
Role of Glycans on Key Cell Surface Receptors That Regulate Cell Proliferation and Cell Death


Abstract
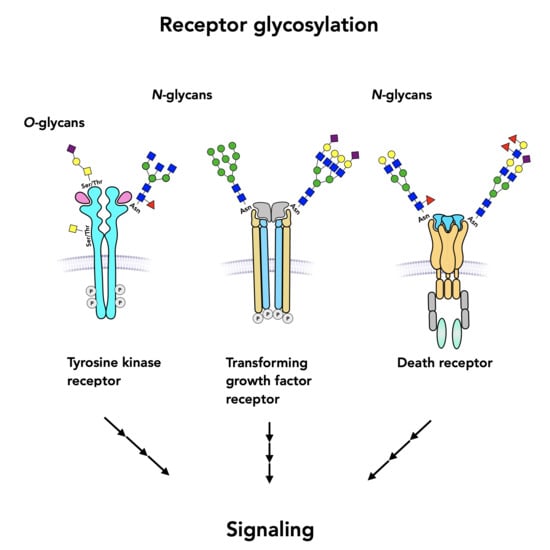
:
1. Introduction
2. Tyrosine Kinase Receptors
2.1. Epidermal Growth Factor Receptor, EGFR
2.2. Hepatocyte Growth Factor Receptor MET
2.3. Fibroblast Growth Factor Receptor, FGFR
2.4. Vascular Endothelial Growth Factor Receptor, VEGFR
2.5. Insulin Receptor and Insulin-Like Growth Factor Receptor
2.6. Receptors for Neurotrophic Factors
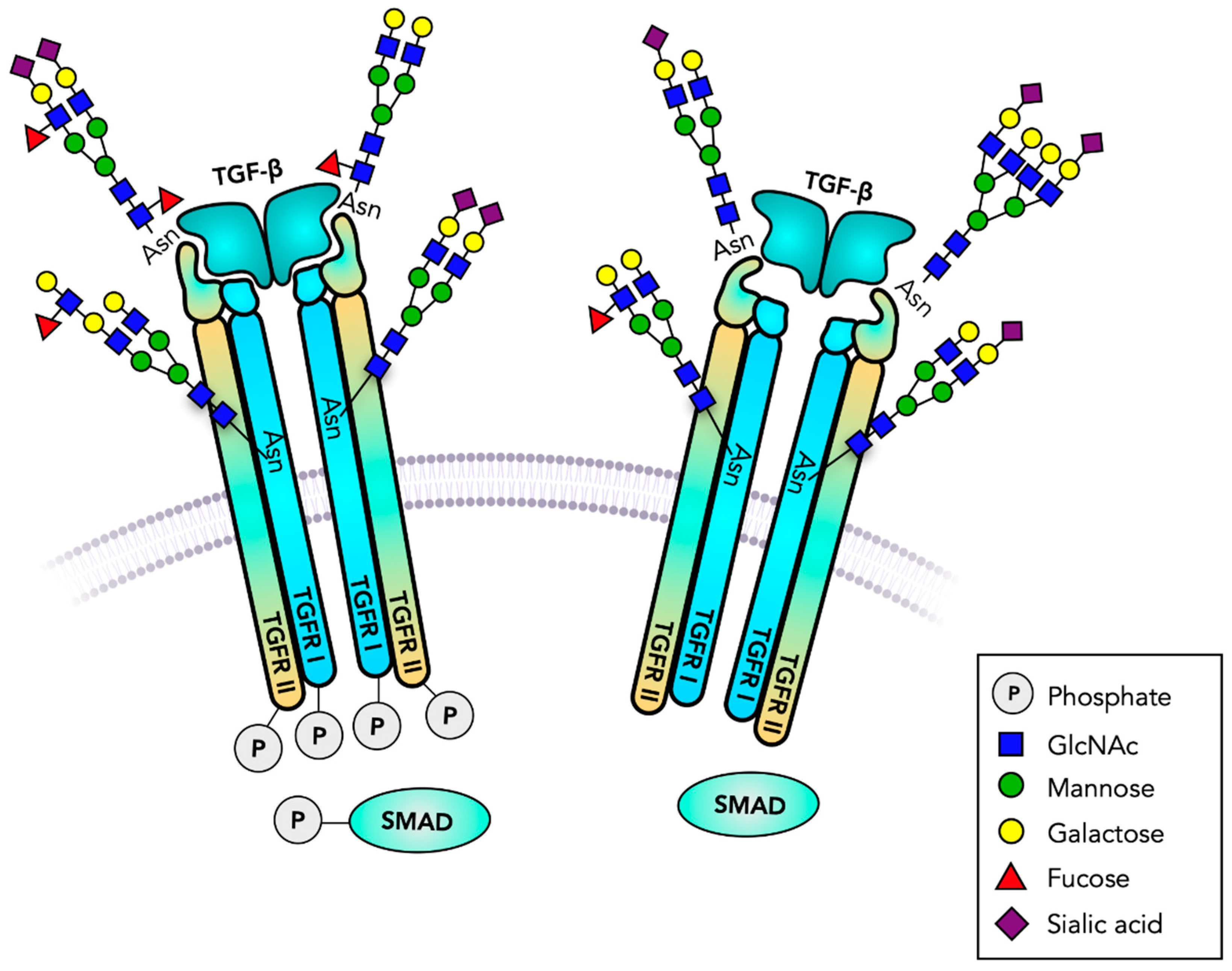
3. Transforming Growth Factor-Beta Receptor, TGFBR
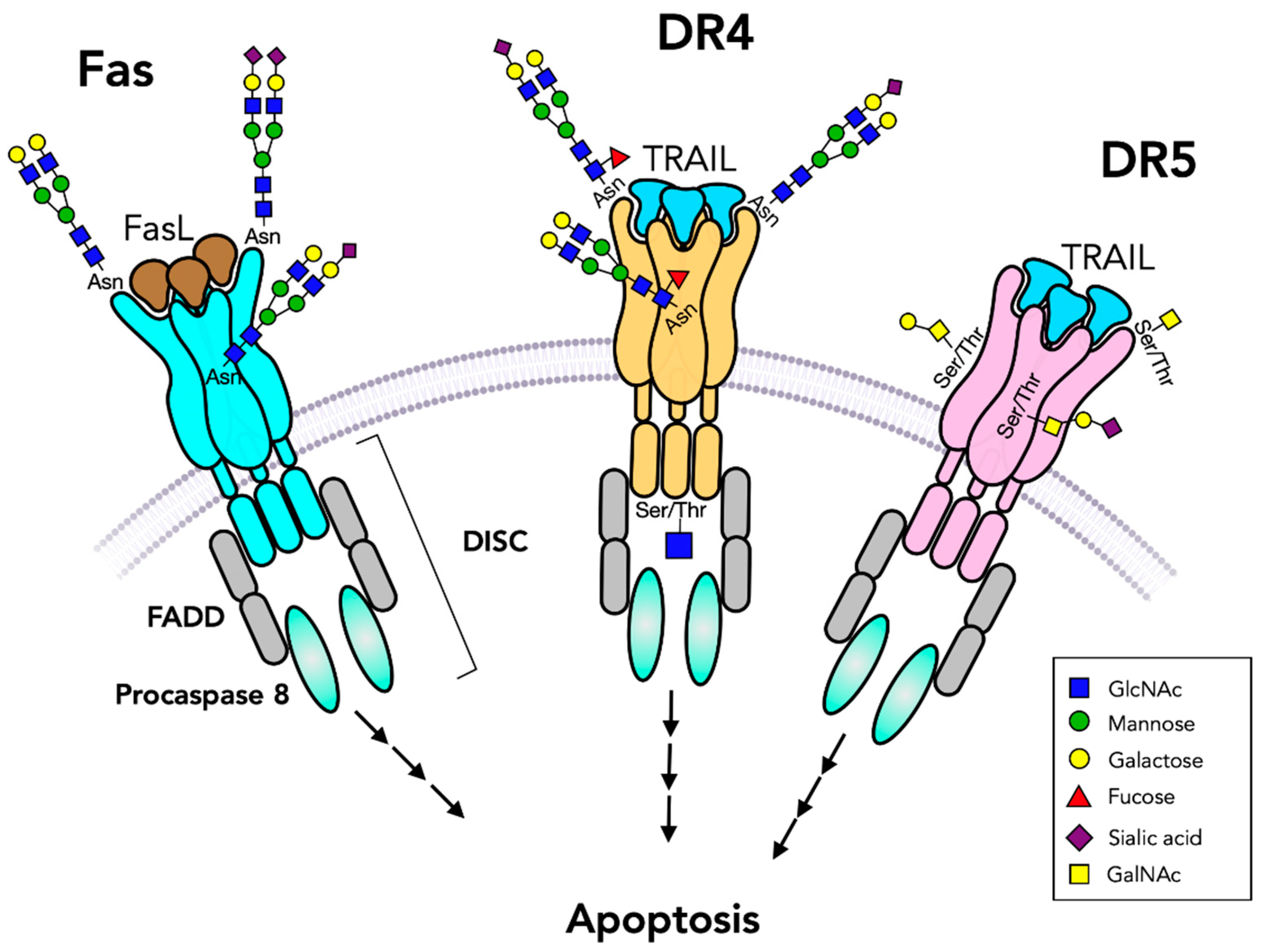
4. Death Receptors
4.1. Fas Glycosylation
4.2. TNF/TRAIL Receptors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sabbah, D.A.; Hajjo, R.; Sweidan, K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors. Curr. Top. Med. Chem. 2020, 20, 815–834. [Google Scholar] [CrossRef]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H. Death receptors. Essays Biochem. 2003, 39, 53–71. [Google Scholar] [PubMed]
- Gong, S.G. Isoforms of receptors of fibroblast growth factors. J. Cell Physiol. 2014, 229, 1887–1895. [Google Scholar] [CrossRef]
- Roghani, M.; Mansukhani, A.; Dell’Era, P.; Bellosta, P.; Basilico, C.; Rifkin, D.B.; Moscatelli, D. Heparin increases the affinity of basic fibroblast growth factor for its receptor but is not required for binding. J. Biol. Chem. 1994, 269, 3976–3984. [Google Scholar] [CrossRef]
- Gill, K.; Macdonald-Obermann, J.L.; Pike, L.J. Epidermal growth factor receptors containing a single tyrosine in their C-terminal tail bind different effector molecules and are signaling-competent. J. Biol. Chem. 2017, 292, 20744–20755. [Google Scholar] [CrossRef] [Green Version]
- Saldaña-Rivera, L.; Bello, M.; Méndez-Luna, D. Structural insight into the binding mechanism of ATP to EGFR and L858R, and T790M and L858R/T790 mutants. J. Biomol. Struct. Dyn. 2019, 37, 4671–4684. [Google Scholar] [CrossRef]
- Russo, A.; Franchina, T.; Ricciardi, G.; Battaglia, A.; Picciotto, M.; Adamo, V. Heterogeneous Responses to Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors (TKIs) in Patients with Uncommon EGFR Mutations: New Insights and Future Perspectives in this Complex Clinical Scenario. Int. J. Mol. Sci. 2019, 20, 1431. [Google Scholar] [CrossRef] [Green Version]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estornes, Y.; Dondelinger, Y.; Weber, K.; Bruggeman, I.; Peall, A.; MacFarlane, M.; Lebecque, S.; Vandenabeele, P.; Bertrand, M.J.M. N-glycosylation of mouse TRAIL-R restrains TRAIL-induced apoptosis. Cell Death Dis. 2018, 9, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouyer, V.; Leteurtre, E.; Zanetta, J.P.; Lesuffleur, T.; Delannoy, P.; Huet, G. Inhibition of the glycosylation and alteration in the intracellular trafficking of mucins and other glycoproteins by GalNAcalpha-O-bn in mucosal cell lines: An effect mediated through the intracellular synthesis of complex GalNAcalpha-O-bn oligosaccharides. Front. Biosci. 2001, 6, D1235–D1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, F.; Isaji, T.; Duan, C.; Yang, J.; Wang, Y.; Fukuda, T.; Gu, J. ST3GAL3, ST3GAL4, and ST3GAL6 differ in their regulation of biological functions via the specificities for the α2,3-sialylation of target proteins. FASEB J. 2020, 34, 881–897. [Google Scholar] [CrossRef] [Green Version]
- Schneider, P.; Bodmer, J.L.; Holler, N.; Mattmann, C.; Scuderi, P.; Terskikh, A.; Peitsch, M.C.; Tschopp, J. Characterization of Fas (Apo-1, CD95)-Fas ligand interaction. J. Biol. Chem. 1997, 272, 18827–18833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partridge, E.A.; Le Roy, C.; Di Guglielmo, G.M.; Pawling, J.; Cheung, P.; Granovsky, M.; Nabi, I.R.; Wrana, J.L.; Dennis, J.W. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 2004, 306, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woronowicz, A.; De Vusser, K.; Laroy, W.; Contreras, R.; Meakin, S.O.; Ross, G.M.; Szewczuk, M.R. Trypanosome trans-sialidase targets TrkA tyrosine kinase receptor and induces receptor internalization and activation. Glycobiology 2004, 14, 987–998. [Google Scholar] [CrossRef] [Green Version]
- Amith, S.R.; Jayanth, P.; Franchuk, S.; Finlay, T.; Seyrantepe, V.; Beyaert, R.; Pshezhetsky, A.V.; Szewczuk, M.R. Neu1 desialylation of sialyl alpha-2,3-linked beta-galactosyl residues of TOLL-like receptor 4 is essential for receptor activation and cellular signaling. Cell Signal. 2010, 22, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J. Neurotrophin Trk Receptors: New Targets for Cancer Therapy. Rev. Physiol. Biochem. Pharmacol. 2018, 174, 67–79. [Google Scholar] [PubMed]
- Jayanth, P.; Amith, S.R.; Gee, K.; Szewczuk, M.R. Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for neurotrophin activation of Trk receptors and cellular signaling. Cell Signal. 2010, 22, 1193–1205. [Google Scholar] [CrossRef]
- Hayes, J.M.; Cosgrave, E.F.; Struwe, W.B.; Wormald, M.; Davey, G.P.; Jefferis, R.; Rudd, P.M. Glycosylation and Fc receptors. Curr. Top. Microbiol. Immunol. 2014, 382, 165–199. [Google Scholar]
