
Store Operated Calcium Entry in Cell Migration and Cancer Metastasis


Abstract
:1. Introduction
2. Cell Migration
3. Ca2+ Signaling
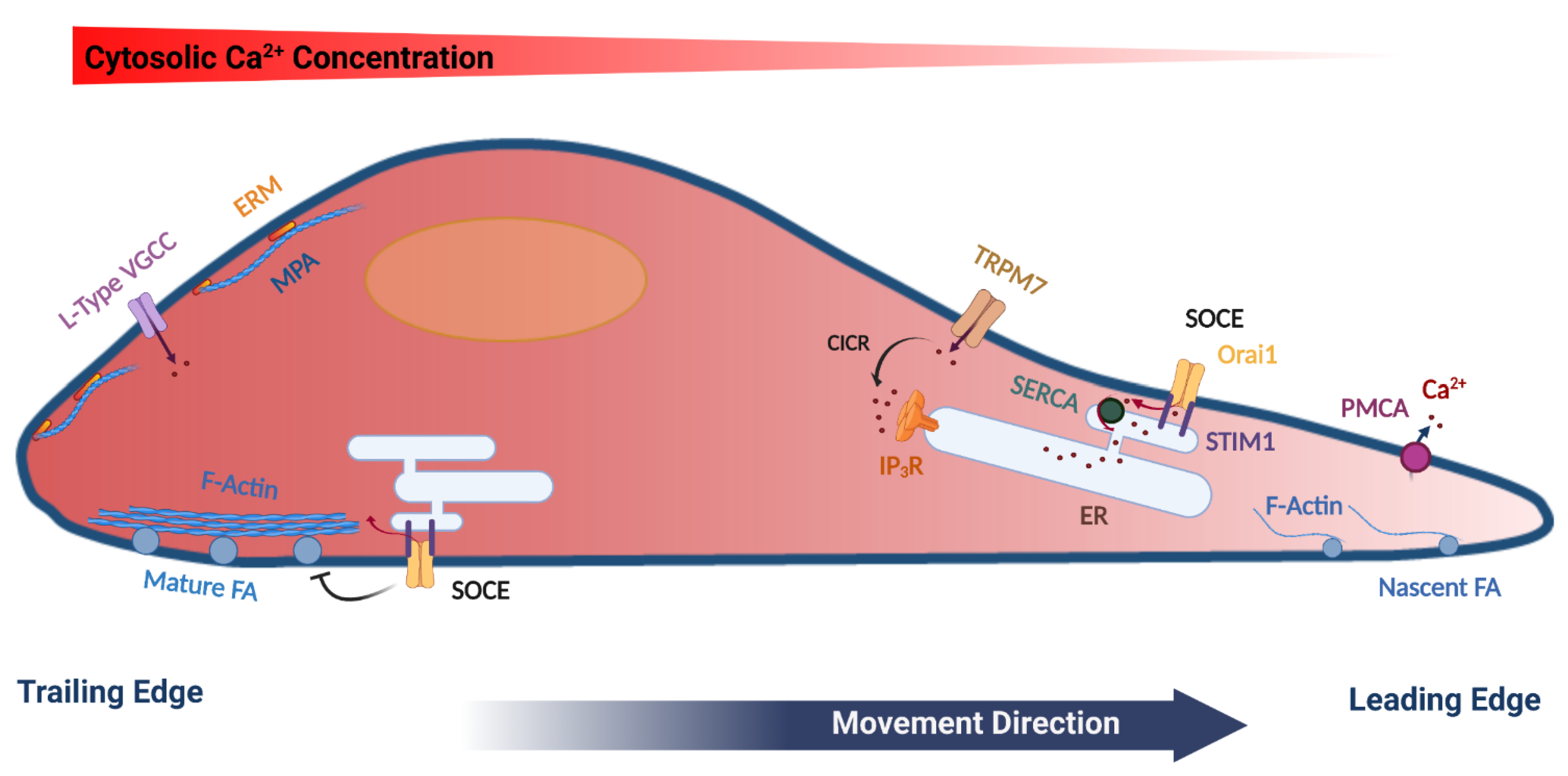
4. Polarized Ca2+ Signals in Migrating Cells
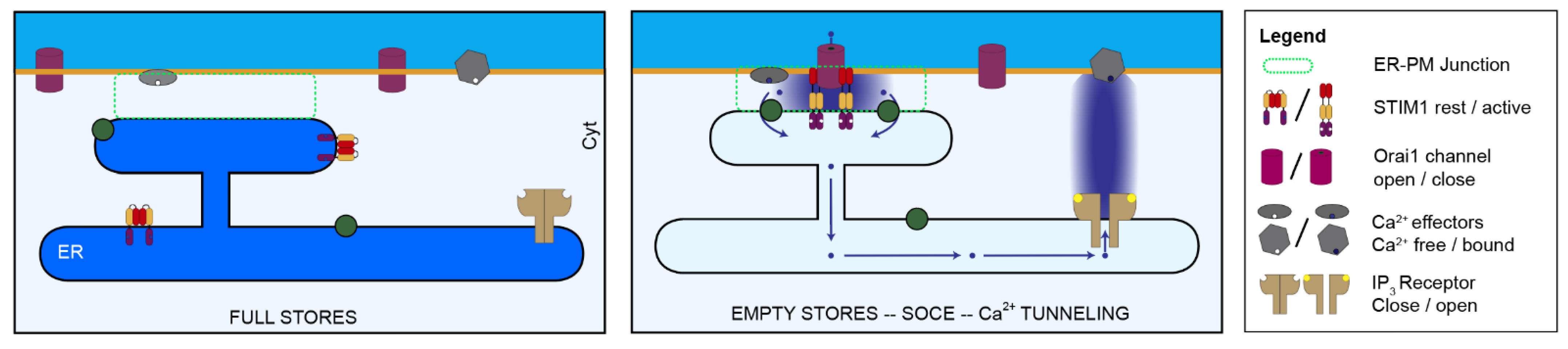
5. Store-Operated Ca2+ Entry (SOCE)
6. SOCE Regulates Cell Migration by Modulating Focal Adhesion Dynamics
7. Additional Mechanisms Involving SOCE in Cell Migration
8. Disruption of Ca2+ Homeostasis in Cancer Cells
9. SOCE Dependent Regulation of Cancer Cell Migration and Metastasis
10. SOCE and the Epithelial to Mesenchymal Transition (EMT)
11. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CBP | Calcium Binding Proteins |
| PMCA | Plasma Membrane Ca2+ ATPase |
| MLCK | Myosin Light Chain Kinase |
| PKA | Protein Kinase A |
| ER | Endoplasmic Reticulum |
| SR | Sarcoplasmic Reticulum |
| SOCE | Store-Operated Calcium Entry |
| STIM1 | Stromal Interation Molecule 1 |
| PM | Plasma Membrane |
| CRAC | Ca2+ Release-Activated Ca2+ channels |
| SOAR | STIM1-Orai1-Activating Region |
| SAM | Sterile α-Motif |
| GpcR | G-Protein Coupled Receptors |
| PLC | Phospholipase C |
| PIP2 | Phosphatidylinositol 4,5-Bisphosphate |
| IP3 | Inositol 1,4,5-Trisphosphate |
| PKC | Protein Kinase C |
| ORAI | Calcium Release-Activated Calcium Channel Protein |
| EB1 | End-Binding Protein 1 |
| MT | Microtubule |
| HBC | Human Breast Cancer |
| ECM | Extracellular Matrix |
| ERM | Ezrin, Radixin and Moesin |
| MPA | Membrane-Proximal F-Actin |
| TRPM7 | Transient Receptor Potential Melastatin Channel 7 |
| CaM | Calmodulin |
| CACNG4 | Calcium Voltage-Gated Channel Auxillary Subunit Gamma 4 |
| pCRC | Primary Colorectal Tumor Cells |
| MEF | Mouse Embryonic Fibroblast |
| PDGF | Platelet-Derived Growth Factor |
| PTK2B | Protein-Rich Tyrosine Kinase 2 Beta |
| FAK | Focal Adhesion Kinase |
| PDGF | Platelet-Derived Growth Factor |
| HUVEC | Human Umbilical Vein Endothelial Cells |
| EMT | Epithelial-to Mesenchymal Transition |
| TGF-β | Transforming Growth Factor-β |
| MMP | Matrix Metalloproteinases |
References
- Shellard, A.; Mayor, R. All Roads Lead to Directional Cell Migration. Trends Cell Biol. 2020, 30, 852–868. [Google Scholar] [CrossRef]
- Kraljevic Pavelic, S.; Sedic, M.; Bosnjak, H.; Spaventi, S.; Pavelic, K. Metastasis: New perspectives on an old problem. Mol. Cancer 2011, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Mo, P.; Yang, S. The store-operated calcium channels in cancer metastasis: From cell migration, invasion to metastatic colonization. Front. Biosci. (Landmark Ed.) 2018, 23, 1241–1256. [Google Scholar]
- Huang, H.K.; Lin, Y.H.; Chang, H.A.; Lai, Y.S.; Chen, Y.C.; Huang, S.C.; Chou, C.Y.; Chiu, W.T. Chemoresistant ovarian cancer enhances its migration abilities by increasing store-operated Ca2+ entry-mediated turnover of focal adhesions. J. Biomed. Sci. 2020, 27, 36. [Google Scholar] [CrossRef]
- Chang, S.J.; Chen, Y.C.; Yang, C.H.; Huang, S.C.; Huang, H.K.; Li, C.C.; Harn, H.I.; Chiu, W.T. Revealing the three dimensional architecture of focal adhesion components to explain Ca2+-mediated turnover of focal adhesions. Biochim. Biophys. Acta Gen Subj. 2017, 1861, 624–635. [Google Scholar] [CrossRef]
- Derouiche, S.; Warnier, M.; Mariot, P.; Gosset, P.; Mauroy, B.; Bonnal, J.L.; Slomianny, C.; Delcourt, P.; Prevarskaya, N.; Roudbaraki, M. Bisphenol A stimulates human prostate cancer cell migration via remodelling of calcium signalling. Springerplus 2013, 2, 54. [Google Scholar] [CrossRef] [Green Version]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridley, A.J. Life at the leading edge. Cell 2011, 145, 1012–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Huang, X.Y. Ca2+ influx through L-type Ca2+ channels controls the trailing tail contraction in growth factor-induced fibroblast cell migration. J. Biol. Chem. 2005, 280, 27130–27137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayor, R.; Etienne-Manneville, S. The front and rear of collective cell migration. Nat. Rev. Mol. Cell Biol. 2016, 17, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrie, R.J.; Doyle, A.D.; Yamada, K.M. Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Fehon, R.G.; McClatchey, A.I.; Bretscher, A. Organizing the cell cortex: The role of ERM proteins. Nat. Rev. Mol. Cell Biol. 2010, 11, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Diz-Muñoz, A.; Krieg, M.; Bergert, M.; Ibarlucea-Benitez, I.; Muller, D.J.; Paluch, E.; Heisenberg, C.P. Control of directed cell migration in vivo by membrane-to-cortex attachment. PLoS Biol. 2010, 8, e1000544. [Google Scholar] [CrossRef]
