in diabetes-2019. Diabetes Ca 2019, 42 (Suppl. 1), S13–S28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, H.; Luo, S.; Huang, G.; Xia, Y.; Xie, Z.; Zhou, Z. Advances in knowledge of candidate genes acting at the β-cell level in the pathogenesis of T1DM. Front. Endocrinol. 2020, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.B.; Cerosaletti, K.; Flanagan, S.E.; Buckner, J.H. Genetic mechanisms highlight shared pathways for the pathogenesis of polygenic type 1 diabetes and monogenic autoimmune diabetes. Curr. Diabetes Rep. 2019, 19, 20. [Google Scholar] [CrossRef] [Green Version]
- Howard, S.G. Exposure to environmental chemicals and type 1 diabetes: An update. J. Epidemiol. Community Health 2019, 73, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Cerna, M. Epigenetic regulation in etiology of type 1 diabetes mellitus. Int. J. Mol. Sci. 2019, 21, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaishi, J.; Saisho, Y. β-cell mass in obesity and type 2 diabetes, and its relation to pancreas fat: A mini-review. Nutrients 2020, 12, 3846. [Google Scholar] [CrossRef]
- Saisho, Y. β-cell dysfunction: Its critical role in prevention and management of type 2 diabetes. World J. Diabetes 2015, 6, 109–124. [Google Scholar] [CrossRef]
- Saisho, Y. Importance of β cell function for the treatment of type 2 diabetes. J. Clin. Med. 2014, 3, 923–943. [Google Scholar] [CrossRef] [Green Version]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β cell dedifferentiation as a mechanism of diabetic b cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of b cell dedifferentiation in human type 2 diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [Green Version]
- Parrettini, S.; Caroli, A.; Torlone, E. Nutrition and metabolic adaptations in physiological and complicated pregnancy: Focus on obesity and gestational diabetes. Front. Endocrinol. 2020, 11, 611929. [Google Scholar] [CrossRef]
- Firdous, P.; Nissar, K.; Ali, S.; Ganai, B.A.; Shabir, U.; Hassan, T.; Masoodi, S.R. Genetic testing of maturity-onset diabetes of the young current status and futures perspectives. Front. Endocrinol. 2018, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Nkonge, K.M.; Nkonge, D.K.; Nkonge, T.N. The epidemiology, molecular pathogenesis, diagnosis and treatment of maturity-onset diabetes of the young (MODY). Clin. Diabetes Endocrinol. 2020, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Fajans, S.S.; Bell, G.I. History, genetics, pathophysiology, and clinical decision making. Diabetes Care 2011, 34, 1878–1884. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.J.; Crimmins, D.L.; Myers, M.G., Jr.; Miralpeix, M.; White, M.F. Pleiotropic insulin signals are engaged by multisite phosphorylation of IRS-1. Mol. Cell Biol. 1993, 13, 7418–7428. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.M. The insulin receptor substrate (IRS) proteins: At the intersection of metabolism and cancer. Cell Cycle 2011, 10, 1750–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Kassouf, T.; Sumara, G. Impact of conventional and atypical MAPKs on the development of metabolic diseases. Biomolecules 2020, 10, 1256. [Google Scholar] [CrossRef]
- Pronk, G.J.; McGlade, J.; Pelicci, G.; Pawson, T.; Bos, J.L. Insulin-induced phosphorylation of the 46- and 52-kDa shc proteins. J. Biol. Chem. 1993, 268, 5748–5753. [Google Scholar] [CrossRef]
- Avruch, J.; Khokhlatchev, A.; Kyriakis, J.M.; Luo, Z.; Tzivion, G.; Vavvas, D.; Zhang, X.F. Ras activation of the Raf kinase: Tyrosine kinase recruitment of the MAP kinase cascade. Recent. Prog. Horm. Res. 2001, 56, 127–155. [Google Scholar] [CrossRef]
- Gehart, H.; Kumpf, S.; Ittner, A.; Ricci, R. MAPK signaling in cellular metabolism: Stress or wellness? EMBO J. 2010, 11, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Sasaoka, T.; Kobayashi, M. The functional significance of Shc in insulin signaling as a substrate of the insulin receptor. Endocr. J. 2000, 47, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waskiewicz, A.J.; Flynn, A.; Proud, C.G.; Cooper, J.A. Mitogen-activated protein kinases activate serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997, 16, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Flynn, A.; Waskiewicz, A.J.; Webb, B.L.; Vries, R.G.; Baines, I.A.; Cooper, J.A.; Proud, C.G. The phosphorylation of eukaryotic initiation factor eIF4E in response to phorbol esters, cell stresses, and cytokines is mediated by distinct MAP kinase pathways. J. Biol. Chem. 1998, 273, 9373–9377. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.; Cohen, P. MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett. 2000, 482, 44–48. [Google Scholar] [CrossRef]
- Ding, Q.; Xia, W.; Liu, J.-C.; Yang, J.-Y.; Lee, D.-F.; Xia, J.; Bartholomeusz, G.; Li, Y.; Pan, Y.; Li, Z.; et al. Erk associates with and primes GSK-3β for its inactivation resulting in upregulation of β-catenin. Mol. Cell 2005, 19, 159–170. [Google Scholar] [CrossRef]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3 β by phosphorylation: New kinase connections in insulin and growth-factor signaling. Biochem. J. 1993, 296, 15–19. [Google Scholar] [CrossRef]
- Ceresa, B.P.; Kao, A.W.; Santeler, S.R.; Pessin, J.E. Inhibition of clathrin-mediated endocytosis selectively attenuates specific insulin receptor signal transduction pathways. Mol. Cell Biol. 1998, 18, 3862–3870. [Google Scholar] [CrossRef] [Green Version]
- Backer, J.M.; Kahn, C.R.; Cahill, D.A.; Ullrich, A.; White, M.F. Receptor-mediated internalization of insulin requires a 12-amino acid sequence in the juxtamembrane region of the insulin receptor β-subunit. J. Biol. Chem. 1990, 265, 16450–16454. [Google Scholar] [CrossRef]
- Backer, J.M.; Shoelson, S.E.; Haring, E.; White, M.F. Insulin receptors internalize by a rapid, saturable pathway requiring receptor autophosphorylation and an intact juxtamembrane region. J. Cell Biol. 1991, 115, 1535–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.; Yu, H.; Choi, E. Insulin receptor endocytosis in the pathophysiology of insulin resistance. Exp. Mol. Med. 2020, 52, 911–920. [Google Scholar] [CrossRef]
- De Fea, K.; Roth, R.A. Modulation of insulin receptor substrate-1 tyrosine phosphorylation and function by mitogen-activated protein kinase. J. Biol. Chem. 1997, 272, 31400–31406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.; Kikuchi, S.; Gao, H.; Brodzik, K.; Nassour, I.; Yopp, A.; Singal, A.G.; Zhu, H.; Yu, H. Mitotic regulators and the SHP2-MAPK pathway promote IR endocytosis and feedback regulation of insulin signaling. Nat. Commun. 2019, 10, 1473. [Google Scholar] [CrossRef]
- Zhao, Y.; Ma, S.; Hu, X.; Feng, M.; Xiang, R.; Li, M.; Liu, C.; Lu, T.; Huang, A.; Chen, J.; et al. JAB1 promotes palmitate-induced insulin resistance via ERK pathway in hepatocytes. J. Physiol. Biochem. 2020, 76, 655–662. [Google Scholar] [CrossRef]
- Tan, Y.; Ichikawa, T.; Li, J.; Si, Q.; Yang, H.; Chen, X.; Goldblatt, C.S.; Meyer, C.J.; Li, X.; Cai, L.; et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes 2011, 60, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Xu, W.; Su, Y.; Gao, K.; Chen, Y.; Ma, L.; Xie, Y. Nicotine induces insulin resistance via downregulation of Nrf2 in cardiomyocyte. Mol. Cell Endocrinol. 2019, 495, 110507. [Google Scholar] [CrossRef]
- Zong, J.; Li, S.; Wang, Y.; Mo, W.; Sun, R.; Yu, M. Bromodomain-containing protein 2 promotes lipolysis via ERK/HSL signaling pathway in white adipose tissue of mice. Gen. Comp. Endocrinol. 2019, 281, 105–116. [Google Scholar] [CrossRef]
- Zang, K.; Wang, J.; Dong, M.; Sun, R.; Wang, Y.; Huang, Y.; Liu, X.; Li, Y.; Wang, F.; Yu, M. Brd2 inhibits adipogenesis via the ERK1/2 signaling pathway in 3T3-L1 adipocytes. PLoS ONE 2013, 8, e78536. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Song, W.; Zushin, P.-J.H.; Liu, B.; Jedrychowski, M.O.; Mina, A.I.; Deng, Z.; Cabarkapa, D.; Hall, J.A.; Palmer, C.J.; et al. Phosphorylation of β-3 adrenergic receptor at serine 247 by ERK MAP kinase drives lipolysis in obese adipocytes. Mol. Metab. 2018, 12, 25–38. [Google Scholar] [CrossRef]
- Guo, L.; Costanzo-Garvey, D.L.; Smith, D.R.; Neilsen, B.K.; Macdonald, R.G.; Lewis, R.E. Kinase suppressor of Ras 2 (KSR2) expression in the brain regulates energy balance and glucose homeostasis. Mol. Metab. 2016, 6, 194–205. [Google Scholar] [CrossRef]
- Banks, A.S.; McAllister, F.E.; Camporez, J.P.G.; Zushin, P.-J.H.; Jurczak, M.J.; Laznik-Bogoslavski, D.; Shulman, G.I.; Gygi, S.O.; Spiegelman, B.M. An ERK/CDK5 axis controls the diabetogenic actions of PPARg. Nature 2015, 517, 391–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nohara, A.; Okada, S.; Ohshima, K.; Pessin, J.E.; Mori, M. Cyclin-dependent kinase-5 is a key molecule in tumor necrosis- a -induced insulin resistance. J. Biol. Chem. 2011, 286, 33457–33465. [Google Scholar] [CrossRef] [Green Version]
- Jiao, P.; Feng, B.; Li, Y.; He, Q.; Xu, H. Hepatic ERK activity plays a role in energy metabolism. Mol. Cell Endocrinol. 2013, 375, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Jiao, P.; Yang, Z.; Xu, H. MEK/ERK pathway mediates insulin-promoted degradation of MKP-3 protein in liver cells. Mol. Cell Endocrinol. 2012, 361, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Fujishiro, M.; Gotoh, Y.; Katagiri, H.; Sakoda, H.; Ogihara, T.; Anai, M.; Onishi, Y.; Ono, H.; Abe, M.; Shojima, N.; et al. Three mitogen-activated protein kinases inhibit insulin signaling by different mechanisms in 3T3-L1 adipocytes. Mol. Endocrinol. 2003, 17, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Boura-Halfon, S.; Zick, Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E581–E591. [Google Scholar] [CrossRef] [Green Version]
- Manowsky, J.; Camargo, R.G.; Kipp, A.P.; Henkel, J.; Püschel, G.P. Insulin-induced cytokine production in macrophages causes insulin resistance in hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E938–E946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Yu, R.; Xiong, Y.; Du, F.; Zhu, S. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis. 2017, 16, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, S.; Zhu, W.; Wang, S.; Xie, C.; Li, X.; Wu, J.; Li, Y.; Chen, Y.; Wang, X.; Meng, Y.; et al. P53 modulates hepatic insulin sensitivity through NF-kB and p38/ERK MAPK pathways. Biochem. Biophys. Res. Commun. 2018, 495, 2139–2144. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Subramaniam, S.; Dramane, G.; Khelifi, D.; Khan, N.A. ERK1 and ERK2 activation modulates diet-induced obesity in mice. Biochimie 2017, 137, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Bost, F.; Aouadi, M.; Caron, L.; Even, P.; Belmonte, N.; Prot, M.; Dani, C.; Hofman, P.; Pagés, G.; Poysségur, J.; et al. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes 2005, 54, 402–411. [Google Scholar] [CrossRef] [Green Version]