- de Haas, P.; Hendriks, W.J.A.J.; Lefeber, D.J.; Cambi, A. Biological and Technical Challenges in Unraveling the Role of N-Glycans in Immune Receptor Regulation. Front. Chem. 2020, 8, 55. [Google Scholar] [CrossRef]
- De Luca, A.; Carotenuto, A.; Rachiglio, A.; Gallo, M.; Maiello, M.R.; Aldinucci, D.; Pinto, A.; Normanno, N. The role of the EGFR signaling in tumor microenvironment. J. Cell Physiol. 2008, 214, 559–567. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.S.; Leahy, D.J. Structure of the extracellular region of HER3 reveals an interdomain tether. Science 2002, 297, 1330–1333. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell. 2000, 103, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Dawson, J.P.; Berger, M.B.; Lin, C.C.; Schlessinger, J.; Lemmon, M.A.; Ferguson, K.M. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Mol. Cell Biol. 2005, 25, 7734–7742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A. Ligand-induced ErbB receptor dimerization. Exp. Cell Res. 2009, 315, 638–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ren, J.; Yu, W.; Li, Q.; Kuwahara, H.; Yin, L.; Carraway, K.L., 3rd; Kufe, D. The epidermal growth factor receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-Src and beta-catenin. J. Biol. Chem. 2001, 276, 35239–35242. [Google Scholar] [CrossRef] [Green Version]
- Taylor, E.S.; Pol-Fachin, L.; Lins, R.D.; Lower, S.K. Conformational stability of the epidermal growth factor (EGF) receptor as influenced by glycosylation, dimerization and EGF hormone binding. Proteins 2017, 5, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Yokoe, S.; Asahi, M.; Lee, S.H.; Li, W.; Osumi, D.; Miyoshi, E.; Taniguchi, N. N-glycan of ErbB family plays a crucial role in dimer formation and tumor promotion. Biochim. Biophys. Acta 2008, 1780, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Yokoe, S.; Takahashi, M.; Asahi, M.; Lee, S.H.; Li, W.; Osumi, D.; Miyoshi, E.; Taniguchi, N. The Asn418-linked N-glycan of ErbB3 plays a crucial role in preventing spontaneous heterodimerization and tumor promotion. Cancer Res. 2007, 67, 1935–1942. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, T.; Ikeda, Y.; Taniguchi, N. The Asn-420-linked sugar chain in human epidermal growth factor receptor suppresses ligand-independent spontaneous oligomerization. Possible role of a specific sugar chain in controllable receptor activation. J. Biol. Chem. 2000, 275, 21988–21994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitson, K.B.; Whitson, S.R.; Red-Brewer, M.L.; McCoy, A.J.; Vitali, A.A.; Walker, F.; Johns, T.G.; Beth, A.H.; Staros, J.V. Functional effects of glycosylation at Asn-579 of the epidermal growth factor receptor. Biochemistry 2005, 44, 14920–14931. [Google Scholar] [CrossRef]
- Sato, Y.; Takahashi, M.; Shibukawa, Y.; Jain, S.K.; Hamaoka, R.; Miyagawa, J.; Yaginuma, Y.; Honke, K.; Ishikawa, M.; Taniguchi, N. Overexpression of N-acetylglucosaminyltransferase III enhances the epidermal growth factor-induced phosphorylation of ERK in HeLaS3 cells by up-regulation of the internalization rate of the receptors. J. Biol. Chem. 2001, 276, 11956–11962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebbaa, A.; Yamamoto, H.; Saito, T.; Meuillet, E.; Kim, P.; Kersey, D.S.; Bremer, E.G.; Taniguchi, N.; Moskal, J.R. Gene transfection-mediated overexpression of beta1,4-N-acetylglucosamine bisecting oligosaccharides in glioma cell line U373 MG inhibits epidermal growth factor receptor function. J. Biol. Chem. 1997, 272, 9275–9279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Zhao, Y.; Isaji, T.; Shibukawa, Y.; Ihara, H.; Takahashi, M.; Ikeda, Y.; Miyoshi, E.; Honke, K.; Taniguchi, N. Beta1,4-N-Acetylglucosaminyltransferase III down-regulates neurite outgrowth induced by costimulation of epidermal growth factor and integrins through the Ras/ERK signaling pathway in PC12 cells. Glycobiology 2004, 14, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.B.; Randolph, M.; Pierce, M. Inhibition of a specific N-glycosylation activity results in attenuation of breast carcinoma cell invasiveness-related phenotypes: Inhibition of epidermal growth factor-induced dephosphorylation of focal adhesion kinase. J. Biol. Chem. 2007, 282, 22150–22162. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gu, J.; Ihara, H.; Miyoshi, E.; Honke, K.; Taniguchi, N. Core fucosylation regulates epidermal growth factor receptor-mediated intracellular signaling. J. Biol. Chem. 2006, 281, 2572–2577. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.; Yokote, H.; Arao, T.; Maegawa, M.; Tanaka, K.; Fujita, Y.; Shimizu, C.; Hanafusa, T.; Fujiwara, Y.; Nishio, K. N-Glycan fucosylation of epidermal growth factor receptor modulates receptor activity and sensitivity to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci. 2008, 99, 1611–1617. [Google Scholar] [CrossRef]
- Lin, W.L.; Lin, Y.S.; Shi, G.Y.; Chang, C.F.; Wu, H.L. Lewisy promotes migration of oral cancer cells by glycosylation of epidermal growth factor receptor. PLoS ONE 2015, 10, e0120162. [Google Scholar] [CrossRef]