- Liu, Y.J.; Le Berre, M.; Lautenschlaeger, F.; Maiuri, P.; Callan-Jones, A.; Heuzé, M.; Takaki, T.; Voituriez, R.; Piel, M. Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 2015, 160, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Cordero, J.J.; Magalhaes, M.A.; Eddy, R.J.; Hodgson, L.; Condeelis, J. Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell Biol. 2013, 14, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Bisaria, A.; Hayer, A.; Garbett, D.; Cohen, D.; Meyer, T. Membrane-proximal F-actin restricts local membrane protrusions and directs cell migration. Science 2020, 368, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.; Condeelis, J. The cofilin activity cycle in lamellipodia and invadopodia. J. Cell Biochem. 2009, 108, 1252–1262. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Courjaret, R.J.; Machaca, K. Expanding the Store-operated Ca2+ Entry microdomain through Ca2+ tunneling. Current Opinion in Physiology 2020, 17, 158–162. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brundage, R.A.; Fogarty, K.E.; Tuft, R.A.; Fay, F.S. Calcium Gradients Underlying Polarization and Chemotaxis of Eosinophils. Science 1991, 254, 703–706. [Google Scholar] [CrossRef]
- Gilbert, S.H.; Perry, K.; Fay, F.S. Mediation of chemoattractant-induced changes in [Ca2+]i and cell shape, polarity, and locomotion by InsP3, DAG, and protein kinase C in newt eosinophils. J. Cell Biol. 1994, 127, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.C.; Seki, A.; Yang, H.W.; Hayer, A.; Carrasco, S.; Malmersjo, S.; Meyer, T. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 2014, 16, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.M.; Lee, M.; Kim, N.; Heo, W.D. Optogenetic toolkit reveals the role of Ca2+ sparklets in coordinated cell migration. Proc. Natl. Acad. Sci. USA 2016, 113, 5952–5957. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Wang, X.; Chen, M.; Ouyang, K.; Song, L.S.; Cheng, H. Calcium flickers steer cell migration. Nature 2009, 457, 901–905. [Google Scholar] [CrossRef] [Green Version]
- Visser, D.; Langeslag, M.; Kedziora, K.M.; Klarenbeek, J.; Kamermans, A.; Horgen, F.D.; Fleig, A.; van Leeuwen, F.N.; Jalink, K. TRPM7 triggers Ca2+ sparks and invadosome formation in neuroblastoma cells. Cell Calcium. 2013, 54, 404–415. [Google Scholar] [CrossRef] [Green Version]
- Giannone, G.; Ronde, P.; Gaire, M.; Haiech, J.; Takeda, K. Calcium oscillations trigger focal adhesion disassembly in human U87 astrocytoma cells. J. Biol. Chem. 2002, 277, 26364–26371. [Google Scholar] [CrossRef] [Green Version]
- Tsai, F.C.; Meyer, T. Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Curr. Biol. 2012, 22, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Middelbeek, J.; Kuipers, A.J.; Henneman, L.; Visser, D.; Eidhof, I.; van Horssen, R.; Wieringa, B.; Canisius, S.V.; Zwart, W.; Wessels, L.F.; et al. TRPM7 is required for breast tumor cell metastasis. Cancer Res. 2012, 72, 4250–4261. [Google Scholar] [CrossRef] [Green Version]
- Minton, K. Cell migration: Coordinating calcium signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 152. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.H.; Falke, J.J. Ca2+ influx is an essential component of the positive-feedback loop that maintains leading-edge structure and activity in macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 16176–16181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, L.S.; Langeslag, M.; ten Klooster, J.P.; Hordijk, P.L.; Jalink, K.; Collard, J.G. Calcium signaling regulates translocation and activation of Rac. J. Biol. Chem. 2003, 278, 39413–39421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kholmanskikh, S.S.; Koeller, H.B.; Wynshaw-Boris, A.; Gomez, T.; Letourneau, P.C.; Ross, M.E. Calcium-dependent interaction of Lis1 with IQGAP1 and Cdc42 promotes neuronal motility. Nat. Neurosci. 2006, 9, 50–57. [Google Scholar] [CrossRef]