- Antonescu, C.N.; Huang, C.; Niu, W.; Liu, Z.; Eyers, P.A.; Heidenreich, K.A.; Bilan, P.J.; Klip, A. Reduction of insulin-stimulated glucose uptake in L6 Emyotubes by the protein kinase inhibitor SB203580 is independent of p38MAPK activity. Endocrinology 2005, 146, 3773–3781. [Google Scholar] [CrossRef]
- Moxham, C.M.; Tabrizchi, A.; Davis, R.J.; Malbon, C.C. Jun N-terminal kinase mediates activation of skeletal muscle glycogen synthase hy insulin in vivo. J. Biol. Chem. 1996, 271, 30765–30773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avruch, J. Insulin signal transduction through protein kinase cascades. Mol. Cell Biochem. 1998, 182, 31–48. [Google Scholar] [CrossRef]
- Belgardt, B.F.; Mauer, J.; Brüning, J.C. Novel roles for JNK1 in metabolism. Aging 2010, 2, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Rondinone, C.M. JNK: Bridging the insulin signaling and inflammatory pathway. Curr. Opin. Investig. Drugs 2005, 6, 979–987. [Google Scholar]
- Hotamisligil, G.S. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes 2005, 54, S73–S78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.J. Signal transduction by the c-Jun N-terminal kinase. Biochem. Soc. Symp. 1999, 64, 1–12. [Google Scholar] [CrossRef]
- Minden, A.; Lin, A.; Smeal, T.; Dérijard, B.; Cobb, M.; Davis, R.; Karin, M. c-Jun N-terminal phosphorylation correlates with activation of the JNK subgroup but not the ERK subgroup of mitogen-activated protein kinases. Mol. Cell Biol. 1994, 14, 6683–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frödin, M.; Gammeltoft, S. Role and regulation of 90 KDa ribosomal S6 kinase (RSK) in signal transduction. Mol. Cell Endocrinol. 1999, 151, 65–77. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhong, S.; Chen, N.; Bode, A.M.; Ma, W.; Dong, Z. UVA induces Ser381 phosphorylation of p90RSK/MAPKAP-K1 via ERK and JNK pathways. J. Biol. Chem. 2001, 276, 14572–14580. [Google Scholar] [CrossRef] [Green Version]
- Pal, M.; Febbraio, M.A.; Lancaster, G.I. The roles of c-Jun NH2-terminal kinases (JNKs) in obesity and and insulin resistance. J. Physiol. 2016, 594, 267–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manieri, E.; Sabio, G. Stress kinases in the modulation of metabolism and energy balance. J. Mol. Endocrinol. 2015, 55, R11–R22. [Google Scholar] [CrossRef]
- Nikolic, I.; Leiva, M.; Sabio, G. The role of stress kinases in metabolic disease. Nat. Rev. Endocrinol. 2020, 16, 697–716. [Google Scholar] [CrossRef]
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal kinase (JN) in obesity and type 2 diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef] [Green Version]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2016, 6, 174–184. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Sumara, G.; Belwal, M.; Ricci, R. “Jnking” atherosclerosis. Cell. Mol. Life Sci. 2005, 62, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Tuncman, G.; Hirosumi, J.; Solinas, G.; Chang, L.; Karin, M.; Hotamisligil, G.S. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2006, 103, 10741–10746. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, C.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH(2)-terminal kinase promotes insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabio, G.; Cavanagh-Kyros, J.; Ko, H.J.; Jung, D.Y.; Gray, S.; Jun, J.Y.; Barrett, T.; Mora, A.; Kim, J.K.; Davis, R.J. Prevention of steatosis by hepatic JNK1. Cell Metab. 2009, 10, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Copps, K.D.; Hancer, N.J.; Opare-Ado, L.; Qiu, W.; Walsh, C.; White, M.F. Irs1 serine 307 phosphorylation promotes insulin sensitivity in mice. Cell Metab. 2010, 11, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Endoplasmir reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Yalcin, A.; Lee, G.Y.; Li, P.; Fan, J.; Arruda, A.P.; Pers, B.M.; Yilmaz, M.; Eguchi, K.; Hotamisligil, G.S. Phenotypic assays identify azoramide as a small-molecule modulator of the unfolded protein response with antidiabetic activity. Sci. Transl. Med. 2015, 7, 292ra98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillén, C. Azoramide: A new drug for the treatment of type 2 diabetes? Ann. Transl. Med. 2016, 4, S45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Inflammation and endoplasmic-reticulum stress in obesity and diabetes. Int. J. Obes. 2008, 32, S52–S54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeschke, A.; Davis, R.J. Metabolic stress signaling mediated by mixed-lineage kinases. Mol. Cell. 2007, 27, 498–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeschke, A.; Czech, M.P.; Davis, R.J. An essential role of the JIP1 scaffold protein for JNK activation in adipose tissue. Genes Dev. 2004, 18, 1976–1980. [Google Scholar] [CrossRef] [Green Version]
- Morel, C.; Standen, C.L.; Jung, D.Y.J.; Gray, S.; Ong, H.; Flavell, R.A.; Kim, J.K.; Davis, R.J. Requirement of JIP1-mediated c-Jun N-terminal kinase activation for obesity-induced insulin resistance. Mol. Cell Biol. 2010, 30, 4616–4625. [Google Scholar] [CrossRef] [Green Version]
- Waeber, G.; Delplanque, J.; Bonny, C.; Mooser, V.; Steinmann, M.; Widmann, C.; Maillard, A.; Miklossy, J.; Dina, C.; Hani, E.H.; et al. The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nat. Genet. 2000, 24, 291–295. [Google Scholar] [CrossRef]
- Burns, K.A.; Heuvel, J.P.V. Modulation of PPAR activity via phosphorylation. Biochim. Biophys. Acta 2007, 1771, 952–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol. Cell 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Tanaka-Yachi, R.; Shirasaki, M.; Otsu, R.; Takahashi-Muto, C.; Inoue, H.; Aoki, Y.; Koike, T.; Kiyose, C. d-Tocopherol promotes thermogenic gene expression via PGC-1a upregulation in 3T3-L1 cells. Biochem. Biophys. Res. Commun. 2018, 506, 53–59. [Google Scholar] [CrossRef]
- Cao, W.; Daniel, K.W.; Robidoux, J.; Puigserver, P.; Medvedev, A.V.; Bai, X.; Floering, L.M.; Spiegelman, B.M.; Collins, S. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell Biol. 2004, 24, 3057–3067. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Lisanti, M.P.; Scherer, P.E. Specific inhibitors of p38 mitogen-activated protein kinase block 3T3-L1 adipogenesis. J. Biol. Chem. 1998, 273, 32111–32120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deak, M.; Clifton, A.D.; Lucocq, L.M.; Alessi, D.R. Mitogen-and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998, 17, 4426–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Sun, C.; Zhou, Y.; Lee, J.; Gokalp, D.; Herrema, H.; Park, S.W.; Davis, R.J.; Ozcan, U. p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 2011, 17, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, S.; Lee, A.S. Requirement of the p38 mitogen-activated protein kinase signaling pathway for the induction of the 78 kDa glucose-regulated protein/immunoglobulin heavy-chain binding protein by azetidine stress: Activating transcription factor 6 as a target for stress-induced phosphorylation. Biochem. J. 2002, 366, 787–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieven, G.L. The biology of p38 kinase: A central role in inflammation. Curr. Top. Med. Chem. 2005, 5, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell. Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potashnik, R.; Bloch-Damti, A.; Bashan, N.; Rudich, A. IRS1 degradation and increased serine phosphorylation cannot predict the degree of metabolic insulin resistance induced by oxidative stress. Diabetologia 2003, 46, 639–648. [Google Scholar] [CrossRef]
- Archuleta, T.L.; Lemieux, A.M.; Saengsirisuwan, V.; Teachey, M.K.; Lindborg, K.A.; Kim, J.S.; Henriksen, E.J. Oxidant stress-induced loss of IRS-1 and IRS-2 proteins in rat skeletal muscle: Role of p38 MAPK. Free Radic. Biol. Med. 2009, 47, 1486–1493. [Google Scholar] [CrossRef] [Green Version]
- Hemi, R.; Yochananov, Y.; Barhod, E.; Kasher-Meron, M.; Karasik, A.; Tirosh, A.; Kanety, H. p38 mitogen-activated protein kinase-dependent transactivation of ErbB receptor family: A novel common mechanism for stress-induced IRS-1 serine phosphorylation and insulin resistance. Diabetes 2011, 60, 1134–1145. [Google Scholar] [CrossRef] [Green Version]
- González-Terán, B.; Matesanz, N.; Nikolic, I.; Verdugo, M.A.; Sreeramkumar, V.; Hernández-Cosido, L.; Mora, A.; Crainiciuc, G.; Sáiz, M.L.; Bernardo, E.; et al. p38g and p38d reprogram liver metabolism by modulating neutrophil infiltration. EMBO J. 2016, 35, 536–552. [Google Scholar] [CrossRef] [Green Version]
- Liao, M.J.; Lin, H.; He, Y.-W.; Zou, C. NFATc3 deficiency protects against high fat diet (HFD)-induced hypothalamus inflammation and apoptosis via p38 and JNK suppression. Biochem. Biophys. Res. Commun. 2010, 499, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Benomar, Y.; Gertler, A.; De Lacy, P.; Crépin, D.; Hamouda, H.O.; Riffault, L.; Taouis, M. Central resistin overexposure induces insulin resistance through Toll-like receptor 4. Diabetes 2013, 62, 102–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.P.; Müller, J.; Cross, M.J. ERK5: Structure, regulation and function. Cell. Signal. 2012, 24, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.A.; Burow, M.E.; Beckman, B.S. MEK5/ERK5 pathway: The first fifteen years. Biochim. Biophys. Acta 2014, 1825, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Woo, C.-H.; Massett, M.P.; Shishido, T.; Itoh, S.; Ding, B.; McClain, C.; Che, W.; Vulapalli, S.R.; Yan, C.; Abe, J.-I. ERK5 activation inhibits inflammatory responses via peroxisome proliferator-activated receptor delta (PPARdelta) stimulation. J. Biol. Chem. 2006, 281, 32164–32174. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Guariglia, S.; Li, W.; Brancho, D.; Wang, Z.V.; Scherer, P.E.; Chow, C.-W. Role of extracellular signal-regulated kinase-5 in adipocyte signaling. J. Biol. Chem. 2014, 289, 6311–6322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Velasco, A.; Zi, M.; Hille, S.S.; Azam, T.; Kaur, N.; Jiang, J.; Nguyen, B.; Sekeres, K.; Binder, P.; Collins, L.; et al. Targeting mir128-3p alleviates myocardial insulin resistance and prevents ischemia-induced heart failure. Elife 2020, 9, e54298. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signaling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Vadas, O.; Burke, J.E.; Zhang, X.; Berndt, A.; Williams, R.L. Structural basis for activation and inhibition of Class I Phosphoinositide 3-Kinases. Sci. Signal. 2011, 4, re2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, H.; Liu, J. Akt activation: A potential strategy to ameliorate insulin resistance. Diabetes Res. Clin. Pract. 2019, 156. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krycer, J.R.; Sharpe, L.J.; Luu, W.; Brown, A.J. The Akt-SREBP nexus: Cell signaling meets lipid metabolism. Trends Endocrinol. Metab. 2010, 21, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Abeyrathna, P.; Su, Y. The critical role of Akt in cardiovascular function. Vasc. Pharmacol. 2015, 74, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Easton, R.M.; Cho, H.; Roovers, K.; Shineman, D.W.; Mizrahi, M.; Forman, M.S.; Lee, V.M.; Szabolcs, M.; De Jong, R.; Oltersdorf, T.; et al. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol. Cell Biol. 2005, 25, 1869–1878. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Cantley, L.C.; Toker, A.; Su, B.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Insulin-PI3K signalling: An evolutionarily insulated metabolic driver of cancer. Nat. Rev. Endocrinol. 2020, 16, 276–283. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, R. Critical nodes in signalling pathway: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Kousteni, S. FoxO1, the transcriptional chief of staff of energy metabolism. Bone 2012, 50, 437–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Monks, B.; Ge, Q.; Birnbaum, M.J. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature 2007, 447, 1012–1016. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, N.; Yan, F.; Jin, H.; Zhou, S.; Shi, J.; Jin, F. Diabetes mellitus and Alzheimer’s disease: GSK-3 as a potential link. Behav. Brain Res. 2018, 339, 57–65. [Google Scholar] [CrossRef]