- Lin, M.C.; Huang, M.J.; Liu, C.H.; Yang, T.L.; Huang, M.C. GALNT2 enhances migration and invasion of oral squamous cell carcinoma by regulating EGFR glycosylation and activity. Oral Oncol. 2014, 50, 478–484. [Google Scholar] [CrossRef]
- Kawai, S.; Kato, S.; Imai, H.; Okada, Y.; Ishioka, C. Suppression of FUT1 attenuates cell proliferation in the HER2-overexpressing cancer cell line NCI-N87. Oncol. Rep. 2013, 29, 13–20. [Google Scholar] [CrossRef]
- Liu, J.J.; Lin, B.; Hao, Y.Y.; Li, F.F.; Liu, D.W.; Qi, Y.; Zhu, L.C.; Zhang, S.L.; Iwamori, M. Lewis(y) antigen stimulates the growth of ovarian cancer cells via regulation of the epidermal growth factor receptor pathway. Oncol. Rep. 2010, 23, 833–841. [Google Scholar] [PubMed]
- Zhang, Z.; Sun, P.; Liu, J.; Fu, L.; Yan, J.; Liu, Y.; Yu, L.; Wang, X.; Yan, Q. Suppression of FUT1/FUT4 expression by siRNA inhibits tumor growth. Biochim. Biophys. Acta 2008, 1783, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Shan, X.; Aziz, F.; Tian, L.L.; Wang, X.Q.; Yan, Q.; Liu, J.W. Ginsenoside Rg3-induced EGFR/MAPK pathway deactivation inhibits melanoma cell proliferation by decreasing FUT4/LeY expression. Int. J. Oncol. 2015, 46, 1667–1676. [Google Scholar] [CrossRef]
- Allahverdian, S.; Wang, A.; Singhera, G.K.; Wong, B.W.; Dorscheid, D.R. Sialyl Lewis X modification of the epidermal growth factor receptor regulates receptor function during airway epithelial wound repair. Clin. Exp. Allergy 2010, 40, 607–618. [Google Scholar] [CrossRef]
- Liu, Y.C.; Yen, H.Y.; Chen, C.Y.; Chen, C.H.; Cheng, P.F.; Juan, Y.H.; Chen, C.H.; Khoo, K.H.; Yu, C.J.; Yang, P.C.; et al. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11332–11337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, H.Y.; Liu, Y.C.; Chen, N.Y.; Tsai, C.F.; Wang, Y.T.; Chen, Y.J.; Hsu, T.L.; Yang, P.C.; Wong, C.H. Effect of sialylation on EGFR phosphorylation and resistance to tyrosine kinase inhibition. Proc. Natl. Acad. Sci. USA 2015, 112, 6955–6960. [Google Scholar] [CrossRef] [Green Version]
- Mozzi, A.; Forcella, M.; Riva, A.; Difrancesco, C.; Molinari, F.; Martin, V.; Papini, N.; Bernasconi, B.; Nonnis, S.; Tedeschi, G.; et al. NEU3 activity enhances EGFR activation without affecting EGFR expression and acts on its sialylation levels. Glycobiology 2015, 25, 855–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, T.; Hata, K.; Yamaguchi, K.; Shiozaki, K.; Koseki, K.; Moriya, S.; Miyagi, T. A crucial role of plasma membrane-associated sialidase in the survival of human cancer cells. Oncogene 2007, 26, 2483–2490. [Google Scholar] [CrossRef] [Green Version]
- Rybak, A.; Zarzecki, M.; Golabiewska, E.; Niechoda, A.; Holownia, A. Sialidase Attenuates Epidermal Growth Factor Response and Abolishes Antiproliferative Effects of Erlotinib in A549Alveolar Epithelial Cells. Adv. Exp. Med. Biol. 2019, 1153, 55–61. [Google Scholar]
- Britain, C.M.; Holdbrooks, A.T.; Anderson, J.C.; Willey, C.D.; Bellis, S.L. Sialylation of EGFR by the ST6Gal I sialyltransferase promotes EGFR activation and resistance to gefitinib-mediated cell death. J. Ovarian Res. 2018, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Britain, C.M.; Bhalerao, N.; Silva, A.D.; Chakraborty, A.; Buchsbaum, D.J.; Crowley, M.R.; Crossman, D.K.; Edwards, Y.J.K.; Bellis, S.L. Glycosyltransferase ST6Gal-I promotes the epithelial to mesenchymal transition in pancreatic cancer cells. J. Biol. Chem. 2020, 296, 100034. [Google Scholar] [CrossRef]
- Park, J.J.; Yi, J.Y.; Jin, Y.B.; Lee, Y.J.; Lee, J.S.; Lee, Y.S.; Ko, Y.G.; Lee, M. Sialylation of epidermal growth factor receptor regulates receptor activity and chemosensitivity to gefitinib in colon cancer cells. Biochem. Pharmacol. 2012, 83, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.T.; Yeh, C.C.; Liu, S.Y.; Huang, M.C.; Lai, I.R. The O-glycosylating enzyme GALNT2 suppresses the malignancy of gastric adenocarcinoma by reducing EGFR activities. Am. J. Cancer Res. 2018, 8, 1739–1751. [Google Scholar]
- Wu, Y.M.; Liu, C.H.; Hu, R.H.; Huang, M.J.; Lee, J.J.; Chen, C.H.; Huang, J.; Lai, H.S.; Lee, P.H.; Hsu, W.M.; et al. Mucin glycosylating enzyme GALNT2 regulates the malignant character of hepatocellular carcinoma by modifying the EGF receptor. Cancer Res. 2011, 71, 7270–7279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Xue, H.; Wei, Y.; Wang, C.; Yu, R.; Wang, C.; Wang, S.; Xu, J.; Qian, M.; Meng, Q.; et al. Mucin O-glycosylating enzyme GALNT2 facilitates the malignant character of glioma by activating the EGFR/PI3K/Akt/mTOR axis. Clin. Sci. (Lond.) 2019, 133, 1167–1184. [Google Scholar] [CrossRef]