- Ohta, Y.; Nishida, E.; Sakai, H. Type II Ca2+/calmodulin-dependent protein kinase binds to actin filaments in a calmodulin-sensitive manner. FEBS Lett. 1986, 208, 423–426. [Google Scholar] [CrossRef] [Green Version]
- Larsson, C. Protein kinase C and the regulation of the actin cytoskeleton. Cell Signal. 2006, 18, 276–284. [Google Scholar] [CrossRef]
- Hoffman, L.; Farley, M.M.; Waxham, M.N. Calcium-calmodulin-dependent protein kinase II isoforms differentially impact the dynamics and structure of the actin cytoskeleton. Biochemistry 2013, 52, 1198–1207. [Google Scholar] [CrossRef]
- Sotelo-Avila, C.; Gooch, W.M., 3rd. Neoplasms associated with the Beckwith-Wiedemann syndrome. Perspect Pediatr. Pathol. 1976, 3, 255–272. [Google Scholar]
- Parker, N.J.; Begley, C.G.; Smith, P.J.; Fox, R.M. Molecular cloning of a novel human gene (D11S4896E) at chromosomal region 11p15.5. Genomics 1996, 37, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Manji, S.S.; Parker, N.J.; Williams, R.T.; van Stekelenburg, L.; Pearson, R.B.; Dziadek, M.; Smith, P.J. STIM1: A novel phosphoprotein located at the cell surface. Biochim. Biophys. Acta 2000, 1481, 147–155. [Google Scholar] [CrossRef]
- Hu, R.J.; Lee, M.P.; Connors, T.D.; Johnson, L.A.; Burn, T.C.; Su, K.; Landes, G.M.; Feinberg, A.P. A 2.5-Mb transcript map of a tumor-suppressing subchromosomal transferable fragment from 11p15.5, and isolation and sequence analysis of three novel genes. Genomics 1997, 46, 9–17. [Google Scholar] [CrossRef]
- Sabbioni, S.; Barbanti-Brodano, G.; Croce, C.M.; Negrini, M. GOK: A gene at 11p15 involved in rhabdomyosarcoma and rhabdoid tumor development. Cancer Res. 1997, 57, 4493–4497. [Google Scholar]
- Oritani, K.; Kincade, P.W. Identification of stromal cell products that interact with pre-B cells. J. Cell Biol. 1996, 134, 771–782. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef]
- Soboloff, J.; Spassova, M.A.; Tang, X.D.; Hewavitharana, T.; Xu, W.; Gill, D.L. Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 2006, 281, 20661–20665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Lopez, E.; Frischauf, I.; Jardin, I.; Derler, I.; Muik, M.; Cantonero, C.; Salido, G.M.; Smani, T.; Rosado, J.A.; Redondo, P.C. STIM1 phosphorylation at Y(316) modulates its interaction with SARAF and the activation of SOCE and I (CRAC). J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Stathopulos, P.B.; Li, G.Y.; Plevin, M.J.; Ames, J.B.; Ikura, M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J. Biol. Chem. 2006, 281, 35855–35862. [Google Scholar] [CrossRef] [Green Version]
- Fahrner, M.; Muik, M.; Schindl, R.; Butorac, C.; Stathopulos, P.; Zheng, L.; Jardin, I.; Ikura, M.; Romanin, C. A coiled-coil clamp controls both conformation and clustering of stromal interaction molecule 1 (STIM1). J. Biol. Chem. 2014, 289, 33231–33244. [Google Scholar] [CrossRef] [Green Version]
- Korzeniowski, M.K.; Manjarres, I.M.; Varnai, P.; Balla, T. Activation of STIM1-Orai1 involves an intramolecular switching mechanism. Sci. Signal 2010, 3, ra82. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Srinivasan, P.; Razavi, S.; Seymour, S.; Meraner, P.; Gudlur, A.; Stathopulos, P.B.; Ikura, M.; Rao, A.; Hogan, P.G. Initial activation of STIM1, the regulator of store-operated calcium entry. Nat. Struct. Mol. Biol. 2013, 20, 973–981. [Google Scholar] [CrossRef] [Green Version]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef]
- Chen, Y.F.; Chen, L.H.; Shen, M.R. The distinct role of STIM1 and STIM2 in the regulation of store-operated Ca2+ entry and cellular function. J. Cell Physiol. 2019, 234, 8727–8739. [Google Scholar] [CrossRef]