- Chakrabarti, P.; Kandror, K.V. FoxO1 controls insulin-dependent adipose triglyceride lipase (ATGL) expression and lipolysis in adipocytes. J. Biol. Chem. 2009, 284, 13296–13300. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, B.; Hedjazifar, S.; Gogg, S.; Hammarstedt, A.; Smith, U. Insulin resistance and impaired adipogenesis. Trends Endocrinol. Metab. 2015, 26, 193–200. [Google Scholar] [CrossRef]
- Amrani, A.; Jafarian-Tehrani, M.; Mormede, P.; Durant, S.; Pleau, J.M.; Haour, F.; Dardenne, M.; Homo-Delarche, F. Interleukin-1 effect on glycemia in the non-obese diabetic mouse at the pre-diabetic stage. J. Endocrinol. 1996, 148, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Kang, H.; Im, S.S. Recent insight into the correlation of SREBP-mediated lipid metabolism and innate immune response. J. Mol. Endocrinol, 61. [CrossRef] [Green Version]
- Lundsgaard, A.M. Glucometabolic consequences of acute and prolongated inhibition of fatty acid oxidation. J. Lipid Res. 2020, 61, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Brozinick, J.T.; Wang, L.-P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Kraegen, E.W.; Cooney, G.J. Free fatty acids and skeletal muscle insulin resistance. Curr. Opin. Lipidol. 2008, 19, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Hesselink, M.K.C.; Schrauwen-Hinderling, V.; Schrauwen, P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2016, 12, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Balland, E.; Cowley, M.A. Hypothalamic Insulin Resistance in Obesity: Effects on Glucose Homeostasis. Neuroendocrinology 2017, 104, 364–381. [Google Scholar] [CrossRef] [PubMed]
- Iskandar, K. PDK-1/FoxO1 pathway in POMC neurons regulates Pomc expression and food intake. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E787–E798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalheira, J.B.; Ribeiro, E.B.; Araujo, E.P.; Guimaraes, R.B.; Telles, M.M.; Torsoni, M.; Gontijo, J.A.R.; Velloso, L.A.; Saad, M.J.A. Selective impairment of insulin signalling in the hypothalamus of obese Zucker rats. Diabetologia 2003, 46, 1629–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, H.; Pocai, A.; Wang, Y.; Sakoda, H.; Asano, T.; Backer, J.M.; Schwartz, G.J.; Rossetti, L. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. J. Clin. Investig. 2008, 118, 2959–2968. [Google Scholar] [CrossRef]
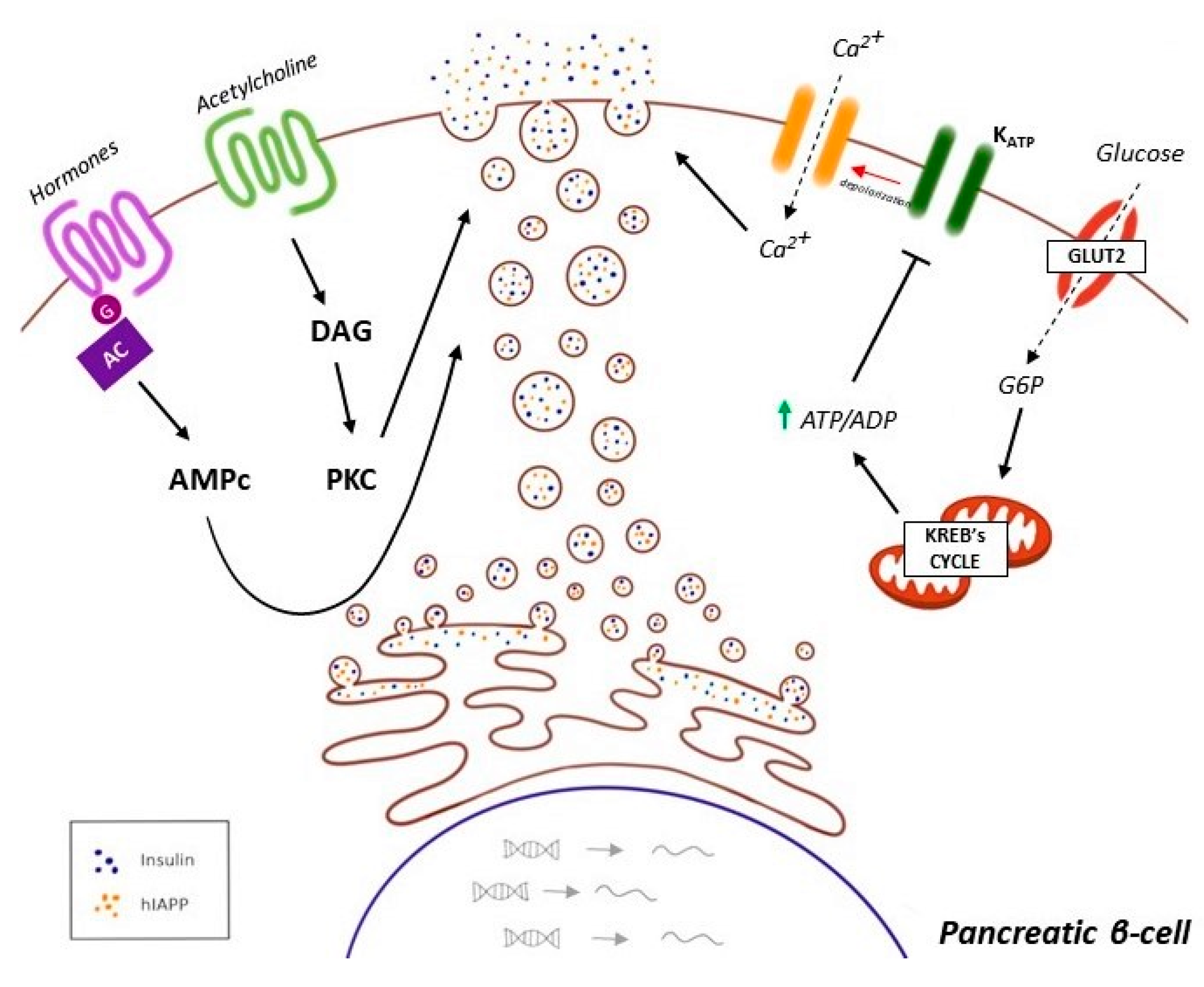
- McTaggart, J.S.; Clark, R.H.; Ashcroft, F.M. The role of the K ATP channel in glucose homeostasis in health and disease: More than meets the islet. K Physiol. 2010, 588, 3201–3209. [Google Scholar] [CrossRef]
- Oliveira, J.M.; Rebuffat, S.A.; Gasa, R.; Gomis, R. Targeting type 2 diabetes: Lessons from a knockout model of insulin receptor substrate 2. Can. J. Physiol. Pharmacol. 2014, 92, 613–620. [Google Scholar] [CrossRef]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006, 444, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erion, D.M.; Shulman, G.I. Diacylglycerol-mediated insulin resistance. Nat. Med. 2010, 16, 400–402. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [Green Version]
- Kubota, N.; Kubota, T.; Kajiwara, E.; Iwamura, T.; Kumagai, H.; Watanabe, T.; Inoue, M.; Takamoto, I.; Sasako, T.; Kumagai, K.; et al. Differential hepatic distribution of insulin receptor substrates causes selective insulin resistance in diabetes and obesity. Nat. Commun. 2016, 7, 12977. [Google Scholar] [CrossRef] [Green Version]
- Lempiäinen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009, 28, 3067–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Guillén, C.; Benito, M. mTORC1 Overactivation as a Key Aging Factor in the Progression to Type 2 Diabetes Mellitus. Front. Endocrinol. 2018, 9, 621. [Google Scholar] [CrossRef] [Green Version]
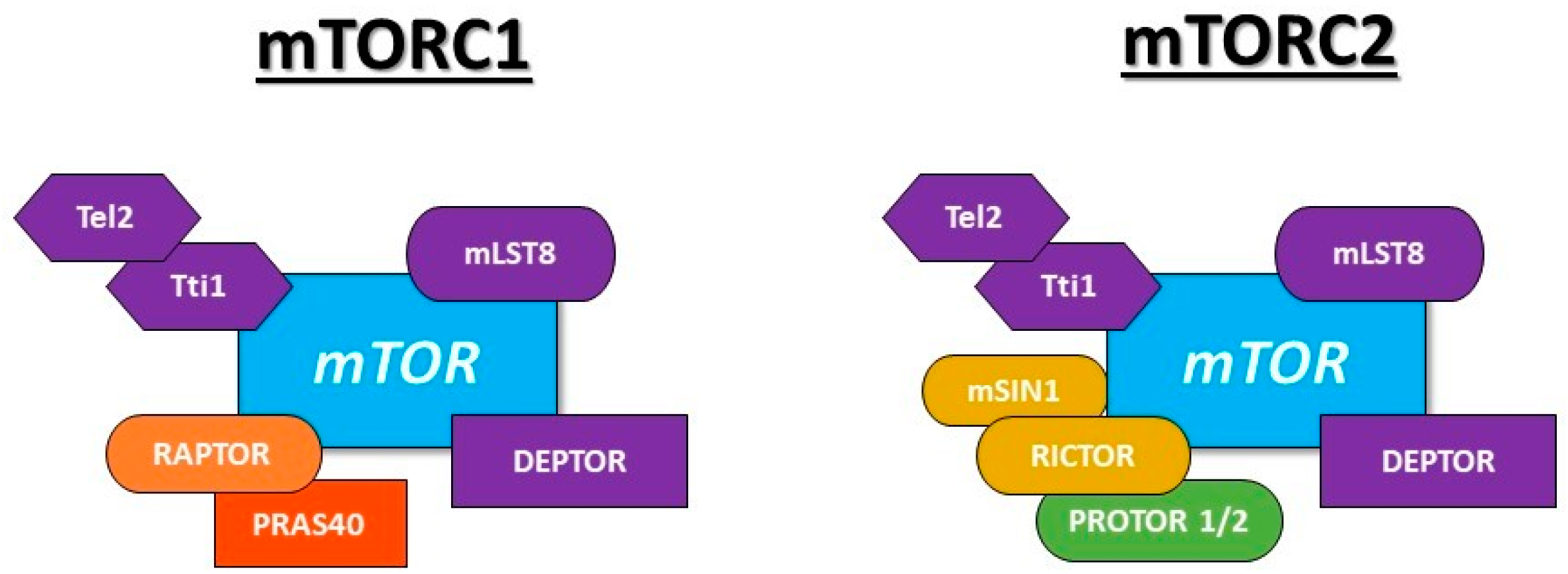
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.P.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GβL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Kaizuka, T.; Hara, T.; Oshiro, N.; Kikkawa, U.; Yonezawa, K.; Takehana, K.; Iemura, S.I.; Natsume, T.; Mizushima, N. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J. Biol. Chem. 2010, 285, 20109–20116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. Sin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawlowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a novel Rictor-binding component of mTOR-complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, X.; Saucedo, L.J.; Ru, B.; Edgar, B.A.; Pan, D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 2003, 5, 578–581. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.N.; Ha, S.H.; Kim, J.; Koh, A.; Lee, C.S.; Kim, J.H.; Jeon, H.; Kim, D.H.; Suh, P.G.; Ryu, S.H. Glycolytic flux signals to mTOR through glyceraldehyde-3-phosphate dehydrogenase-mediated regulation of Rheb. Mol. Cell Biol. 2009, 29, 3991–4001. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. An emerging role of mTOR in lipid biosynthesis. Curr. Biol. 2009, 19, R1046–R1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Chen, J. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Ganley, I.G.; Lam, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signalling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Ardestani, A.; Lupse, B.; Kido, Y.; Leibowitz, G.; Maedler, K. mTORC1 Signaling: A double-edged sword in diabetic b cells. Cell Metab. 2018, 27, 314–331. [Google Scholar] [CrossRef] [Green Version]
- Rachdi, L.; Balcazar, N.; Osorio-Duque, F.; Elghazi, L.; Weiss, A.; Gould, A.; Chang-Chen, K.J.; Gambello, M.J.; Bernal-Mizrachi, E. Disruption of Tsc2 in pancreatic β cells induces β cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc. Natl. Acad. Sci. USA 2008, 105, 9250–9255. [Google Scholar] [CrossRef] [Green Version]
- Blandino-Rosano, M.; Barbaresso, R.; Jiménez-Palomares, M.; Bozadjieva, N.; Werneck-de-Castro, J.P.; Hatanaka, M.; Mirmira, R.G.; Sonenberd, N.; Ming, L.; Rüegg, M.A.; et al. Loss of mTORC1 signalling impairs β-cell homeostasis and insulin processing. Nat. Commun. 2017, 8, 16014. [Google Scholar] [CrossRef]
- Boutouja, F.; Stiehm, C.M.; Platta, H.W. mTOR: A cellular regulator interface in health and disease. Cells 2019, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Shigeyama, Y.; Kobayashi, T.; Kido, Y.; Hashimoto, N.; Asahara, S.; Matsuda, T.; Takeda, A.; Inoue, T.; Shibutani, Y.; Koyanagi, M.; et al. Biphasic response of pancreatic β-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol. Cell Biol. 2008, 28, 2971–2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnevalli, L.S.; Masuda, K.; Frigerio, F.; Le Bacquer, O.; Um, S.H.; Gandin, V.; Topisirovic, I.; Sonenberg, N.; Thomas, G.; Kozma, S.C. S6K1 plays a critical role in early adipocyte differentiation. Dev. Cell. 2010, 18, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Um, S.H.; Frigerio, F.; Watanabe, M.; Picard, F.; Joaquin, M.; Sticker, M.; Fumagalli, S.; Allegrini, P.R.; Kozma, S.C.; Auwerx, J.; et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004, 431, 200–205. [Google Scholar] [CrossRef]
- Khamzina, L.; Veilleux, A.; Bergeron, S.; Marette, A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: Possible involvement in obesity-linked insulin resistance. Endocrinology 2005, 146, 1473–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titchenell, P.M.; Lazar, M.A.; Birnbaum, M.J. Unraveling the regulation of hepatic metabolism by insulin. Trends Endocrinol. Metab. 2017, 28, 497–505. [Google Scholar] [CrossRef]
- Suhara, T.; Baba, Y.; Shimada, B.K.; Higa, J.K.; Matsui, T. The mTOR sinaling pathway in myocardial dysfunction in type 2 diabetes mellitus. Curr. Diabetes Rep. 2017, 17, 38. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, J.; Bao, J.-K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Arias, E. Chaperone-mediated autophagy in protein quality control. Curr. Opin. Cell Biol. 2011, 23, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Glick, D.; Barth, S.; Macleod, F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell. Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell. Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell. Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Wei, J.; Childress, C.; Shaw, J.H.; Peng, K.; Shao, G.; Yang, W.; Lin, Q. The E3 ubiquitin ligase NEDD4 is an LC3-interactive protein and regulates autophagy. Autophagy 2017, 13, 522–537. [Google Scholar] [CrossRef] [Green Version]
- Riahi, Y.; Wikstrom, J.D.; Bachar-Wikstrom, E.; Polin, N.; Zucker, H.; Lee, M.S.; Quan, W.; Haataja, L.; Liu, M.; Arvan, P.; et al. Autophagy is a major regulator of β cell insulin homeostasis. Diabetologia 2016, 59, 1480–1491. [Google Scholar] [CrossRef] [Green Version]
- Bartolome, A.; Guillén, C.; Benito, M. Autophagy plays a protective role in endoplasmic reticulum stress-mediated pancreatic β cell death. Autophagy 2012, 8, 1757–1768. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.; Chung, K.W.; Kim, J.W.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; Jeong, Y.T.; et al. Loss of autophagy diminishes pancreatic β cell mass and function with resultant hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, J.F.; Costes, S.; Gurlo, T.; Glabe, C.G.; Butler, P.C. Autophagy defends pancreatic β cells from human islet amyloid polypeptide-induced toxicity. J. Clin. Investig. 2014, 124, 3489–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigihara, N.; Fukunaka, A.; Hara, A.; Komiya, K.; Honda, A.; Uchida, T.; Abe, H.; Toyofuku, Y.; Tamaki, M.; Ogihara, T.; et al. Human IAPP-induced pancreatic β cell toxicity and its regulation by autophagy. J. Clin. Investig. 2014, 124, 3634–3644. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, K.; Kim, M.J.; Lim, H.; Kim, K.H.; Kim, S.W.; Lee, E.S.; Kim, H.H.; Kim, S.J.; Hur, K.Y.; et al. An autophagy enhancer ameliorates diabetes of human IAPP-transgenic mice through clearance of amyloidogenic oligomer. Nat. Commun. 2021, 12, 183. [Google Scholar] [CrossRef]