- Zhen, Y.; Caprioli, R.M.; Staros, J.V. Characterization of glycosylation sites of the epidermal growth factor receptor. Biochemistry 2003, 42, 5478–5492. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.D.; Davies, M.J.; Bailey, D.; Renouf, D.V.; Hounsell, E.F. Analysis of the glycosylation patterns of the extracellular domain of the epidermal growth factor receptor expressed in Chinese hamster ovary fibroblasts. Growth Factors 1996, 13, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Sato, C.; Kim, J.H.; Abe, Y.; Saito, K.; Yokoyama, S.; Kohda, D. Characterization of the N-oligosaccharides attached to the atypical Asn-X-Cys sequence of recombinant human epidermal growth factor receptor. J. Biochem. 2000, 127, 65–72. [Google Scholar] [CrossRef]
- Ferreira, I.G.; Pucci, M.; Venturi, G.; Malagolini, N.; Chiricolo, M.; Dall’Olio, F. Glycosylation as a Main Regulator of Growth and Death Factor Receptors Signaling. Int. J. Mol. Sci. 2018, 19, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azimzadeh Irani, M.; Kannan, S.; Verma, C. Role of N-glycosylation in EGFR ectodomain ligand binding. Proteins 2017, 85, 1529–1549. [Google Scholar] [CrossRef]
- Lau, K.S.; Dennis, J.W. N-Glycans in cancer progression. Glycobiology 2008, 18, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Fukuda, T.; Isaji, T.; Lu, J.; Gu, W.; Lee, H.H.; Ohkubo, Y.; Kamada, Y.; Taniguchi, N.; Miyoshi, E.; et al. Loss of α1,6-fucosyltransferase suppressed liver regeneration: Implication of core fucose in the regulation of growth factor receptor-mediated cellular signaling. Sci. Rep. 2015, 5, 8264. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Fukuda, T.; Isaji, T.; Lu, J.; Im, S.; Hang, Q.; Gu, W.; Hou, S.; Ohtsubo, K.; Gu, J. Loss of α1,6-fucosyltransferase inhibits chemical-induced hepatocellular carcinoma and tumorigenesis by down-regulating several cell signaling pathways. FASEB J. 2015, 29, 3217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinger, M.; Farhan, H.; Just, H.; Drobny, H.; Himmler, G.; Loibner, H.; Mudde, G.C.; Freissmuth, M.; Sexl, V. Antibodies directed against Lewis-Y antigen inhibit signaling of Lewis-Y modified ErbB receptors. Cancer Res. 2004, 64, 1087–1093. [Google Scholar] [CrossRef] [Green Version]
- Park, J.J.; Lee, M. Increasing the α 2, 6 sialylation of glycoproteins may contribute to metastatic spread and therapeutic resistance in colorectal cancer. Gut Liver. 2013, 7, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Li, J.; Feng, C.H.; Chen, S.K.; Liu, Y.P.; Duan, C.Y.; Li, H.; Xia, X.M.; He, T.; Wei, M.; et al. c-Met function requires N-linked glycosylation modification of pro-Met. J. Cell Biochem. 2013, 114, 816–822. [Google Scholar] [CrossRef]
- Hyuga, M.; Hyuga, S.; Kawasaki, N.; Ohta, M.; Itoh, S.; Niimi, S.; Kawanishi, T.; Hayakawa, T. Enhancement of hepatocyte growth factor-induced cell scattering in N-acetylglucosaminyltransferase III-transfected HepG2 cells. Biol. Pharm. Bull. 2004, 27, 781–785. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, T.; Orita, T.; Katsuya, K.; Yamasaki, Y.; Akiyama, K.; Li, H.; Yamamoto, T.; Saito, Y.; Nakamura, M. MUC20 suppresses the hepatocyte growth factor-induced Grb2-Ras pathway by binding to a multifunctional docking site of met. Mol. Cell Biol. 2004, 24, 7456–7468. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Tang, F.; Liu, P.; Zhong, T.; Yuan, F.; He, Q.; von Itzstein, M.; Li, H.; Weng, L.; Yu, X. Structural and Functional Insight Into the Glycosylation Impact Upon the HGF/c-Met Signaling Pathway. Front. Cell Dev. Biol. 2020, 8, 490. [Google Scholar] [CrossRef]
- Niemann, H.H. Structural basis of MET receptor dimerization by the bacterial invasion protein InlB and the HGF/SF splice variant NK1. Biochim. Biophys. Acta 2013, 1834, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Dong, S.M.; Kim, S.Y.; Na, E.Y.; Shin, M.S.; Pi, J.H.; Kim, B.J.; Bae, J.H.; Hong, Y.K.; Lee, K.S.; et al. Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer Res. 1999, 59, 307–310. [Google Scholar]
- Cao, J.; Shen, C.; Wang, H.; Shen, H.; Chen, Y.; Nie, A.; Yan, G.; Lu, H.; Liu, Y.; Yang, P. Identification of N-glycosylation sites on secreted proteins of human hepatoellular carcinoma cells with a complementary proteomics approach. J. Proteome Res. 2009, 8, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Contessa, J.N.; Bhojani, M.S.; Freeze, H.H.; Ross, B.D.; Rehemtulla, A.; Lawrence, T.S. Molecular imaging of N-linked glycosylation suggests glycan biosynthesis is a novel target for cancer therapy. Clin. Cancer Res. 2010, 16, 3205–3214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Liu, Q.; Pan, S.; Huang, Y.; Qi, Y.; Li, S.; Xiao, Y.; Jia, L. The HOTAIR/miR-214/ST6GAL1 crosstalk modulates colorectal cancer procession through mediating sialylated c-Met via JAK2/STAT3 cascade. J. Exp. Clin. Cancer Res. 2019, 38, 455. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Zhu, C.H.; Tang, S.; Shen, A.J.; Ai, J.; Li, J.; Geng, M.Y.; Ding, J. Alpha2,6-hyposialylation of c-Met abolishes cell motility of ST6Gal-I-knockdown HCT116 cells. Acta Pharmacol. Sin. 2009, 30, 1039–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, C.; Osório, H.; Pinto, M.T.; Campos, D.; Oliveira, M.J.; Reis, C.A. Expression of ST3GAL4 leads to SLe(x) expression and induces c-Met activation and an invasive phenotype in gastric carcinoma cells. PLoS ONE 2013, 8, e66737. [Google Scholar] [CrossRef]