- Parvez, S.; Beck, A.; Peinelt, C.; Soboloff, J.; Lis, A.; Monteilh-Zoller, M.; Gill, D.L.; Fleig, A.; Penner, R. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J. 2008, 22, 752–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Stathopulos, P.; Li, G.Y.; Ikura, M. Biophysical characterization of the EF-hand and SAM domain containing Ca2+ sensory region of STIM1 and STIM2. Biochem. Biophys. Res. Commun. 2008, 369, 240–246. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Zheng, L.; Ikura, M. Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J. Biol. Chem. 2009, 284, 728–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, Y.; Zhou, Y.; Hendron, E.; Mancarella, S.; Andrake, M.D.; Rothberg, B.S.; Soboloff, J.; Gill, D.L. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun. 2014, 5, 3183. [Google Scholar] [CrossRef]
- Bird, G.S.; Hwang, S.Y.; Smyth, J.T.; Fukushima, M.; Boyles, R.R.; Putney, J.W., Jr. STIM1 is a calcium sensor specialized for digital signaling. Curr. Biol. 2009, 19, 1724–1729. [Google Scholar] [CrossRef] [Green Version]
- Oh-Hora, M.; Yamashita, M.; Hogan, P.G.; Sharma, S.; Lamperti, E.; Chung, W.; Prakriya, M.; Feske, S.; Rao, A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat. Immunol. 2008, 9, 432–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miederer, A.M.; Alansary, D.; Schwär, G.; Lee, P.H.; Jung, M.; Helms, V.; Niemeyer, B.A. A STIM2 splice variant negatively regulates store-operated calcium entry. Nat. Commun. 2015, 6, 6899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, A.; Yen, M.; Sadaghiani, A.M.; Malmersjö, S.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. Alternative splicing converts STIM2 from an activator to an inhibitor of store-operated calcium channels. J. Cell Biol. 2015, 209, 653–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakriya, M. Store-operated Orai channels: Structure and function. Curr. Top Membr. 2013, 71, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.; Yuan, J.P.; Kim, M.S.; Choi, Y.J.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol. Cell 2008, 32, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Miao, Y.; Zheng, X.; Gong, Y.; Zhang, J.; Zou, F.; Cai, C. STIM1 and STIM2 differently regulate endogenous Ca2+ entry and promote TGF-β-induced EMT in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 488, 74–80. [Google Scholar] [CrossRef]
- Yang, Y.; Jiang, Z.; Wang, B.; Chang, L.; Liu, J.; Zhang, L.; Gu, L. Expression of STIM1 is associated with tumor aggressiveness and poor prognosis in breast cancer. Pathol. Res. Pract. 2017, 213, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Umemura, M.; Baljinnyam, E.; Feske, S.; De Lorenzo, M.S.; Xie, L.H.; Feng, X.; Oda, K.; Makino, A.; Fujita, T.; Yokoyama, U.; et al. Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration. PLoS ONE 2014, 9, e89292. [Google Scholar] [CrossRef]
- Suyama, E.; Wadhwa, R.; Kaur, K.; Miyagishi, M.; Kaul, S.C.; Kawasaki, H.; Taira, K. Identification of metastasis-related genes in a mouse model using a library of randomized ribozymes. J. Biol. Chem. 2004, 279, 38083–38086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Calcium in tumour metastasis: New roles for known actors. Nat. Rev.Cancer 2011, 11, 609–618. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Pan, T.; Li, L.; Li, J.; Yang, H. STIM1 silencing inhibits the migration and invasion of A549 cells. Mol. Med. Rep. 2017, 16, 3283–3289. [Google Scholar] [CrossRef] [Green Version]
- Stanisz, H.; Saul, S.; Muller, C.S.; Kappl, R.; Niemeyer, B.A.; Vogt, T.; Hoth, M.; Roesch, A.; Bogeski, I. Inverse regulation of melanoma growth and migration by Orai1/STIM2-dependent calcium entry. Pigment. Cell Melanoma Res. 2014, 27, 442–453. [Google Scholar] [CrossRef]
- Zuccolo, E.; Laforenza, U.; Ferulli, F.; Pellavio, G.; Scarpellino, G.; Tanzi, M.; Turin, I.; Faris, P.; Lucariello, A.; Maestri, M.; et al. Stim and Orai mediate constitutive Ca2+ entry and control endoplasmic reticulum Ca2+ refilling in primary cultures of colorectal carcinoma cells. Oncotarget 2018, 9, 31098–31119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Liu, X.; Feng, B.; Liu, N.; Wu, Q.; Han, Y.; Nie, Y.; Wu, K.; Shi, Y.; Fan, D. STIM1, a direct target of microRNA-185, promotes tumor metastasis and is associated with poor prognosis in colorectal cancer. Oncogene 2015, 34, 4808–4820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.; Shen, Q.; Zhang, S.; Huang, H.; Meng, X.; Zheng, X.; Yao, Z.; He, Z.; Lu, S.; Cai, C.; et al. Calcium-sensing stromal interaction molecule 2 upregulates nuclear factor of activated T cells 1 and transforming growth factor-β signaling to promote breast cancer metastasis. Breast Cancer Res. 2019, 21, 99. [Google Scholar] [CrossRef]