- Rivera, J.F.; Gurlo, T.; Daval, M.; Huang, C.J.; Matveyenko, A.V.; Butler, P.C.; Costes, S. Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic β-cells: Protective role of p62-positive cytoplasmic inclusions. Cell Death Differ. 2011, 18, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Hernández, M.G.; Aguilar, A.G.; Burillo, J.; Oca, R.G.; Manca, M.A.; Novials, A.; Vizan, G.; Guillén, C.; Benito, M. Pancreatic β cells overexpressing hIAPP impaired mitophagy and unbalanced mitochondrial dynamics. Cell Death Dis. 2018, 9, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhansali, S.; Bhansali, A.; Walia, R.; Saikia, U.N.; Dhawan, V. Alterations in Mitochondrial Oxidative Stress and Mitophagy in Subjects with Prediabetes and Type 2 Diabetes Mellitus. Cell Death Dis. 2018, 9, 481. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.H.; Wei, Y.H. Role of mitochondrial dysfunction and dysregulation of Ca2+ homeostasis in the pathophysiology of insulin resistance and type 2 diabetes. J. Biomed. Sci. 2017, 24, 70. [Google Scholar] [CrossRef]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [Green Version]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Poletti, F.; Giorgi, C.; Pandolfi, P.P.; Pinton, P. Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell. Death Differ. 2013, 20, 1631–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanemura, M.; Ohmura, Y.; Deguchi, T.; Machida, T.; Tsukamoto, R.; Wada, H.; Kobayashi, S.; Marubashi, S.; Eguchi, H.; Ito, T.; et al. Rapamycin causes upregulation of autophagy and impairs islets function both in vitro and in vivo. Am. J. Transpl. 2012, 12, 102–114. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kuramoto, K.; Wang, N.; Situ, S.; Priyadarshini, M.; Zhang, W.; Cordoba-Chacon, J.; Layden, B.T.; He, C. Autophagy Differentially Regulates Insulin Production and Insulin Sensitivity. Cell. Rep. 2018, 23, 3286–3299. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wang, N.; Rocchi, A.; Zhang, W.; Vassar, R.; Zhou, Y.; He, C. Identification of natural products with neuronal and metabolic benefits through autophagy induction. Autophagy 2017, 13, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.S.; Blower, M.D. The Endoplasmic Reticulum: Structure, Function and Response to Cellular Signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voeltz, G.K.; Rolls, M.M.; Rapoport, T.A. Structural Organization of the Endoplasmic Reticulum. EMBO Rep. 2002, 3, 944–950. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The Mammalian Unfolded Protein Response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.P.; Weissman, J.S. Oxidative Protein Folding In Eukaryotes: Mechanisms and Consequences. J. Cell. Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Van Anken, E.; Braakman, I. Versatility of the endoplasmic reticulum protein folding factory. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 191–228. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Ron, D. Endoplasmic Reticulum Stress Signaling in Disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef]
- Mohan, S.; Brown, L.; Ayyappan, P. Endoplasmic reticulum stress: A master regulator of metabolic syndrome. Eur. J. Pharmacol. 2019, 860, 172553. [Google Scholar] [CrossRef]
- Chami, M.; Checler, F. Alterations of the Endoplasmic Reticulum (ER) Calcium Signaling Molecular Components in Alzheimer’s Disease. Cells 2020, 9, 2577. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.; Chae, H. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulation Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Cap, S.S.; Kaufman, R. Targeting endoplasmic reticulum stress in metabolic disease. Expert Opin. Targets.
- Cnop, M.; Toivonen, S.; Igoillo-Esteve, M.; Salpea, P. Endoplasmic reticulum stress and eIF2α phosphorylation: The Achilles heel of pancreatic β cells. Mol. Metab. 2017, 6, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Xu, W.; Wang, C.; Hua, J. X-box binding protein 1 (XBP1) function in diseases. Cell. Biol. Int. 2020. [Google Scholar] [CrossRef]
- Kaneko, M.; Imaizumi, K.; Saito, A.; Kanemoto, S.; Asada, R.; Matsuhisa, K.; Ohtake, Y. ER Stress and Disease: Toward Prevention and Treatment. Biol. Pharm. Bull. 2017, 40, 1337–1343. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Ariyasu, D.; Yoshida, H.; Hasegawa, Y. Endoplasmic Reticulum (ER) Stress and Endocrine Disorders. Int. J. Mol. Sci. 2017, 18, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakes, S.A.; Papa, F.R. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 101169. [Google Scholar] [CrossRef] [PubMed]
- Danilova, T.; Lindahl, M. Emerging Roles for Mesencephalic Astrocyte-Derived Neurotrophic Factor (MANF) in Pancreatic Β Cells and Diabetes. Front. Physiol. 2018, 9, 1457. [Google Scholar] [CrossRef]
- Wang, Y.; Vera, L.; Fischer, W.H.; Montminy, M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 2009, 460, 534–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zhou, R.; Zhang, C.; He, S.; Su, Z. Mitochondria-Associated Endoplasmic Reticulum Membranes in the Pathogenesis of Type 2 Diabetes Mellitus. Front. Cell. Dev. Biol. 2020, 8, 571554. [Google Scholar] [CrossRef]
- Boden, G. Endoplasmic reticulum stress: Another link between obesity and insulin resistance/inflammation? Diabetes 2009, 58, 518–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, S.E. Neurotransmitters. Curr. Biol. 2005, 15, R154–R158. [Google Scholar] [CrossRef] [Green Version]
- Sigel, E.; Steinmann, M.E. Structure, function, and modulation of GABA(A) receptors. J. Biol. Chem. 2012, 287, 40224–40231. [Google Scholar] [CrossRef] [Green Version]
- Sickmann, H.M.; Waagepetersen, H.S.; Schousboe, A.; Benie, A.J.; Bouman, S.D. Brain glycogen and its role in supporting glutamate and GABA homeostasis in a type 2 diabetes rat model. Neurochem. Int. 2012, 60, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Buseel, F.C.G.; Backes, W.H.; Hofman, P.A.M.; Puts, N.A.J.; Edden, R.A.E.; Boxtel, M.P.J.; Schram, M.T.; Stehouwer, C.D.A.; Wildberger, J.E.; Jansen, J.F.A. Increased GABA concentrations in type 2 diabetes mellitus are related to lower cognitive functioning. Medicine 2016, 95, e4803. [Google Scholar] [CrossRef] [PubMed]
- Thielen, J.W.; Gancheva, S.; Hong, D.; Rankouhi, S.R.; Chen, B.; Apostolopoulou, M.; Anadol-Schmitz, E.; Roden, M.; Norris, D.G.; Tendolkar, I. Higher GABA concentration in the medial prefrontal cortex of Type 2 diabetes patients is associated with episodic memory dysfunction. Hum. Brain. Mapp. 2019, 40, 4287–4295. [Google Scholar] [CrossRef]
- García-Tornadú, I.; Ornstein, A.M.; Chamson-Reig, A.; Wheeler, M.B.; Hill, D.J.; Arany, E.; Rubinstein, M.; Becu-Villalobos, D. Disruption of the dopamine d2 receptor impairs insulin secretion and causes glucose intolerance. Endocrinology 2010, 151, 1441–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, E.; Santhosh, K.T.; Paulose, C.S. Dopaminergic regulation of glucose-induced insulin secretion through dopamine D2 receptors in the pancreatic islets in vitro. IUBMB Life 2006, 58, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Morgan, N.G.; Montague, W. Studies on the mechanism of inhibition of glucose-stimulated insulin secretion by noradrenaline in rat islets of Langerhans. Biochem. J. 1985, 226, 571–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zern, R.T.; Bird, J.L.; Feldman, J.M. Effect of increased pancreatic islet norepinephrine, dopamine and serotonin concentration on insulin secretion in the golden hamster. Diabetologia 1980, 18, 341–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, D.L. Amylin. Headache 2017, 57, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.E.; Lutz, T.A. Amylin and Leptin: Co-Regulators of Energy Homeostasis and Neuronal Development. Trends Endocrinol. Metab. 2017, 28, 153–164. [Google Scholar] [CrossRef]
- Boyle, C.N.; Lutz, T.A.; Le Foll, C. Amylin—Its Role in the Homeostatic and Hedonic Control of Eating and Recent Developments of Amylin Analogs to Treat Obesity. Mol. Metab. 2018, 8, 203–210. [Google Scholar] [CrossRef]
- Verchere, C.B.; D’Alessio, D.A.; Palmiter, R.D.; Weir, G.C.; Bonner-Weir, S.; Baskin, D.G.; Kahn, S.E. Islet Amyloid Formation Associated with Hyperglycemia in Transgenic Mice with Pancreatic Β Cell Expression of Human Islet Amyloid Polypeptide. Proc. Natl. Acad. Sci. USA 1996, 93, 3492–3496. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, S.G.; Gromada, J.; Urano, F. Endoplasmic Reticulum Stress and Pancreatic β-Cell Death. Trends Endocrinol. Metab. 2011. [Google Scholar] [CrossRef] [Green Version]
- Saghir, A.E.; Farrugia, G.; Vassallo, N. The Human Islet Amyloid Polypeptide in Protein Misfolding Disorders: Mechanisms of Aggregation and Interaction with Biomembranes. Chem. Phys. Lipids 2020, 105010. [Google Scholar] [CrossRef]
- Raleigh, D.; Zhang, X.; Hastoy, B.; Clark, A. The β-Cell Assassin: IAPP Cytotoxicity. J. Mol. Endocrinol. 2017, 59, R121–R140. [Google Scholar] [CrossRef] [PubMed]
- Matveyenko, A.V.; Gurlo, T.; Daval, M.; Butler, A.E.; Butler, P.C. Successful versus Failed Adaptation to High-Fat Diet-Induced Insulin Resistance: The Role of IAPP-Induced β-Cell Endoplasmic Reticulum Stress. Diabetes 2009, 58, 906–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.M.; Zhang, Q.; Zheng, M.; Fan, Z.H.; Li, Y.H.; Zhang, D.; Zhang, Z.; Yuan, S.S.; Wang, Y.Y.; Zhou, P.; et al. Protective Effects of a G. Lucidum Proteoglycan on INS-1 Cells against IAPP-Induced Apoptosis via Attenuating Endoplasmic Reticulum Stress and Modulating CHOP/JNK Pathways. Int. J. Biol. Macromol. 2018, 106, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Cadavez, L.; Montane, J.; Alcarraz-Vizán, G.; Visa, M.; Vidal-Fàbrega, L.; Servitja, J.M.; Novials, A. Chaperones Ameliorate Β Cell Dysfunction Associated with Human Islet Amyloid Polypeptide Overexpression. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Montane, J.; De Pablo, S.; Castaño, C.; Rodríguez-Comas, J.; Cadavez, L.; Obach, M.; Visa, M.; Alcarraz-Vizán, G.; Sanchez-Martinez, M.; Nonell-Canals, A.; et al. Amyloid-Induced b-Cell Dysfunction and Islet Inflammation Are Ameliorated by 4-Phenylbutyrate (PBA) Treatment. FASEB J. 2017, 31, 5296–5306. [Google Scholar] [CrossRef] [Green Version]
- Wali, J.; Masters, S.; Thomas, H. Linking Metabolic Abnormalities to Apoptotic Pathways in Β Cells in Type 2 Diabetes. Cells 2013, 2, 266–283. [Google Scholar] [CrossRef] [Green Version]
- Sassano, M.L.; van Vliet, A.R.; Agostinis, P. Mitochondria-Associated Membranes as Networking Platforms and Regulators of Cancer Cell Fate. Front. Oncol. 2017. [Google Scholar] [CrossRef]