- Liu, S.Y.; Shun, C.T.; Hung, K.Y.; Juan, H.F.; Hsu, C.L.; Huang, M.C.; Lai, I.R. Mucin glycosylating enzyme GALNT2 suppresses malignancy in gastric adenocarcinoma by reducing MET phosphorylation. Oncotarget 2016, 7, 11251. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.M.; Liu, C.H.; Huang, M.J.; Lai, H.S.; Lee, P.H.; Hu, R.H.; Huang, M.C. C1GALT1 enhances proliferation of hepatocellular carcinoma cells via modulating MET glycosylation and dimerization. Cancer Res. 2013, 73, 5580–5590. [Google Scholar] [CrossRef] [Green Version]
- Levine, K.M.; Ding, K.; Chen, L.; Oesterreich, S. FGFR4: A promising therapeutic target for breast cancer and other solid tumors. Pharmacol. Ther. 2020, 214, 107590. [Google Scholar] [CrossRef] [PubMed]
- Duchesne, L.; Tissot, B.; Rudd, T.R.; Dell, A.; Fernig, D.G. N-glycosylation of fibroblast growth factor receptor 1 regulates ligand and heparan sulfate co-receptor binding. J. Biol. Chem. 2006, 281, 27178–27189. [Google Scholar] [CrossRef] [Green Version]
- Feige, J.J.; Baird, A. Glycosylation of the basic fibroblast growth factor receptor. The contribution of carbohydrate to receptor function. J. Biol. Chem. 1988, 263, 14023–14029. [Google Scholar] [CrossRef]
- Tuominen, H.; Heikinheimo, P.; Loo, B.M.; Kataja, K.; Oker-Blom, C.; Uutela, M.; Jalkanen, M.; Goldman, A. Expression and glycosylation studies of human FGF receptor 4. Protein Expr. Purif. 2001, 21, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Triantis, V.; Saeland, E.; Bijl, N.; Oude-Elferink, R.P.; Jansen, P.L. Glycosylation of fibroblast growth factor receptor 4 is a key regulator of fibroblast growth factor 19-mediated down-regulation of cytochrome P450 7A1. Hepatology 2010, 52, 656–666. [Google Scholar] [CrossRef]
- Winterpacht, A.; Hilbert, K.; Stelzer, C.; Schweikardt, T.; Decker, H.; Segerer, H.; Spranger, J.; Zabel, B. A novel mutation in FGFR-3 disrupts a putative N-glycosylation site and results in hypochondroplasia. Physiol. Genomics. 2000, 2, 9–12. [Google Scholar] [CrossRef]
- Hatch, N.E.; Hudson, M.; Seto, M.L.; Cunningham, M.L.; Bothwell, M. Intracellular retention, degradation, and signaling of glycosylation-deficient FGFR2 and craniosynostosis syndrome-associated FGFR2C278F. J. Biol. Chem. 2006, 281, 27292–27305. [Google Scholar] [CrossRef] [Green Version]
- Sugihara, K.; Shibata, T.K.; Takata, K.; Kimura, T.; Kanayama, N.; Williams, R.; Hatakeyama, S.; Akama, T.O.; Kuo, C.W.; Khoo, K.H.; et al. Attenuation of fibroblast growth factor signaling by poly-N-acetyllactosamine type glycans. FEBS Lett. 2013, 587, 3195–3201. [Google Scholar] [CrossRef] [Green Version]
- Hung, J.S.; Huang, J.; Lin, Y.C.; Huang, M.J.; Lee, P.H.; Lai, H.S.; Liang, J.T.; Huang, M.C. C1GALT1 overexpression promotes the invasive behavior of colon cancer cells through modifying O-glycosylation of FGFR2. Oncotarget 2014, 5, 2096–2106. [Google Scholar] [CrossRef] [Green Version]
- Chandler, K.B.; Leon, D.R.; Meyer, R.D.; Rahimi, N.; Costello, C.E. Site-Specific N-Glycosylation of Endothelial Cell Receptor Tyrosine Kinase VEGFR-2. J. Proteome Res. 2017, 16, 677–688. [Google Scholar] [CrossRef] [Green Version]
- Chandler, K.B.; Leon, D.R.; Kuang, J.; Meyer, R.D.; Rahimi, N.; Costello, C.E. N-Glycosylation regulates ligand-dependent activation and signaling of vascular endothelial growth factor receptor 2 (VEGFR2). J. Biol. Chem. 2019, 294, 13117–13130. [Google Scholar] [CrossRef]
- Hahm, Y.H.; Lee, J.Y.; Ahn, Y.H. Investigation of Site-Specific Differences in Glycan Microheterogeneity by N-Glycopeptide Mapping of VEGFR-IgG Fusion Protein. Molecules 2019, 24, 3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Shibuya, M. The 230 kDa mature form of KDR/Flk-1 (VEGF receptor-2) activates the PLC-gamma pathway and partially induces mitotic signals in NIH3T3 fibroblasts. Oncogene 1997, 14, 2079–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamaki, R.; Ogawa, K.; Kizuka, Y.; Komi, Y.; Kojima, S.; Kotani, N.; Honke, K.; Honda, T.; Taniguchi, N.; Kitazume, S. Glycosylation controls cooperative PECAM-VEGFR2-β3 integrin functions at the endothelial surface for tumor angiogenesis. Oncogene 2018, 37, 4287–4299. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Fukuda, T.; Li, W.; Gao, C.X.; Kondo, A.; Matsumoto, A.; Miyoshi, E.; Taniguchi, N.; Gu, J. Requirement of Fut8 for the expression of vascular endothelial growth factor receptor-2: A new mechanism for the emphysema-like changes observed in Fut8-deficient mice. J. Biochem. 2009, 145, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Lee, J., Jr.; Chen, C.H.; Chen, Y.H.; Huang, M.J.; Huang, J.; Hung, J.S.; Chen, M.T.; Huang, M.C. COSMC is overexpressed in proliferating infantile hemangioma and enhances endothelial cell growth via VEGFR2. PLoS ONE 2013, 8, e56211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weroha, S.J.; Haluska, P. The insulin-like growth factor system in cancer. Endocrinol. Metab. Clin. N. Am. 2012, 41, 335–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masnikosa, R.; Baricević, I.; Jones, D.R.; Nedić, O. Characterisation of insulin-like growth factor receptors and insulin receptors in the human placenta using lectin affinity methods. Growth Horm. IGF Res. 2006, 16, 174–184. [Google Scholar] [CrossRef]