- Zhou, Y.; Gu, P.; Li, J.; Li, F.; Zhu, J.; Gao, P.; Zang, Y.; Wang, Y.; Shan, Y.; Yang, D. Suppression of STIM1 inhibits the migration and invasion of human prostate cancer cells and is associated with PI3K/Akt signaling inactivation. Oncol. Rep. 2017, 38, 2629–2636. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhang, S.; Niu, H.; Ye, Y.; Hu, F.; Chen, S.; Li, X.; Luo, X.; Jiang, S.; Liu, Y.; et al. STIM1 accelerates cell senescence in a remodeled microenvironment but enhances the epithelial-to-mesenchymal transition in prostate cancer. Sci. Rep. 2015, 5, 11754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Guerrero, A.M.; Tomas-Martin, P.; Pascual-Caro, C.; Macartney, T.; Rojas-Fernandez, A.; Ball, G.; Alessi, D.R.; Pozo-Guisado, E.; Martin-Romero, F.J. Regulation of membrane ruffling by polarized STIM1 and ORAI1 in cortactin-rich domains. Sci. Rep. 2017, 7, 383. [Google Scholar] [CrossRef] [Green Version]
- Zang, J.; Zuo, D.; Shogren, K.L.; Gustafson, C.T.; Zhou, Z.; Thompson, M.A.; Guo, R.; Prakash, Y.S.; Lu, L.; Guo, W.; et al. STIM1 expression is associated with osteosarcoma cell survival. Chin. J. Cancer Res. 2019, 31, 203–211. [Google Scholar] [CrossRef]
- Xu, J.M.; Zhou, Y.; Gao, L.; Zhou, S.X.; Liu, W.H.; Li, X.A. Stromal interaction molecule 1 plays an important role in gastric cancer progression. Oncol. Rep. 2016, 35, 3496–3504. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Wang, H.; Huang, H.; Sun, L.; Dong, S.; Huang, N.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Elevated Orai1 and STIM1 expressions upregulate MACC1 expression to promote tumor cell proliferation, metabolism, migration, and invasion in human gastric cancer. Cancer Lett. 2016, 381, 31–40. [Google Scholar] [CrossRef]
- Chen, Y.F.; Chiu, W.T.; Chen, Y.T.; Lin, P.Y.; Huang, H.J.; Chou, C.Y.; Chang, H.C.; Tang, M.J.; Shen, M.R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, C.; Rymarczyk, G.; Ding, L.; Kirber, M.T.; Bolotina, V.M. Role of molecular determinants of store-operated Ca2+ entry (Orai1, phospholipase A2 group 6, and STIM1) in focal adhesion formation and cell migration. J. Biol. Chem. 2012, 287, 40745–40757. [Google Scholar] [CrossRef] [Green Version]
- Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Abdullaev, I.F.; Bisaillon, J.M.; Singer, H.A.; Trebak, M. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: Role in proliferation and migration. FASEB J. 2009, 23, 2425–2437. [Google Scholar] [CrossRef] [Green Version]
- Bisaillon, J.M.; Motiani, R.K.; Gonzalez-Cobos, J.C.; Potier, M.; Halligan, K.E.; Alzawahra, W.F.; Barroso, M.; Singer, H.A.; Jourd’heuil, D.; Trebak, M. Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration. Am. J. Physiol. Cell Physiol. 2010, 298, C993-1005. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.Y.; Lin, Y.H.; Chang, H.A.; Yeh, T.Y.; Chang, Y.H.; Chen, Y.F.; Chen, Y.C.; Li, C.C.; Chiu, W.T. STIM1 Knockout Enhances PDGF-Mediated Ca2+ Signaling through Upregulation of the PDGFR(-)PLCgamma(-)STIM2 Cascade. Int. J. Mol. Sci. 2018, 19, 1799. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.W.; Lai, C.S.; Chen, Y.F.; Chiu, W.T.; Chen, H.C.; Shen, M.R. STIM1-dependent Ca2+ signaling regulates podosome formation to facilitate cancer cell invasion. Sci. Rep. 2017, 7, 11523. [Google Scholar] [CrossRef] [Green Version]
- Suganuma, N.; Ito, S.; Aso, H.; Kondo, M.; Sato, M.; Sokabe, M.; Hasegawa, Y. STIM1 regulates platelet-derived growth factor-induced migration and Ca2+ influx in human airway smooth muscle cells. PLoS ONE 2012, 7, e45056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, R.S.; Lim, J.Y.; Turgut, A.; Servage, K.; Zhang, J.; Orth, K.; Sosale, N.; Lazzara, M.; Allegood, J.; Casanova, J.E. Calcium-stimulated disassembly of focal adhesions mediated by an ORP3/IQSec1 complex. Elife 2020, 9. [Google Scholar] [CrossRef]
- Tsai, F.C.; Kuo, G.H.; Chang, S.W.; Tsai, P.J. Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. Biomed Res. Int. 2015, 2015, 409245. [Google Scholar] [CrossRef] [Green Version]