- Bartolomé, A.; Kimura-Koyanagi, M.; Asahara, S.I.; Guillén, C.; Inoue, H.; Teruyama, K.; Shimizu, S.; Kanno, A.; García-Aguilar, A.; Koike, M.; et al. Pancreatic β-Cell Failure Mediated by MTORC1 Hyperactivity and Autophagic Impairment. Diabetes 2014, 63, 2996–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in Lysosomal and Proteasomal Markers in Parkinson’s Disease: Relationship to Alpha-Synuclein Inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Zhang, J.; Jiao, A.; Wang, B.; Chen, B.; Lin, J. Resveratrol Attenuates HIAPP Amyloid Formation and Restores the Insulin Secretion Ability in HIAPP-INS1 Cell Line via Enhancing Autophagy. Can. J. Physiol. Pharmacol. 2019, 97, 82–89. [Google Scholar] [CrossRef]
- Burillo, J.; Fernández-Rhodes, M.; Piquero, M.; López-Alvarado, P.; Menéndez, J.C.; Jiménez, B.; González-Blanco, C.; Marqués, P.; Guillén, C.; Benito, M. Human Amylin Aggregates Release within Exosomes as a Protective Mechanism in Pancreatic β Cells: Pancreatic β-Hippocampal Cell Communication. Biochim. Biophys. Acta Mol. Cell Res. 2021, 118971. [Google Scholar] [CrossRef] [PubMed]
- Kegulian, N.C.; Sankhagowit, S.; Apostolidou, M.; Jayasinghe, S.A.; Malmstadt, N.; Butler, P.C.; Langen, R. Membrane Curvature-Sensing and Curvature-Inducing Activity of Islet Amyloid Polypeptide and Its Implications for Membrane Disruption. J. Biol. Chem. 2015, 290, 25782–25793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.L.; Chen, T.; Wong, Y.S.; Xu, G.; Fan, R.R.; Zhao, H.L.; Chan, J.C.N. Involvement of Mitochondrial Dysfunction in Human Islet Amyloid Polypeptide-Induced Apoptosis in INS-1E Pancreatic Β Cells: An Effect Attenuated by Phycocyanin. Int. J. Biochem. Cell Biol. 2011, 43, 525–534. [Google Scholar] [CrossRef]
- Li, X.L.; Wong, Y.S.; Xu, G.; Chan, J.C.N. Selenium-Enriched Spirulina Protects INS-1E Pancreatic Β Cells from Human Islet Amyloid Polypeptide-Induced Apoptosis through Suppression of ROS-Mediated Mitochondrial Dysfunction and PI3/AKT Pathway. Eur. J. Nutr. 2015, 54, 509–522. [Google Scholar] [CrossRef]
- Kilmer, P.D. Role of Pancreatic β-Cell Death and Inflammation in Diabetes. J. Theorypract. Crit. 2010, 11, 369–373. [Google Scholar] [CrossRef]
- Masters, S.L.; O’Neill, L.A.J. Disease-Associated Amyloid and Misfolded Protein Aggregates Activate the Inflammasome. Trends Mol. Med. 2011. [Google Scholar] [CrossRef]
- Meier, D.T.; Morcos, M.; Samarasekera, T.; Zraika, S.; Hull, R.L.; Kahn, S.E. Islet Amyloid Formation Is an Important Determinant for Inducing Islet Inflammation in High-Fat-Fed Human IAPP Transgenic Mice. Diabetologia 2014, 57, 1884–1888. [Google Scholar] [CrossRef]
- Westwell-Roper, C.; Dai, D.L.; Soukhatcheva, G.; Potter, K.J.; van Rooijen, N.; Ehses, J.A.; Verchere, C.B. IL-1 Blockade Attenuates Islet Amyloid Polypeptide-Induced Proinflammatory Cytokine Release and Pancreatic Islet Graft Dysfunction. J. Immunol. 2011, 187, 2755–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westwell-Roper, C.Y.; Ehses, J.A.; Verchere, C.B. Resident Macrophages Mediate Islet Amyloid Polypeptide-Induced Islet IL-1β Production and β-Cell Dysfunction. Diabetes 2014, 63, 1698–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westwell-Roper, C.Y.; Chehroudi, C.A.; Denroche, H.C.; Courtade, J.A.; Ehses, J.A.; Verchere, C.B. IL-1 Mediates Amyloid-Associated Islet Dysfunction and Inflammation in Human Islet Amyloid Polypeptide Transgenic Mice. Diabetologia 2015, 58, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Templin, A.T.; Mellati, M.; Meier, D.T.; Esser, N.; Hogan, M.F.; Castillo, J.J.; Akter, R.; Raleigh, D.P.; Zraika, S.; Hull, R.L.; et al. Low Concentration IL-1β Promotes Islet Amyloid Formation by Increasing HIAPP Release from Humanised Mouse Islets in Vitro. Diabetologia 2020, 63, 2385–2395. [Google Scholar] [CrossRef]
- Hui, Q.; Asadi, A.; Park, Y.J.; Kieffer, T.J.; Ao, Z.; Warnock, G.L.; Marzban, L. Amyloid Formation Disrupts the Balance between Interleukin-1β and Interleukin-1 Receptor Antagonist in Human Islets. Mol. Metab. 2017, 6, 833–844. [Google Scholar] [CrossRef]
- Aftabizadeh, M.; Tatarek-Nossol, M.; Andreetto, E.; El Bounkari, O.; Kipp, M.; Beyer, C.; Latz, E.; Bernhagen, J.; Kapurniotu, A. Blocking Inflammasome Activation Caused by β-Amyloid Peptide (Aβ) and Islet Amyloid Polypeptide (IAPP) through an IAPP Mimic. ACS Chem. Neurosci. 2019, 10, 3703–3717. [Google Scholar] [CrossRef]
- Morikawa, S.; Kaneko, N.; Okumura, C.; Taguchi, H.; Kurata, M.; Yamamoto, T.; Osawa, H.; Nakanishi, A.; Zako, T.; Masumoto, J. IAPP/Amylin Deposition, Which Is Correlated with Expressions of ASC and IL-1β in β-Cells of Langerhans’ Islets, Directly Initiates NLRP3 Inflammasome Activation. Int. J. Immunopathol. Pharm. 2018, 32. [Google Scholar] [CrossRef]
- Wang, E.; Zhu, H.; Wang, X.; Gower, A.C.; Wallack, M.; Blusztajn, J.K.; Kowall, N.; Qiu, W.Q. Amylin Treatment Reduces Neuroinflammation and Ameliorates Abnormal Patterns of Gene Expression in the Cerebral Cortex of an Alzheimer’s Disease Mouse Model. J. Alzheimer’s Dis. 2017, 56, 47–61. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Xue, X.; Wang, E.; Wallack, M.; Na, H.; Hooker, J.M.; Kowall, N.; Tao, Q.; Stein, T.D.; Wolozin, B.; et al. Amylin Receptor Ligands Reduce the Pathological Cascade of Alzheimer’s Disease. Neuropharmacology 2017, 119, 170–181. [Google Scholar] [CrossRef]
- Fu, W.; Vukojevic, V.; Patel, A.; Soudy, R.; MacTavish, D.; Westaway, D.; Kaur, K.; Goncharuk, V.; Jhamandas, J. Role of Microglial Amylin Receptors in Mediating Β Amyloid (Aβ)-Induced Inflammation. J. Neuroinflammation 2017, 14. [Google Scholar] [CrossRef] [Green Version]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012. [Google Scholar] [CrossRef] [Green Version]
- Ray Dorsey, E.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.Y.J.; Collado-Mateo, D.; et al. Global, Regional, and National Burden of Parkinson’s Disease, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Ott, A.; Stolk, R.P.; Van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M.B. Diabetes Mellitus and the Risk of Dementia: The Rotterdam Study. Neurology 1999. [Google Scholar] [CrossRef]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of Dementia in Diabetes Mellitus: A Systematic Review. Lancet Neurol. 2006. [Google Scholar] [CrossRef]
- Gudala, K.; Bansal, D.; Schifano, F.; Bhansali, A. Diabetes Mellitus and Risk of Dementia: A Meta-Analysis of Prospective Observational Studies. J. Diabetes Investig. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biessels, G.J.; Despa, F. Cognitive Decline and Dementia in Diabetes Mellitus: Mechanisms and Clinical Implications. Nat. Rev. Endocrinol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, L.; Rosset, I.; Roriz-Cruz, M. Global Epidemiology of Dementia: Alzheimer’s and Vascular Types. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease: Mechanism of Disease. N. Engl. J. Med. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubert, L.; Pichierri, S.; Hommet, C.; Camus, V.; Berrut, G.; De Decker, L. Association between Comorbidity Burden and Rapid Cognitive Decline in Individuals with Mild to Moderate Alzheimer’s Disease. J. Am. Geriatr. Soc. 2015. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheime’s Disease. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Recuero, M.; Serrano, E.; Bullido, M.J.; Valdivieso, F. Aβ Production as Consequence of Cellular Death of a Human Neuroblastoma Overexpressing APP. FEBS Lett. 2004. [Google Scholar] [CrossRef] [Green Version]
- Lammich, S.; Kojro, E.; Postina, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Haass, C.; Fahrenholz, F. Constitutive and Regulated α-Secretase Cleavage of Alzheimer’s Amyloid Precursor Protein by a Disintegrin Metalloprotease. Proc. Natl. Acad. Sci. USA 1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Multhaup, G.; Huber, O.; Buée, L.; Galas, M.C. Amyloid Precursor Protein (APP) Metabolites APP Intracellular Fragment (AICD), Aβ42, and Tau in Nuclear Roles. J. Biol. Chem. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.M.; Selkoe, D.J. Aβ Oligomers—A Decade of Discovery. J. Neurochem. 2007. [Google Scholar] [CrossRef]
- Wang, J.Z.; Xia, Y.Y.; Grundke-Iqbal, I.; Iqbal, K. Abnormal Hyperphosphorylation of Tau: Sites, Regulation, and Molecular Mechanism of Neurofibrillary Degeneration. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1). [Google Scholar] [CrossRef]
- Freude, S.; Schilbach, K.; Schubert, M. The Role of IGF-1 Receptor and Insulin Receptor Signaling for the Pathogenesis of Alzheimers Disease: From Model Organisms to Human Disease. Curr. Alzheimer’s Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; De La Monte, S.M. Impaired Insulin and Insulin-like Growth Factor Expression and Signaling Mechanisms in Alzheimer’s Disease—Is This Type 3 Diabetes? J. Alzheimer’s Dis. 2005. [Google Scholar] [CrossRef] [Green Version]
- De La Monte, S.M.; Wands, J.R. Review of Insulin and Insulin-like Growth Factor Expression, Signaling, and Malfunction in the Central Nervous System: Relevance to Alzheimer’s Disease. J. Alzheimer’s Dis. 2005. [Google Scholar] [CrossRef] [Green Version]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated Brain Insulin Resistance in Alzheimer’s Disease Patients Is Associated with IGF-1 Resistance, IRS-1 Dysregulation, and Cognitive Decline. J. Clin. Investig. 2012. [Google Scholar] [CrossRef] [Green Version]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; De La Monte, S.M. Insulin and Insulin-like Growth Factor Expression and Function Deteriorate with Progression of Alzheimer’s Disease: Link to Brain Reductions in Acetylcholine. J. Alzheimer’s Dis. 2005. [Google Scholar] [CrossRef]
- Pandini, G.; Pace, V.; Copani, A.; Squatrito, S.; Milardi, D.; Vigneri, R. Insulin Has Multiple Antiamyloidogenic Effects on Human Neuronal Cells. Endocrinology 2013, 154, 375–387. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Alldred, M.J.; Ginsberg, S.D.; Ohno, M. Mechanisms Underlying Insulin Deficiency-Induced Acceleration of β-Amyloidosis in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, J.R.; Lyra e Silva, N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razolli, D.; Carvalho, B.M.; Frazão, R.; et al. Alzheimer-associated Aβ Oligomers Impact the Central Nervous System to Induce Peripheral Metabolic Deregulation. EMBO Mol. Med. 2015, 7, 190–210. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guénette, S. Insulin-Degrading Enzyme Regulates the Levels of Insulin, Amyloid β-Protein, and the β-Amyloid Precursor Protein Intracellular Domain in Vivo. Proc. Natl. Acad. Sci. USA 2003. [Google Scholar] [CrossRef] [Green Version]