- Robajac, D.; Masnikosa, R.; Miković, Ž.; Nedić, O. Gestation-associated changes in the glycosylation of placental insulin and insulin-like growth factor receptors. Placenta 2016, 39, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Cui, X.; Wang, H.; Liu, J.; Qin, H.; Liu, S.; Yan, Q. FUT8 drives the proliferation and invasion of trophoblastic cells via IGF-1/IGF-1R signaling pathway. Placenta 2019, 75, 45–53. [Google Scholar] [CrossRef]
- Itkonen, H.M.; Mills, I.G. N-linked glycosylation supports cross-talk between receptor tyrosine kinases and androgen receptor. PLoS ONE 2013, 8, e65016. [Google Scholar] [CrossRef]
- Forbes, K.; Shah, V.K.; Siddals, K.; Gibson, J.M.; Aplin, J.D.; Westwood, M. Statins inhibit insulin-like growth factor action in first trimester placenta by altering insulin-like growth factor 1 receptor glycosylation. Mol. Hum. Reprod. 2015, 21, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddals, K.W.; Allen, J.; Sinha, S.; Canfield, A.E.; Kalra, P.A.; Gibson, J.M. Apposite insulin-like growth factor (IGF) receptor glycosylation is critical to the maintenance of vascular smooth muscle phenotype in the presence of factors promoting osteogenic differentiation and mineralization. J. Biol. Chem. 2011, 286, 16623–16630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girnita, L.; Wang, M.; Xie, Y.; Nilsson, G.; Dricu, A.; Wejde, J.; Larsson, O. Inhibition of N-linked glycosylation down-regulates insulin-like growth factor-1 receptor at the cell surface and kills Ewing’s sarcoma cells: Therapeutic implications. Anticancer Drug Des. 2000, 15, 67–72. [Google Scholar] [PubMed]
- Klaver, E.; Zhao, P.; May, M.; Flanagan-Steet, H.; Freeze, H.H.; Gilmore, R.; Wells, L.; Contessa, J.; Steet, R. Selective inhibition of N-linked glycosylation impairs receptor tyrosine kinase processing. Dis. Model Mech. 2019, 12, dmm039602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, J.G.; Liang, J.; Antonopoulos, A.; Giovannone, N.; Kang, S.; Mondala, T.S.; Head, S.R.; King, S.L.; Tani, Y.; Brackett, D.; et al. Loss of GCNT2/I-branched glycans enhances melanoma growth and survival. Nat. Commun. 2018, 9, 3368. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.L.; Chou, C.H.; Jeng, Y.M.; Lu, M.Y.; Yang, Y.L.; Jou, S.T.; Lin, D.T.; Chang, H.H.; Lin, K.H.; Hsu, W.M.; et al. GALNT2 suppresses malignant phenotypes through IGF-1 receptor and predicts favorable prognosis in neuroblastoma. Oncotarget 2014, 5, 12247–12259. [Google Scholar] [CrossRef] [Green Version]
- Haxho, F.; Haq, S.; Szewczuk, M.R. Biased G protein-coupled receptor agonism mediates Neu1 sialidase and matrix metalloproteinase-9 crosstalk to induce transactivation of insulin receptor signaling. Cell Signal. 2018, 43, 71–84. [Google Scholar] [CrossRef]
- Hinek, A.; Bodnaruk, T.D.; Bunda, S.; Wang, Y.; Liu, K. Neuraminidase-1, a subunit of the cell surface elastin receptor, desialylates and functionally inactivates adjacent receptors interacting with the mitogenic growth factors PDGF-BB and IGF-2. Am. J. Pathol. 2008, 173, 1042–1056. [Google Scholar] [CrossRef] [Green Version]
- Arabkhari, M.; Bunda, S.; Wang, Y.; Wang, A.; Pshezhetsky, A.V.; Hinek, A. Desialylation of insulin receptors and IGF-1 receptors by neuraminidase-1 controls the net proliferative response of L6 myoblasts to insulin. Glycobiology 2010, 20, 603–616. [Google Scholar] [CrossRef] [Green Version]
- Sidorova, Y.A.; Volcho, K.P.; Salakhutdinov, N.F. Neuroregeneration in Parkinson’s Disease: From Proteins to Small Molecules. Curr. Neuropharmacol. 2019, 17, 268–287. [Google Scholar] [CrossRef]
- Baeza-Raja, B.; Eckel-Mahan, K.; Zhang, L.; Vagena, E.; Tsigelny, I.F.; Sassone-Corsi, P.; Ptácek, L.J.; Akassoglou, K. p75 neurotrophin receptor is a clock gene that regulates oscillatory components of circadian and metabolic networks. J. Neurosci. 2013, 33, 10221–10234. [Google Scholar] [CrossRef] [PubMed]
- Colley, K.J.; Kitajima, K.; Sato, C. Polysialic acid: Biosynthesis, novel functions and applications. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 498–532. [Google Scholar] [CrossRef]
- Youker, R.T.; Bruns, J.R.; Costa, S.A.; Rbaibi, Y.; Lanni, F.; Kashlan, O.B.; Teng, H.; Weisz, O.A. Multiple motifs regulate apical sorting of p75 via a mechanism that involves dimerization and higher-order oligomerization. Mol. Biol. Cell. 2013, 24, 1996–2007. [Google Scholar] [CrossRef]