- Courjaret, R.; Dib, M.; Machaca, K. Spatially restricted subcellular Ca2+ signaling downstream of store-operated calcium entry encoded by a cortical tunneling mechanism. Sci. Rep. 2018, 8, 11214. [Google Scholar] [CrossRef]
- Petersen, O.H.; Courjaret, R.; Machaca, K. Ca2+ tunnelling through the ER lumen as a mechanism for delivering Ca2+ entering via store-operated Ca2+ channels to specific target sites. J. Physiol. 2017, 595, 2999–3014. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.W.; Machaca, K. IP3 receptors and store-operated Ca2+ entry: A license to fill. Curr. Opin. Cell Biol. 2019, 57, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Okeke, E.; Parker, T.; Dingsdale, H.; Concannon, M.; Awais, M.; Voronina, S.; Molgo, J.; Begg, M.; Metcalf, D.; Knight, A.E.; et al. Epithelial-mesenchymal transition, IP3 receptors and ER-PM junctions: Translocation of Ca2+ signalling complexes and regulation of migration. Biochem. J. 2016, 473, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Franco, S.J.; Rodgers, M.A.; Perrin, B.J.; Han, J.; Bennin, D.A.; Critchley, D.R.; Huttenlocher, A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell Biol. 2004, 6, 977–983. [Google Scholar] [CrossRef]
- Casas-Rua, V.; Tomas-Martin, P.; Lopez-Guerrero, A.M.; Alvarez, I.S.; Pozo-Guisado, E.; Martin-Romero, F.J. STIM1 phosphorylation triggered by epidermal growth factor mediates cell migration. Biochim. Biophys. Acta 2015, 1853, 233–243. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Guerrero, A.M.; Espinosa-Bermejo, N.; Sanchez-Lopez, I.; Macartney, T.; Pascual-Caro, C.; Orantos-Aguilera, Y.; Rodriguez-Ruiz, L.; Perez-Oliva, A.B.; Mulero, V.; Pozo-Guisado, E.; et al. RAC1-Dependent ORAI1 Translocation to the Leading Edge Supports Lamellipodia Formation and Directional Persistence. Sci. Rep. 2020, 10, 6580. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Hubrack, S.Z.; Chakraborty, S.; Sun, L.; Alcantara-Adap, E.; Kulkarni, R.; Billing, A.M.; Graumann, J.; Taylor, C.W.; Machaca, K. Remodeling of ER-plasma membrane contact sites but not STIM1 phosphorylation inhibits Ca2+ influx in mitosis. Proc. Natl. Acad. Sci. USA 2019, 116, 10392–10401. [Google Scholar] [CrossRef] [Green Version]
- Samtleben, S.; Jaepel, J.; Fecher, C.; Andreska, T.; Rehberg, M.; Blum, R. Direct imaging of ER calcium with targeted-esterase induced dye loading (TED). J. Vis. Exp. 2013, e50317. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Bong, A.H.L.; Monteith, G.R. Calcium signaling and the therapeutic targeting of cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1786–1794. [Google Scholar] [CrossRef]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.G.; Vignjevic, D.M. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Shim, K.N.; Li, J.M.; Estrema, C.; Ornelas, T.A.; Nguyen, F.; Liu, S.; Ramamoorthy, S.L.; Ho, S.; Carethers, J.M.; et al. Molecular mechanisms underlying Ca2+-mediated motility of human pancreatic duct cells. Am. J. Physiol. Cell Physiol. 2010, 299, C1493–C1503. [Google Scholar] [CrossRef] [Green Version]
- Su, L.T.; Agapito, M.A.; Li, M.; Simonson, W.T.; Huttenlocher, A.; Habas, R.; Yue, L.; Runnels, L.W. TRPM7 regulates cell adhesion by controlling the calcium-dependent protease calpain. J. Biol. Chem. 2006, 281, 11260–11270. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.P.; Luan, Y.; You, C.X.; Chen, X.H.; Luo, R.C.; Li, R. TRPM7 regulates the migration of human nasopharyngeal carcinoma cell by mediating Ca2+ influx. Cell Calcium 2010, 47, 425–432. [Google Scholar] [CrossRef]
- Gao, H.; Chen, X.; Du, X.; Guan, B.; Liu, Y.; Zhang, H. EGF enhances the migration of cancer cells by up-regulation of TRPM7. Cell Calcium 2011, 50, 559–568. [Google Scholar] [CrossRef]
- Rybarczyk, P.; Gautier, M.; Hague, F.; Dhennin-Duthille, I.; Chatelain, D.; Kerr-Conte, J.; Pattou, F.; Regimbeau, J.M.; Sevestre, H.; Ouadid-Ahidouch, H. Transient receptor potential melastatin-related 7 channel is overexpressed in human pancreatic ductal adenocarcinomas and regulates human pancreatic cancer cell migration. Int. J. Cancer 2012, 131, E851–E861. [Google Scholar] [CrossRef]