- De La Monte, S.M. Contributions of Brain Insulin Resistance and Deficiency in Amyloid-Related Neurodegeneration in Alzheimers Disease. Drugs 2012. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Helmerhorst, E.; Taddei, K.; Plewright, B.; Van Bronswijk, W.; Martins, R. Alzheimer’s Β-Amyloid Peptides Compete for Insulin Binding to the Insulin Receptor. J. Neurosci. 2002. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.Q.; Wu, D.K.; Liu, J.K. MTOR and Tau Phosphorylated Proteins in the Hippocampal Tissue of Rats with Type 2 Diabetes and Alzheimer’s Disease. Mol. Med. Rep. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbondante, S.; Baglietto-Vargas, D.; Rodriguez-Ortiz, C.J.; Estrada-Hernandez, T.; Medeiros, R.; LaFerla, F.M. Genetic Ablation of Tau Mitigates Cognitive Impairment Induced by Type 1 Diabetes. Am. J. Pathol. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Z.; Jiao, Z.; Sun, X.; Zhao, Y.; Ren, J.; Xu, G. Effects of Streptozotocin-Induced Diabetes on Tau Phosphorylation in the Rat Brain. Brain Res. 2011. [Google Scholar] [CrossRef]
- Lu, Y.; Jiang, X.; Liu, S.; Li, M. Changes in Cerebrospinal Fluid Tau and β-Amyloid Levels in Diabetic and Prediabetic Patients: A Meta-Analysis. Front. Aging Neurosci. 2018. [Google Scholar] [CrossRef]
- Hong, M.; Lee, V.M.Y. Insulin and Insulin-like Growth Factor-1 Regulate Tau Phosphorylation in Cultured Human Neurons. J. Biol. Chem. 1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monte, S. Brain Insulin Resistance and Deficiency as Therapeutic Targets in Alzheimers Disease. Curr. Alzheimer Res. 2012. [Google Scholar] [CrossRef]
- Clodfelder-Miller, B.J.; Zmijewska, A.A.; Johnson, G.V.W.; Jope, R.S. Tau Is Hyperphosphorylated at Multiple Sites in Mouse Brain in Vivo after Streptozotocin-Induced Insulin Deficiency. Diabetes 2006, 55, 3320–3325. [Google Scholar] [CrossRef] [Green Version]
- Diehl, T.; Mullins, R.; Kapogiannis, D. Insulin Resistance in Alzheimer’s Disease. Transl. Res. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macauley, S.L.; Stanley, M.; Caesar, E.E.; Yamada, S.A.; Raichle, M.E.; Perez, R.; Mahan, T.E.; Sutphen, C.L.; Holtzman, D.M. Hyperglycemia Modulates Extracellular Amyloid-β Concentrations and Neuronal Activity in Vivo. J. Clin. Investig. 2015, 125, 2463–2467. [Google Scholar] [CrossRef]
- Valente, T.; Gella, A.; Fernàndez-Busquets, X.; Unzeta, M.; Durany, N. Immunohistochemical Analysis of Human Brain Suggests Pathological Synergism of Alzheimer’s Disease and Diabetes Mellitus. Neurobiol. Dis. 2010. [Google Scholar] [CrossRef]
- Arab, L.; Sadeghi, R.; Walker, D.; Lue, L.-F.; Sabbagh, M. Consequences of Aberrant Insulin Regulation in the Brain: Can Treating Diabetes Be Effective for Alzheimers Disease. Curr. Neuropharmacol. 2011. [Google Scholar] [CrossRef] [Green Version]
- Chuah, Y.K.; Basir, R.; Talib, H.; Tie, T.H.; Nordin, N. Receptor for Advanced Glycation End Products and Its Involvement in Inflammatory Diseases. Int. J. Inflamm. 2013. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Bell, R.; Sagare, A.; Zlokovic, B. Clearance of Amyloid-β Peptide Across the Blood-Brain Barrier: Implication for Therapies in Alzheimers Disease. CNS Neurol. Disord. Drug Targets 2009. [Google Scholar] [CrossRef]
- Li, X.H.; Lv, B.L.; Xie, J.Z.; Liu, J.; Zhou, X.W.; Wang, J.Z. AGEs Induce Alzheimer-like Tau Pathology and Memory Deficit via RAGE-Mediated GSK-3 Activation. Neurobiol. Aging 2012. [Google Scholar] [CrossRef]
- Kong, Y.; Wang, F.; Wang, J.; Liu, C.; Zhou, Y.; Xu, Z.; Zhang, C.; Sun, B.; Guan, Y. Pathological Mechanisms Linking Diabetes Mellitus and Alzheimer’s Disease: The Receptor for Advanced Glycation End Products (RAGE). Front. Aging Neurosci. 2020, 12, 217. [Google Scholar] [CrossRef] [PubMed]
- Batkulwar, K.; Godbole, R.; Banarjee, R.; Kassaar, O.; Williams, R.J.; Kulkarni, M.J. Advanced Glycation End Products Modulate Amyloidogenic APP Processing and Tau Phosphorylation: A Mechanistic Link between Glycation and the Development of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Timberlake, M.A.; Dwivedi, Y. Altered Expression of Endoplasmic Reticulum Stress Associated Genes in Hippocampus of Learned Helpless Rats: Relevance to Depression Pathophysiology. Front. Pharm. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerakis, Y.; Hetz, C. Emerging Roles of ER Stress in the Etiology and Pathogenesis of Alzheimer’s Disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [Green Version]
- Alberdi, E.; Wyssenbach, A.; Alberdi, M.; Sánchez-Gómez, M.V.; Cavaliere, F.; Rodríguez, J.J.; Verkhratsky, A.; Matute, C. Ca2+-Dependent Endoplasmic Reticulum Stress Correlates with Astrogliosis in Oligomeric Amyloid β-Treated Astrocytes and in a Model of Alzheimer’s Disease. Aging Cell 2013. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.O.; Lacor, P.N.; Ferreira, I.L.; Resende, R.; Auberson, Y.P.; Klein, W.L.; Oliveira, C.R.; Rego, A.C.; Pereira, C.M.F. Endoplasmic Reticulum Stress Occurs Downstream of GluN2B Subunit of N-Methyl-d-Aspartate Receptor in Mature Hippocampal Cultures Treated with Amyloid-β Oligomers. Aging Cell 2012. [Google Scholar] [CrossRef]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s Disease with IPSCs Reveals Stress Phenotypes Associated with Intracellular Aβ and Differential Drug Responsiveness. Cell Stem Cell 2013. [Google Scholar] [CrossRef] [Green Version]
- Tseng, B.P.; Green, K.N.; Chan, J.L.; Blurton-Jones, M.; LaFerla, F.M. Aβ Inhibits the Proteasome and Enhances Amyloid and Tau Accumulation. Neurobiol. Aging 2008, 29, 1607–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, L.; Ohno, M. PERK Mediates EIF2α Phosphorylation Responsible for BACE1 Elevation, CREB Dysfunction and Neurodegeneration in a Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2014. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the Translation Initiation Factor EIF2α Increases BACE1 Levels and Promotes Amyloidogenesis. Neuron 2008. [Google Scholar] [CrossRef] [Green Version]
- Baleriola, J.; Walker, C.A.; Jean, Y.Y.; Crary, J.F.; Troy, C.M.; Nagy, P.L.; Hengst, U. Axonally Synthesized ATF4 Transmits a Neurodegenerative Signal across Brain Regions. Cell 2014. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of EIF2α Kinases Alleviates Alzheimer’s Disease-Related Plasticity and Memory Deficits. Nat. Neurosci. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 Controls Diverse Cell Type- and Condition-Specific Transcriptional Regulatory Networks. Mol. Cell 2007. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, S.; Schuck, F.; Grösgen, S.; Riemenschneider, M.; Hartmann, T.; Postina, R.; Grimm, M.; Endres, K. Unfolded Protein Response Signaling by Transcription Factor XBP-1 Regulates ADAM10 and Is Affected in Alzheimer’s Disease. FASEB J. 2014. [Google Scholar] [CrossRef]
- Gerakis, Y.; Dunys, J.; Bauer, C.; Checler, F. Aβ42 Oligomers Modulate β-Secretase through an XBP-1s-Dependent Pathway Involving HRD1. Sci. Rep. 2016. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, M.; Koike, H.; Saito, R.; Kitamura, Y.; Okuma, Y.; Nomura, Y. Loss of HRD1-Mediated Protein Degradation Causes Amyloid Precursor Protein Accumulation and Amyloid-β Generation. J. Neurosci. 2010. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.C.; Chu, J.; Lin, L.; Song, J.; Ning, L.N.; Luo, H.B.; Yang, S.S.; Shi, Y.; Wang, Q.; Qu, N.; et al. SIL1 Rescued Bip Elevation-Related Tau Hyperphosphorylation in ER Stress. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Van Haastert, E.S.; Nijholt, D.A.T.; Rozemuller, A.J.M.; Eikelenboom, P.; Scheper, W. The Unfolded Protein Response Is Activated in Pretangle Neurons in Alzheimer’s Disease Hippocampus. Am. J. Pathol. 2009. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Joe, Y.; Kim, H.J.; Kim, Y.-S.; Jeong, S.O.; Pae, H.-O.; Ryter, S.W.; Surh, Y.-J.; Chung, H.T. Endoplasmic Reticulum Stress–Induced IRE1α Activation Mediates Cross-Talk of GSK-3β and XBP-1 To Regulate Inflammatory Cytokine Production. J. Immunol. 2015. [Google Scholar] [CrossRef] [Green Version]
- Duran-Aniotz, C.; Cornejo, V.H.; Espinoza, S.; Ardiles, Á.O.; Medinas, D.B.; Salazar, C.; Foley, A.; Gajardo, I.; Thielen, P.; Iwawaki, T.; et al. IRE1 Signaling Exacerbates Alzheimer’s Disease Pathogenesis. Acta Neuropathol. 2017. [Google Scholar] [CrossRef]
- Shen, Y.X.; Sun, A.M.; Fang, S.; Feng, L.J.; Li, Q.; Hou, H.L.; Liu, C.; Wang, H.P.; Shen, J.L.; Luo, J.; et al. Hrd1 Facilitates Tau Degradation and Promotes Neuron Survival. Curr. Mol. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Govindarajulu, M.; Jones, E.; Suppiramaniam, V.; Moore, T.; Dhanasekaran, M. Mitochondrial Dysfunction and the Role of Mitophagy in Alzheimer’s Disease. Alzheimer´s Disease Treatment 2018, 1–16. [Google Scholar] [CrossRef]
- Bhatia, V.; Sharma, S. Role of Mitochondrial Dysfunction, Oxidative Stress and Autophagy in Progression of Alzheimer’s Disease. J. Neurol. Sci. 2020, 421, 117253. [Google Scholar] [CrossRef]
- Nixon, R.A. The Calpains in Aging and Aging-Related Diseases. Ageing Res. Rev. 2003, 2, 407–418. [Google Scholar] [CrossRef]
- Gibson, G.E.; Chen, H.L.; Xu, H.; Qiu, L.; Xu, Z.; Denton, T.T.; Shi, Q. Deficits in the Mitochondrial Enzyme α-Ketoglutarate Dehydrogenase Lead to Alzheimer’s Disease-like Calcium Dysregulation. Neurobiol. Aging 2012, 33, 1121.e13–1121.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The Amyloid β-Peptide Is Imported into Mitochondria via the TOM Import Machinery and Localized to Mitochondrial Cristae. Proc. Natl. Acad. Sci. USA 2008. [Google Scholar] [CrossRef] [Green Version]
- Takuma, K.; Fang, F.; Zhang, W.; Yan, S.; Fukuzaki, E.; Du, H.; Sosunov, A.; McKhann, G.; Funatsu, Y.; Nakamichi, N.; et al. RAGE-Mediated Signaling Contributes to Intraneuronal Transport of Amyloid-β and Neuronal Dysfunction. Proc. Natl. Acad. Sci. USA 2009. [Google Scholar] [CrossRef] [Green Version]
- Atamna, H.; Frey, W.H. A Role for Heme in Alzheimer’s Disease: Heme Binds Amyloid β and Has Altered Metabolism. Proc. Natl. Acad. Sci. USA 2004. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of Amyloid Precursor Protein in the Mitochondrial Import Channels of Human Alzheimer’s Disease Brain Is Associated with Mitochondrial Dysfunction. J. Neurosci. 2006. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Kerr, P.M.; Javadov, S.; Woodfield, K.Y. Elucidating the Molecular Mechanism of the Permeability Transition Pore and Its Role in Reperfusion Injury of the Heart. Biochim. Biophys. Acta Bioenerg. 1998. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.K.; Carlson, E.A.; Yan, S.S. Mitochondrial Permeability Transition Pore Is a Potential Drug Target for Neurodegeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöneich, C.; Pogocki, D.; Hug, G.L.; Bobrowski, K. Free Radical Reactions of Methionine in Peptides: Mechanisms Relevant to β-Amyloid Oxidation and Alzheimer’s Disease. J. Am. Chem. Soc. 2003. [Google Scholar] [CrossRef]