- Paratcha, G.; Ledda, F. GDNF and GFRalpha: A versatile molecular complex for developing neurons. Trends Neurosci. 2008, 31, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, J.R.; Bouzas-Rodriguez, J.; Tauszig-Delamasure, S.; Mehlen, P. RET modulates cell adhesion, via its cleavage by caspase in sympathetic neurons. J. Biol. Chem. 2011, 286, 14628–14638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihara, Y.; Sakamoto, Y.; Mihara, M.; Shimizu, K.; Taniguchi, N. Overexpression of N-acetylglucosaminyltransferase III disrupts the tyrosine phosphorylation of Trk with resultant signaling dysfunction in PC12 cells treated with nerve growth factor. J. Biol. Chem. 1997, 272, 9629–9634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Li, J.; Geng, M. N-acetylglucosaminyltransferase V modifies TrKA protein, regulates the receptor function. Cell Mol. Neurobiol. 2008, 28, 663–670. [Google Scholar] [CrossRef]
- Ultsch, M.H.; Wiesmann, C.; Simmons, L.C.; Henrich, J.; Yang, M.; Reilly, D.; Bass, S.H.; de Vos, A.M. Crystal structures of the neurotrophin-binding domain of TrkA, TrkB and TrkC. J. Mol. Biol. 1999, 290, 149–159. [Google Scholar] [CrossRef]
- Wehrman, T.; He, X.; Raab, B.; Dukipatti, A.; Blau, H.; Garcia, K.C. Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron 2007, 53, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Watson, F.L.; Porcionatto, M.A.; Bhattacharyya, A.; Stiles, C.D.; Segal, R.A. TrkA glycosylation regulates receptor localization and activity. J. Neurobiol. 1999, 39, 323–336. [Google Scholar] [CrossRef]
- Bennmann, D.; Kannicht, C.; Fisseau, C.; Jacobs, K.; Navarette-Santos, A.; Hofmann, B.; Horstkorte, R. Glycation of the high affinity NGF-receptor and RAGE leads to reduced ligand affinity. Mech. Ageing Dev. 2015, 150, 1–11. [Google Scholar] [CrossRef]
- Woronowicz, A.; Amith, S.R.; De Vusser, K.; Laroy, W.; Contreras, R.; Basta, S.; Szewczuk, M.R. Dependence of neurotrophic factor activation of Trk tyrosine kinase receptors on cellular sialidase. Glycobiology 2007, 17, 10–24. [Google Scholar] [CrossRef] [Green Version]
- Chiricozzi, E.; Pomè, D.Y.; Maggioni, M.; Di Biase, E.; Parravicini, C.; Palazzolo, L.; Loberto, N.; Eberini, I.; Sonnino, S. Role of the GM1 ganglioside oligosaccharide portion in the TrkA-dependent neurite sprouting in neuroblastoma cells. J. Neurochem. 2017, 143, 645–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haniu, M.; Talvenheimo, J.; Le, J.; Katta, V.; Welcher, A.; Rohde, M.F. Extracellular domain of neurotrophin receptor trkB: Disulfide structure, N-glycosylation sites, and ligand binding. Arch. Biochem. Biophys. 1995, 322, 256–264. [Google Scholar] [CrossRef]
- Bolk, S.; Pelet, A.; Hofstra, R.M.; Angrist, M.; Salomon, R.; Croaker, D.; Buys, C.H.; Lyonnet, S.; Chakravarti, A. A human model for multigenic inheritance: Phenotypic expression in Hirschsprung disease requires both the RET gene and a new 9q31 locus. Proc. Natl. Acad. Sci. USA 2000, 97, 268–273. [Google Scholar] [CrossRef] [Green Version]
- Werner, P.; Paluru, P.; Simpson, A.M.; Latney, B.; Iyer, R.; Brodeur, G.M.; Goldmuntz, E. Mutations in NTRK3 suggest a novel signaling pathway in human congenital heart disease. Hum. Mutat. 2014, 35, 1459–1468. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Diestel, U.; Resch, M.; Meinhardt, K.; Weiler, S.; Hellmann, T.V.; Mueller, T.D.; Nickel, J.; Eichler, J.; Muller, Y.A. Identification of a Novel TGF-β-Binding Site in the Zona Pellucida C-terminal (ZP-C) Domain of TGF-β-Receptor-3 (TGFR-3). PLoS ONE 2013, 8, e67214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Hasegawa, Y.; Gao, C.; Kuroki, Y.; Taniguchi, N. N-glycans of growth factor receptors: Their role in receptor function and disease implications. Clin. Sci. (Lond.) 2016, 130, 1781–1792. [Google Scholar] [CrossRef]
- Kim, Y.W.; Park, J.; Lee, H.J.; Lee, S.Y.; Kim, S.J. TGF-β sensitivity is determined by N-linked glycosylation of the type II TGF-β receptor. Biochem. J. 2012, 445, 403–411. [Google Scholar] [CrossRef]
- Zhang, J.; Ten Dijke, P.; Wuhrer, M.; Zhang, T. Role of glycosylation in TGF-β signaling and epithelial-to-mesenchymal transition in cancer. Protein Cell 2021, 12, 89–106. [Google Scholar] [CrossRef]
- Tu, C.F.; Wu, M.Y.; Lin, Y.C.; Kannagi, R.; Yang, R.B. FUT8 promotes breast cancer cell invasiveness by remodeling TGF-β receptor core fucosylation. Breast Cancer Res. 2017, 19, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Wang, D.; Wu, T.; Dong, C.; Shen, N.; Sun, Y.; Sun, Y.; Xie, H.; Wang, N.; Shan, L. Blocking core fucosylation of TGF-β1 receptors downregulates their functions and attenuates the epithelial-mesenchymal transition of renal tubular cells. Am. J. Physiol. Renal Physiol. 2011, 300, F1017–F1025. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M.; Takimoto, R.; Tamura, F.; Yoshida, M.; Ono, M.; Murase, K.; Sato, Y.; Osuga, T.; Sato, T.; Iyama, S.; et al. Fucosylated TGF-β receptors transduces a signal for epithelial-mesenchymal transition in colorectal cancer cells. Br. J. Cancer. 2014, 110, 156–163. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gu, J.; Miyoshi, E.; Honke, K.; Taniguchi, N. Phenotype changes of Fut8 knockout mouse: Core fucosylation is crucial for the function of growth factor receptor(s). Methods Enzymol. 2006, 417, 11–22. [Google Scholar]