- Wondergem, R.; Ecay, T.W.; Mahieu, F.; Owsianik, G.; Nilius, B. HGF/SF and menthol increase human glioblastoma cell calcium and migration. Biochem. Biophys. Res. Commun. 2008, 372, 210–215. [Google Scholar] [CrossRef]
- Waning, J.; Vriens, J.; Owsianik, G.; Stüwe, L.; Mally, S.; Fabian, A.; Frippiat, C.; Nilius, B.; Schwab, A. A novel function of capsaicin-sensitive TRPV1 channels: Involvement in cell migration. Cell Calcium 2007, 42, 17–25. [Google Scholar] [CrossRef]
- Monet, M.; Lehen’kyi, V.; Gackiere, F.; Firlej, V.; Vandenberghe, M.; Roudbaraki, M.; Gkika, D.; Pourtier, A.; Bidaux, G.; Slomianny, C.; et al. Role of cationic channel TRPV2 in promoting prostate cancer migration and progression to androgen resistance. Cancer Res. 2010, 70, 1225–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhennin-Duthille, I.; Gautier, M.; Faouzi, M.; Guilbert, A.; Brevet, M.; Vaudry, D.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: Correlation with pathological parameters. Cell Physiol. Biochem. 2011, 28, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure. Cancers 2018, 10, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteith, G.R.; Davis, F.M.; Roberts-Thomson, S.J. Calcium channels and pumps in cancer: Changes and consequences. J. Biol. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef] [Green Version]
- Pera, E.; Kaemmerer, E.; Milevskiy, M.J.G.; Yapa, K.; O’Donnell, J.S.; Brown, M.A.; Simpson, F.; Peters, A.A.; Roberts-Thomson, S.J.; Monteith, G.R. The voltage gated Ca2+-channel Cav3.2 and therapeutic responses in breast cancer. Cancer Cell Int. 2016, 16, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwar, N.; Carmine-Simmen, K.; Nair, R.; Wang, C.; Moghadas-Jafari, S.; Blaser, H.; Tran-Thanh, D.; Wang, D.; Wang, P.; Wang, J.; et al. Amplification of a calcium channel subunit CACNG4 increases breast cancer metastasis. EBioMedicine 2020, 52, 102646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vashisht, A.; Trebak, M.; Motiani, R.K. STIM and Orai proteins as novel targets for cancer therapy. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C457-469. [Google Scholar] [CrossRef] [Green Version]
- Jardin, I.; Rosado, J.A. STIM and calcium channel complexes in cancer. Biochim. Biophys. Acta 2016, 1863, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Guan, X. Cancer metastases: Challenges and opportunities. Acta Pharm. Sin. B 2015, 5, 402–418. [Google Scholar] [CrossRef] [Green Version]
- Chiang, S.P.; Cabrera, R.M.; Segall, J.E. Tumor cell intravasation. Am. J. Physiol. Cell Physiol. 2016, 311, C1–C14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef]
- Schwab, A.; Stock, C. Ion channels and transporters in tumour cell migration and invasion. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130102. [Google Scholar] [CrossRef] [Green Version]
- Prevarskaya, N.; Ouadid-Ahidouch, H.; Skryma, R.; Shuba, Y. Remodelling of Ca2+ transport in cancer: How it contributes to cancer hallmarks? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Poletto, V. May the remodeling of the Ca2+ toolkit in endothelial progenitor cells derived from cancer patients suggest alternative targets for anti-angiogenic treatment? Biochim. Biophys. Acta 2015, 1853, 1958–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccolo, E.; Bottino, C.; Diofano, F.; Poletto, V.; Codazzi, A.C.; Mannarino, S.; Campanelli, R.; Fois, G.; Marseglia, G.L.; Guerra, G.; et al. Constitutive Store-Operated Ca2+ Entry Leads to Enhanced Nitric Oxide Production and Proliferation in Infantile Hemangioma-Derived Endothelial Colony-Forming Cells. Stem. Cells Dev. 2016, 25, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Pan, H.; Yao, J.; Zhou, Y.; Han, W. SOCE and cancer: Recent progress and new perspectives. Int. J. Cancer 2016, 138, 2067–2077. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Van Rossum, D.B.; Patterson, R.L.; Ma, H.T.; Gill, D.L. The cellular and molecular basis of store-operated calcium entry. Nat. Cell Biol. 2002, 4, E263–E272. [Google Scholar] [CrossRef] [PubMed]
- Flourakis, M.; Lehen’kyi, V.; Beck, B.; Raphael, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef] [Green Version]