- Schafer, M.J.; Alldred, M.J.; Lee, S.H.; Calhoun, M.E.; Petkova, E.; Mathews, P.M.; Ginsberg, S.D. Reduction of β-Amyloid and γ-Secretase by Calorie Restriction in Female Tg2576 Mice. Neurobiol. Aging 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briston, T.; Hicks, A.R. Mitochondrial Dysfunction and Neurodegenerative Proteinopathies: Mechanisms and Prospects for Therapeutic Intervention. Biochem. Soc. Trans. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, A.; Nisbet, R.; Grimm, A.; Götz, J. March Separate, Strike Together—Role of Phosphorylated TAU in Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal Interaction of VDAC1 with Amyloid Β and Phosphorylated Tau Causes Mitochondrial Dysfunction in Alzheimer’s Disease. Hum. Mol. Genet. 2012. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired Mitochondrial Dynamics and Abnormal Interaction of Amyloid Β with Mitochondrial Protein Drp1 in Neurons from Patients with Alzheimer’s Disease: Implications for Neuronal Damage. Hum. Mol. Genet. 2011. [Google Scholar] [CrossRef]
- Manczak, M.; Sesaki, H.; Kageyama, Y.; Reddy, P.H. Dynamin-Related Protein 1 Heterozygote Knockout Mice Do Not Have Synaptic and Mitochondrial Deficiencies. Biochim. Biophys. Acta Mol. Basis Dis. 2012. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.H.; Cai, Q. Mitochondrial Transport in Neurons: Impact on Synaptic Homeostasis and Neurodegeneration. Nat. Rev. Neurosci. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal Mutant APP and Amyloid Β-Induced Cognitive Decline, Dendritic Spine Loss, Defective Autophagy, Mitophagy and Mitochondrial Abnormalities in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2018. [Google Scholar] [CrossRef] [Green Version]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal Phosphorylated Tau Induced Cognitive Decline, Dendritic Spine Loss and Mitochondrial Abnormalities in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-Mediated Mitophagy in Mutant HAPP Neurons and Alzheimer’s Disease Patient Brains. Hum. Mol. Genet. 2015. [Google Scholar] [CrossRef]
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 Enhancement Is Able to Compensate Mitophagy Alterations Found in Sporadic Alzheimer’s Disease. Hum. Mol. Genet. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsetti, V.; Florenzano, F.; Atlante, A.; Bobba, A.; Ciotti, M.T.; Natale, F.; Della Valle, F.; Borreca, A.; Manca, A.; Meli, G.; et al. NH2-Truncated Human Tau Induces Deregulated Mitophagy in Neurons by Aberrant Recruitment of Parkin and UCHL-1: Implications in Alzheimer’s Disease. Hum. Mol. Genet. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau Accumulation Impairs Mitophagy via Increasing Mitochondrial Membrane Potential and Reducing Mitochondrial Parkin. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, G.; Schwarz, T.L. Pink1- And PARK2-Mediated Local Mitophagy in Distal Neuronal Axons. Autophagy 2015. [Google Scholar] [CrossRef]
- Nie, D.; Di Nardo, A.; Han, J.M.; Baharanyi, H.; Kramvis, I.; Huynh, T.; Dabora, S.; Codeluppi, S.; Pandolfi, P.P.; Pasquale, E.B.; et al. Tsc2-Rheb Signaling Regulates EphA-Mediated Axon Guidance. Nat. Neurosci. 2010. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Lu, Y.; Lee, J.K.; Samara, R.; Willenberg, R.; Sears-Kraxberger, I.; Tedeschi, A.; Park, K.K.; Jin, D.; Cai, B.; et al. PTEN Deletion Enhances the Regenerative Ability of Adult Corticospinal Neurons. Nat. Neurosci. 2010. [Google Scholar] [CrossRef] [Green Version]
- Stoica, L.; Zhu, P.J.; Huang, W.; Zhou, H.; Kozma, S.C.; Costa-Mattioli, M. Selective Pharmacogenetic Inhibition of Mammalian Target of Rapamycin Complex I (MTORC1) Blocks Long-Term Synaptic Plasticity and Memory Storage. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef] [Green Version]
- Henry, F.E.; Hockeimer, W.; Chen, A.; Mysore, S.P.; Sutton, M.A. Mechanistic Target of Rapamycin Is Necessary for Changes in Dendritic Spine Morphology Associated with Long-Term Potentiation. Mol. Brain 2017. [Google Scholar] [CrossRef] [Green Version]
- Russo, E.; Follesa, P.; Citraro, R.; Camastra, C.; Donato, A.; Isola, D.; Constanti, A.; De Sarro, G.; Donato, G. The MTOR Signaling Pathway and Neuronal Stem/Progenitor Cell Proliferation in the Hippocampus Are Altered during the Development of Absence Epilepsy in a Genetic Animal Model. Neurol. Sci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Dibbens, L.M.; De Vries, B.; Donatello, S.; Heron, S.E.; Hodgson, B.L.; Chintawar, S.; Crompton, D.E.; Hughes, J.N.; Bellows, S.T.; Klein, K.M.; et al. Mutations in DEPDC5 Cause Familial Focal Epilepsy with Variable Foci. Nat. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Neff, F.; Flores-Dominguez, D.; Ryan, D.P.; Horsch, M.; Schröder, S.; Adler, T.; Afonso, L.C.; Aguilar-Pimentel, J.A.; Becker, L.; Garrett, L.; et al. Rapamycin Extends Murine Lifespan but Has Limited Effects on Aging. J. Clin. Investig. 2013. [Google Scholar] [CrossRef] [Green Version]
- Ozcelik, S.; Fraser, G.; Castets, P.; Schaeffer, V.; Skachokova, Z.; Breu, K.; Clavaguera, F.; Sinnreich, M.; Kappos, L.; Goedert, M.; et al. Rapamycin Attenuates the Progression of Tau Pathology in P301S Tau Transgenic Mice. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tramutola, A.; Triplett, J.C.; Di Domenico, F.; Niedowicz, D.M.; Murphy, M.P.; Coccia, R.; Perluigi, M.; Butterfield, D.A. Alteration of MTOR Signaling Occurs Early in the Progression of Alzheimer Disease (AD): Analysis of Brain from Subjects with Pre-Clinical AD, Amnestic Mild Cognitive Impairment and Late-Stage AD. J. Neurochem. 2015, 133, 739–749. [Google Scholar] [CrossRef]
- Orr, M.E.; Salinas, A.; Buffenstein, R.; Oddo, S. Mammalian Target of Rapamycin Hyperactivity Mediates the Detrimental Effects of a High Sucrose Diet on Alzheimer’s Disease Pathology. Neurobiol. Aging 2014. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing Autophagy by Rapamycin before, but Not after, the Formation of Plaques and Tangles Ameliorates Cognitive Deficits. PLoS ONE 2011. [Google Scholar] [CrossRef] [Green Version]
- Bitel, C.L.; Kasinathan, C.; Kaswala, R.H.; Klein, W.L.; Frederikse, P.H. Amyloid-β and Tau Pathology of Alzheimer’s Disease Induced by Diabetes in a RABBIT Animal Model. J. Alzheimer’s Dis. 2012. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, D.; Zhang, W.; Liu, J.; Chen, S.; Hua, B. Investigation of Pi3K/PKB/MTOR/S6K1 Signaling Pathway in Relationship of Type 2 Diabetes and Alzheimer’s Disease. Int. J. Clin. Exp. Med. 2015, 8, 18581–18590. [Google Scholar]
- De la Monte, S.M. Type 3 Diabetes Is Sporadic Alzheimer-s Disease: Mini-Review. Eur. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid Β Oligomers Induce Impairment of Neuronal Insulin Receptors. FASEB J. 2008. [Google Scholar] [CrossRef] [Green Version]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An Anti-Diabetes Agent Protects the Mouse Brain from Defective Insulin Signaling Caused by Alzheimer’s Disease-Associated Aβ Oligomers. J. Clin. Investig. 2012. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Toledo, J.B.; Lee, E.B.; Arvanitakis, Z.; Kazi, H.; Han, L.Y.; Louneva, N.; Lee, V.M.Y.; Kim, S.F.; Trojanowski, J.Q.; et al. Abnormal Serine Phosphorylation of Insulin Receptor Substrate 1 Is Associated with Tau Pathology in Alzheimer’s Disease and Tauopathies. Acta Neuropathol. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschoal, V.A.; Amano, M.T.; Belchior, T.; Magdalon, J.; Chimin, P.; Andrade, M.L.; Ortiz-Silva, M.; Castro, É.; Yamashita, A.S.; Rosa Neto, J.C.; et al. MTORC1 Inhibition with Rapamycin Exacerbates Adipose Tissue Inflammation in Obese Mice and Dissociates Macrophage Phenotype from Function. Immunobiology 2017. [Google Scholar] [CrossRef] [PubMed]
- Dasuri, K.; Zhang, L.; Kim, S.O.K.F.; Bruce-Keller, A.J.; Keller, J.N. Dietary and Donepezil Modulation of MTOR Signaling and Neuroinflammation in the Brain. Biochim. Biophys. Acta Mol. Basis Dis. 2016. [Google Scholar] [CrossRef]
- Wang, J.P.; Zhang, M.Y. Role for Target of Rapamycin (MTOR) Signal Pathway in Regulating Neuronal Injury after Intracerebral Hemorrhage. Cell. Physiol. Biochem. 2017. [Google Scholar] [CrossRef]
- Dello Russo, C.; Lisi, L.; Tringali, G.; Navarra, P. Involvement of MTOR Kinase in Cytokine-Dependent Microglial Activation and Cell Proliferation. Biochem. Pharm. 2009. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, C.; Yao, Y.; Chen, L.; Liu, G.; Zhang, R.; Liu, Q.; Shi, F.D.; Hao, J. MTORC1 Pathway Disruption Ameliorates Brain Inflammation Following Stroke via a Shift in Microglia Phenotype from M1 Type to M2 Type. FASEB J. 2016. [Google Scholar] [CrossRef] [Green Version]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-α Mediates PKR-Dependent Memory Impairment and Brain IRS-1 Inhibition Induced by Alzheimer’s β-Amyloid Oligomers in Mice and Monkeys. Cell Metab. 2013. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Hoeffer, C.A.; Capetillo-Zarate, E.; Yu, F.; Wong, H.; Lin, M.T.; Tampellini, D.; Klann, E.; Blitzer, R.D.; Gouras, G.K. Dysregulation of the MTOR Pathway Mediates Impairment of Synaptic Plasticity in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2010. [Google Scholar] [CrossRef]
- Hoeffer, C.A.; Klann, E. MTOR Signaling: At the Crossroads of Plasticity, Memory and Disease. Trends Neurosci. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobert, D.; Topolnik, L.; Azzi, M.; Huang, L.; Badeaux, F.; DesGroseillers, L.; Sossin, W.S.; Lacaille, J.C. Forskolin Induction of Late-LTP and up-Regulation of 5′ TOP MRNAs Translation via MTOR, ERK, and PI3K in Hippocampal Pyramidal Cells. J. Neurochem. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antion, M.D.; Hou, L.; Wong, H.; Hoeffer, C.A.; Klann, E. MGluR-Dependent Long-Term Depression Is Associated with Increased Phosphorylation of S6 and Synthesis of Elongation Factor 1A but Remains Expressed in S6K-Deficient Mice. Mol. Cell Biol. 2008. [Google Scholar] [CrossRef] [Green Version]
- Bateup, H.S.; Takasaki, K.T.; Saulnier, J.L.; Denefrio, C.L.; Sabatini, B.L. Loss of Tsc1 in Vivo Impairs Hippocampal MGluR-LTD and Increases Excitatory Synaptic Function. J. Neurosci. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Zhao, M.; Han, Y.; Zhang, H. GABAergic Inhibitory Interneuron Deficits in Alzheimer’s Disease: Implications for Treatment. Front. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Barthet, G.; Mulle, C. Presynaptic Failure in Alzheimer’s Disease. Prog. Neurobiol. 2020, 194, 101801. [Google Scholar] [CrossRef]
- Sha, Z. Important Factors in the Formation and Clearance of Protein Aggregation. J. Dev. Drugs 2014. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive Involvement of Autophagy in Alzheimer Disease: An Immuno-Electron Microscopy Study. J. Neuropathol. Exp. Neurol. 2005. [Google Scholar] [CrossRef] [Green Version]
- Button, R.W.; Luo, S.; Rubinsztein, D.C. Autophagic Activity in Neuronal Cell Death. Neurosci. Bull. 2015. [Google Scholar] [CrossRef] [Green Version]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The Autophagy-Related Protein Beclin 1 Shows Reduced Expression in Early Alzheimer Disease and Regulates Amyloid β Accumulation in Mice. J. Clin. Investig. 2008. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular Interplay between Mammalian Target of Rapamycin (MTOR), Amyloid-β, and Tau: Effects on Cognitive Impairments. J. Biol. Chem. 2010. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Gestwicki, J.E.; Murphy, L.O.; Klionsky, D.J. Potential Therapeutic Applications of Autophagy. Nat. Rev. Drug Discov. 2007. [Google Scholar] [CrossRef]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Aβ Secretion and Plaque Formation Depend on Autophagy. Cell. Rep. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Su, Y.; Wang, J.; Sun, S.; Wang, T.; Qiao, X.; Run, X.; Li, H.; Liang, Z. Rapamycin Decreases Tau Phosphorylation at Ser214 through Regulation of CAMP-Dependent Kinase. Neurochem. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Magrì, A.; Medina, D.X.; Wisely, E.V.; López-Aranda, M.F.; Silva, A.J.; Oddo, S. MTOR Regulates Tau Phosphorylation and Degradation: Implications for Alzheimer’s Disease and Other Tauopathies. Aging Cell 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolan, P.J.; Johnson, G.V.W. A Caspase Cleaved Form of Tau Is Preferentially Degraded through the Autophagy Pathway. J. Biol. Chem. 2010. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, P.R.; Martins, R.N. Autophagy Modulates Aβ Accumulation and Formation of Aggregates in Yeast. Mol. Cell Neurosci. 2020. [Google Scholar] [CrossRef]
- Haung Yu, W.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy—A Novel β-Amyloid Peptide-Generating Pathway Activated in Alzheimer’s Disease. J. Cell. Biol. 2005. [Google Scholar] [CrossRef]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal Cathepsins and Their Regulation in Aging and Neurodegeneration. Ageing Res. Rev. 2016. [Google Scholar] [CrossRef]
- Bi, D.; Yao, L.; Lin, Z.; Chi, L.; Li, H.; Xu, H.; Du, X.; Liu, Q.; Hu, Z.; Lu, J.; et al. Unsaturated Mannuronate Oligosaccharide Ameliorates Β-amyloid Pathology through Autophagy in Alzheimer’s Disease Cell Models. Carbohydr. Polym. 2021, 251. [Google Scholar] [CrossRef] [PubMed]
- Jantrapirom, S.; Nimlamool, W.; Chattipakorn, N.; Chattipakorn, S.; Temviriyanukul, P.; Inthachat, W.; Govitrapong, P.; Potikanond, S. Liraglutide Suppresses Tau Hyperphosphorylation, Amyloid Β Accumulation through Regulating Neuronal Insulin Signaling and BACE-1 Activity. Int. J. Mol. Sci. 2020, 21, 1725. [Google Scholar] [CrossRef]
- Huang, J.; Huang, N.; Xu, S.; Luo, Y.; Li, Y.; Jin, H.; Yu, C.; Shi, J.; Jin, F. Signaling Mechanisms Underlying Inhibition of Neuroinflammation by Resveratrol in Neurodegenerative Diseases. J. Nutr. Biochem. 2021. [Google Scholar] [CrossRef]
- Broderick, T.L.; Rasool, S.; Li, R.; Zhang, Y.; Anderson, M.; Al-Nakkash, L.; Plochocki, J.H.; Geetha, T.; Babu, J.R. Neuroprotective Effects of Chronic Resveratrol Treatment and Exercise Training in the 3xtg-Ad Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7337. [Google Scholar] [CrossRef]
- Rahman, M.H.; Akter, R.; Bhattacharya, T.; Abdel-Daim, M.M.; Alkahtani, S.; Arafah, M.W.; Al-Johani, N.S.; Alhoshani, N.M.; Alkeraishan, N.; Alhenaky, A.; et al. Resveratrol and Neuroprotection: Impact and Its Therapeutic Potential in Alzheimer’s Disease. Front. Pharm. 2020, 11. [Google Scholar] [CrossRef]
- Luo, F.; Sandhu, A.F.; Rungratanawanich, W.; Williams, G.E.; Akbar, M.; Zhou, S.; Song, B.J.; Wang, X. Melatonin and Autophagy in Aging-Related Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 7174. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Hand, G.R.; Briner, A.; Li, C.; Götz, J. Exosomes Induce Endolysosomal Permeabilization as a Gateway by Which Exosomal Tau Seeds Escape into the Cytosol. Acta Neuropathol. 2021. [Google Scholar] [CrossRef]
- Bilousova, T.; Bilousova, T.; Simmons, B.J.; Knapp, R.R.; Elias, C.J.; Campagna, J.; Melnik, M.; Chandra, S.; Focht, S.; Zhu, C.; et al. Dual Neutral Sphingomyelinase-2/Acetylcholinesterase Inhibitors for the Treatment of Alzheimer’s Disease. ACS Chem. Biol. 2020, 15, 1671–1684. [Google Scholar] [CrossRef] [PubMed]
- Li, T.R.; Wang, X.N.; Sheng, C.; Li, Y.X.; Li, F.Z.T.; Sun, Y.; Han, Y. Extracellular Vesicles as an Emerging Tool for the Early Detection of Alzheimer’s Disease. Mech. Ageing Dev. 2019. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Wan, J.; Liu, A.; Sun, J. Melatonin Regulates Aβ Production/Clearance Balance and Aβ Neurotoxicity: A Potential Therapeutic Molecule for Alzheimer’s Disease. Biomed. Pharmacother. 2020. [Google Scholar] [CrossRef]
- Ojala, J.; Alafuzoff, I.; Herukka, S.K.; van Groen, T.; Tanila, H.; Pirttilä, T. Expression of Interleukin-18 Is Increased in the Brains of Alzheimer’s Disease Patients. Neurobiol. Aging 2009. [Google Scholar] [CrossRef]
- Cartier, L.; Hartley, O.; Dubois-Dauphin, M.; Krause, K.H. Chemokine Receptors in the Central Nervous System: Role in Brain Inflammation and Neurodegenerative Diseases. Brain Res. Rev. 2005. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Rydel, R.; Brigham, E.F.; Yang, L.B.; Hampel, H.; Murphy, G.M.; Brachova, L.; Yan, S.D.; Walker, D.G.; Shen, Y.; et al. Inflammatory Repertoire of Alzheimer’s Disease and Nondemented Elderly Microglia in Vitro. Glia 2001. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 Inflammasome Is Involved in the Innate Immune Response to Amyloid-β. Nat. Immunol. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature 2013. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; De Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid Appearance and Local Toxicity of Amyloid-β Plaques in a Mouse Model of Alzheimer’s Disease. Nature 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varnum, M.M.; Ikezu, T. The Classification of Microglial Activation Phenotypes on Neurodegeneration and Regeneration in Alzheimer’s Disease Brain. Arch. Immunol. Ther. Exp. 2012. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.T.; Liu, L.; Li, Y.; Mrak, R.E.; Barger, S.W. Interleukin-1 Mediates Alzheimer and Lewy Body Pathologies. J. Neuroinflammation 2006. [Google Scholar] [CrossRef] [Green Version]
- Murray, C.A.; Lynch, M.A. Evidence That Increased Hippocampal Expression of the Cytokine Interleukin-1β Is a Common Trigger for Age- and Stress-Induced Impairments in Long-Term Potentiation. J. Neurosci. 1998. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, W. Islet Amyloid Polypeptide: Another Key Molecule in Alzheimer’s Pathogenesis? Prog. Neurobiol. 2017. [Google Scholar] [CrossRef]
- Yang, Y.; Song, W. Molecular Links between Alzheimer’s Disease and Diabetes Mellitus. Neuroscience 2013. [Google Scholar] [CrossRef]
- Maiese, K. Targeting the Core of Neurodegeneration: FoxO, MTOR, and SIRT1. Neural Regen. Res. 2021, 16, 448–455. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morales-Scheihing, D.; Butler, P.C.; Soto, C. Type 2 Diabetes as a Protein Misfolding Disease. Trends Mol. Med. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matveyenko, A.V.; Butler, P.C. Islet Amyloid Polypeptide (IAPP) Transgenic Rodents as Models for Type 2 Diabetes. ILAR J. 2006, 47, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Raimundo, A.F.; Félix, F.; Andrade, R.; García-Conesa, M.T.; González-Sarrías, A.; Gilsa-Lopes, J.; Raimundo, A.; Ribeiro, R.; Rodriguez-Mateos, A. Combined Effect of Interventions with Pure or Enriched Mixtures of (Poly)Phenols and Anti-Diabetic Medication in Type 2 Diabetes Management: A Meta-Analysis of Randomized Controlled Human Trials. Eur. J. Nutr. 2020. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, M.E.; Paulsson, J.F.; Schultz, S.W.; Ingelsson, M.; Westermark, P.; Westermark, G.T. In Vivo Seeding and Cross-Seeding of Localized Amyloidosis: A Molecular Link between Type 2 Diabetes and Alzheimer Disease. Am. J. Pathol. 2015. [Google Scholar] [CrossRef]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin Deposition in the Brain: A Second Amyloid in Alzheimer Disease? Ann. Neurol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Armiento, V.; Spanopoulou, A.; Kapurniotu, A. Peptide-Based Molecular Strategies To Interfere with Protein Misfolding, Aggregation, and Cell Degeneration. Angew. Chem. Int. Ed. Engl. 2020. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.M.; Velkova, A.; Tatarek-Nossol, M.; Andreetto, E.; Kapurniotu, A. IAPP Mimic Blocks Aβ Cytotoxic Self-Assembly: Cross-Suppression of Amyloid Toxicity of Aβ and IAPP Suggests a Molecular Link between Alzheimer’s Disease and Type Ⅱ Diabetes. Angew. Chem. Int. Ed. Engl. 2007. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gonzalez, I.; Edwards, G.; Salvadores, N.; Shahnawaz, M.; Diaz-Espinoza, R.; Soto, C. Molecular Interaction between Type 2 Diabetes and Alzheimer’s Disease through Cross-Seeding of Protein Misfolding. Mol. Psychiatry 2017. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, P.; Solomon, T.; Sahoo, B.R.; Ignasiak, K.; Gaskin, S.; Rowles, J.; Verdile, G.; Howard, M.J.; Bond, C.S.; Ramamoorthy, A.; et al. Amylin and Β Amyloid Proteins Interact to Form Amorphous Heterocomplexes with Enhanced Toxicity in Neuronal Cells. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Martinez-Valbuena, I.; Valenti-Azcarate, R.; Amat-Villegas, I.; Riverol, M.; Marcilla, I.; de Andrea, C.E.; Sánchez-Arias, J.A.; del Mar Carmona-Abellan, M.; Marti, G.; Erro, M.E.; et al. Amylin as a Potential Link between Type 2 Diabetes and Alzheimer Disease. Ann. Neurol. 2019, 86, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Grizzanti, J.; Corrigan, R.; Servizi, S.; Casadesus, G. Amylin Signaling in Diabetes and Alzheimer’s Disease: Therapy or Pathology? J. Neurol. Neuromedicine 2019, 4, 12–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Yang, S.; Wang, C.; Zhang, J.; Huo, L.; Cheng, Y.; Wang, C.; Jia, Z.; Ren, L.; Kang, L.; et al. Multiple Target of HAmylin on Rat Primary Hippocampal Neurons. Neuropharmacology 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burillo, J.; Marqués, P.; Jiménez, B.; González-Blanco, C.; Benito, M.; Guillén, C. Insulin Resistance and Diabetes Mellitus in Alzheimer’s Disease. Cells 2021, 10, 1236. https://doi.org/10.3390/cells10051236
Burillo J, Marqués P, Jiménez B, González-Blanco C, Benito M, Guillén C. Insulin Resistance and Diabetes Mellitus in Alzheimer’s Disease. Cells. 2021; 10(5):1236. https://doi.org/10.3390/cells10051236
Chicago/Turabian StyleBurillo, Jesús, Patricia Marqués, Beatriz Jiménez, Carlos González-Blanco, Manuel Benito, and Carlos Guillén. 2021. "Insulin Resistance and Diabetes Mellitus in Alzheimer’s Disease" Cells 10, no. 5: 1236. https://doi.org/10.3390/cells10051236
APA StyleBurillo, J., Marqués, P., Jiménez, B., González-Blanco, C., Benito, M., & Guillén, C. (2021). Insulin Resistance and Diabetes Mellitus in Alzheimer’s Disease. Cells, 10(5), 1236. https://doi.org/10.3390/cells10051236