- Hooper, R.; Zhang, X.; Webster, M.; Go, C.; Kedra, J.; Marchbank, K.; Gill, D.L.; Weeraratna, A.T.; Trebak, M.; Soboloff, J. Novel Protein Kinase C-Mediated Control of Orai1 Function in Invasive Melanoma. Mol. Cell Biol. 2015, 35, 2790–2798. [Google Scholar] [CrossRef] [Green Version]
- Yen, M.; Lewis, R.S. Numbers count: How STIM and Orai stoichiometry affect store-operated calcium entry. Cell Calcium 2019, 79, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Hodeify, R.; Selvaraj, S.; Wen, J.; Arredouani, A.; Hubrack, S.; Dib, M.; Al-Thani, S.N.; McGraw, T.; Machaca, K. A STIM1-dependent ’trafficking trap’ mechanism regulates Orai1 plasma membrane residence and Ca2+ influx levels. J. Cell Sci. 2015, 128, 3143–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, P.G.; Lewis, R.S.; Rao, A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 2010, 28, 491–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Cancerous Cells | Cell Lines | Perturbation | Result | Ref. |
|---|---|---|---|---|
| Non-small-cell lung cancer (NSCLC) | A549 SK-MES-1 | STIM1 knockdown | Reduced proliferation | [75] |
| A549 | STIM1 knockdown | Reduced migration & metastasis | [79] | |
| H1299 | STIM1 knockdown | Slightly decreased migration | [24] | |
| Melanoma | SK-Mel-2 SK-Mel-24 | STIM1/Orai1 knockdown | Reduced migration & metastasis | [76] |
| B16F0 cells | STIM1 knockdown | Enhanced migration | [77] | |
| non-commercial WM3734 melanoma cell lines non-commercial WM3734 melanoma cell lines non-commercial WM3734 melanoma cell lines WM3734 non-commercial | STIM2/Orai1 knockdown | Reduced migration & invasiveness | [80] | |
| Colorectal Cancer | Primary liver metastasis | STIM1/Orai1/Orai3 knockdown | No effect on migration | [81] |
| SW620 | STIM1 knockdown | Reduced migration & invasiveness | [82] | |
| SW480 | STIM1 overexpression | Enhanced migration & invasiveness | ||
| Breast Cancer | MDA-MB-231 | STIM1/Orai1 knockdown | Reduced migration | [73] |
| MDA-MB-231 | STIM2 knockdown | Reduced migration | [83] | |
| MCF-7 | STIM1/STIM2 overexpression | Enhanced migration & invasiveness | [74] | |
| Prostate Cancer | PC-3 DU-145 | STIM1 knockdown | Reduced migration and invasion | [84] |
| DU145 PC3 | STIM1/Orai1 overexpression | Enhance migration & cell growth | [85] | |
| Osteosarcoma | U2OS | STIM1/Orai1 knockdown | Reduced migration | [86] |
| 143B U2OS | STIM1 knockout | Reduced migration | [87] | |
| Gastric Cancer | MKN-45 SGC-7901 | STIM1knockdown | Reduce migration & invasiveness | [88] |
| MKN-45 BGC-803 | STIM1/Orai1 knockdown | Reduce migration & invasiveness | [89] | |
| Cervical Cancer | SiHa CaSki | STIM1 knockdown and overexpression | KD reduced migration Overexpression increased migration & invasion | [90] |
| Human oesophageal cancer | (KYSE-30) | Orai1 knockdown | Reduced migration | [91] |
| Cell Lines | Method | Result | Ref. |
|---|---|---|---|
| Human embryonic kidney cells (HEK293) | STIM1/Orai1 knockdown | Reduced migration | [92] |
| Vascular smooth muscle cells (VSMCs) | STIM1/STIM2/Orai1/Orai2/Orai3 knockdown | STIM1/Orai1 reduced migration No effect for STIM2/Orai2/Orai3 | [93,94] |
| Mouse embryonic fibroblasts (MEF) | STIM1 knockout | Enhanced migration | [95] |
| STIM1 knockdown | Reduced invasion | [96] | |
| Human umbilical vein endothelial cells (HUVEC) | STIM1 knockdown | Enhanced migration | [24] |
| STIM1 overexpression | Decreased migration | ||
| Primary human bronchial smooth muscle cells | STIM1/STIM2/Orai1 knockdown | STIM1/Orai1 reduced migration STIM2 no effect | [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammad, A.S.; Machaca, K. Store Operated Calcium Entry in Cell Migration and Cancer Metastasis. Cells 2021, 10, 1246. https://doi.org/10.3390/cells10051246
Hammad AS, Machaca K. Store Operated Calcium Entry in Cell Migration and Cancer Metastasis. Cells. 2021; 10(5):1246. https://doi.org/10.3390/cells10051246
Chicago/Turabian StyleHammad, Ayat S., and Khaled Machaca. 2021. "Store Operated Calcium Entry in Cell Migration and Cancer Metastasis" Cells 10, no. 5: 1246. https://doi.org/10.3390/cells10051246
APA StyleHammad, A. S., & Machaca, K. (2021). Store Operated Calcium Entry in Cell Migration and Cancer Metastasis. Cells, 10(5), 1246. https://doi.org/10.3390/cells10051246