- Venkatachalam, M.A.; Weinberg, J.M. New wrinkles in old receptors: Core fucosylation is yet another target to inhibit TGF-β signaling. Kidney Int. 2013, 84, 11–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Maeno, T.; Ota, F.; Ueno, M.; Korekane, H.; Takamatsu, S.; Shirato, K.; Matsumoto, A.; Kobayashi, S.; Yoshida, K.; et al. Sensitivity of heterozygous α1,6-fucosyltransferase knock-out mice to cigarette smoke-induced emphysema: Implication of aberrant transforming growth factor-β signaling and matrix metalloproteinase gene expression. J. Biol. Chem. 2012, 287, 16699–166708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Inoue, S.; Gu, J.; Miyoshi, E.; Noda, K.; Li, W.; Mizuno-Horikawa, Y.; Nakano, M.; Asahi, M.; Takahashi, M.; et al. Dysregulation of TGF-beta1 receptor activation leads to abnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc. Natl. Acad. Sci. USA 2005, 102, 15791–15796. [Google Scholar] [CrossRef] [Green Version]
- Li, F.F.; Liu, J.J.; Liu, D.W.; Lin, B.; Hao, Y.Y.; Cong, J.P.; Zhu, L.C.; Gao, S.; Zhang, S.L.; Iwamori, M. Lewis Y regulates signaling molecules of the transforming growth factor β pathway in ovarian carcinoma-derived RMG-I cells. Int. J. Oncol. 2012, 40, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Mori, K.; Matsumoto, H.; Kiso, S.; Yoshida, Y.; Shinzaki, S.; Hiramatsu, N.; Ishii, M.; Moriwaki, K.; Kawada, N.; et al. N-Acetylglucosaminyltransferase V regulates TGF-β response in hepatic stellate cells and the progression of steatohepatitis. Glycobiology 2012, 22, 778–787. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Isaji, T.; Lu, Y.; Gu, W.; Kondo, M.; Fukuda, T.; Du, Y.; Gu, J. Roles of N-acetylglucosaminyltransferase III in epithelial-to-mesenchymal transition induced by transforming growth factor β1 (TGF-β1) in epithelial cell lines. J. Biol. Chem. 2012, 287, 16563–16574. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Meng, F.; Wu, S.; Kreike, B.; Sethi, S.; Chen, W.; Miller, F.R.; Wu, G. Engagement of I-branching {beta}-1, 6-N-acetylglucosaminyltransferase 2 in breast cancer metastasis and TGF-{beta} signaling. Cancer Res. 2011, 71, 4846–4856. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Isaji, T.; Im, S.; Fukuda, T.; Hashii, N.; Takakura, D.; Kawasaki, N.; Gu, J. β-Galactoside α2,6-sialyltranferase 1 promotes transforming growth factor-β-mediated epithelial-mesenchymal transition. J. Biol. Chem. 2014, 289, 34627–34641. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhang, C.; Zhang, K.; Chen, Q.; Wu, S.; Huang, H.; Huang, T.; Zhang, N.; Wang, X.; Li, W.; et al. ppGalNAc-T4-catalyzed O-Glycosylation of TGF-β type Ⅱ receptor regulates breast cancer cells metastasis. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Ashkenazi, A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene 2010, 29, 4752–4765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinkel, S.; Gross, A.; Yang, E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 2006, 13, 1351–1359. [Google Scholar] [CrossRef]
- Seyrek, K.; Richter, M.; Lavrik, I.N. Decoding the sweet regulation of apoptosis: The role of glycosylation and galectins in apoptotic signaling pathways. Cell Death Differ. 2019, 26, 981–993. [Google Scholar] [CrossRef]
- Micheau, O. Regulation of TNF-Related Apoptosis-Inducing Ligand Signaling by Glycosylation. Int. J. Mol. Sci. 2018, 19, 715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yang, X.; Nguyen, A.H.; Brockhausen, I. Requirement of N-glycosylation for the secretion of recombinant extracellular domain of human Fas in HeLa cells. Int. J. Biochem. Cell Biol. 2007, 39, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Shatnyeva, O.M.; Kubarenko, A.V.; Weber, C.E.; Pappa, A.; Schwartz-Albiez, R.; Weber, A.N.; Krammer, P.H.; Lavrik, I.N. Modulation of the CD95-induced apoptosis: The role of CD95 N-glycosylation. PLoS ONE 2011, 6, e19927. [Google Scholar] [CrossRef] [Green Version]
- Orlinick, J.R.; Elkon, K.B.; Chao, M.V. Separate domains of the human fas ligand dictate self-association and receptor binding. J. Biol. Chem. 1997, 272, 32221–32229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlier, E.; Condé, C.; Zhang, J.; Deneubourg, L.; Di Valentin, E.; Rahmouni, S.; Chariot, A.; Agostinis, P.; Pang, P.C.; Haslam, S.M.; et al. SHIP-1 inhibits CD95/APO-1/Fas-induced apoptosis in primary T lymphocytes and T leukemic cells by promoting CD95 glycosylation independently of its phosphatase activity. Leukemia 2010, 24, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.G.; Balmaña, M.; Macedo, J.A.; Poças, J.; Fernandes, Â.; de-Freitas-Junior, J.C.M.; Pinho, S.S.; Gomes, J.; Magalhães, A.; Gomes, C.; et al. Glycosylation in cancer: Selected roles in tumour progression, immune modulation and metastasis. Cell Immunol. 2018, 333, 46–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, M.E.; Hellbardt, S.; Schwartz-Albiez, R.; Westendorp, M.O.; Walczak, H.; Moldenhauer, G.; Grell, M.; Krammer, P.H. Cell surface sialylation plays a role in modulating sensitivity towards APO-1-mediated apoptotic cell death. Cell Death Differ. 1995, 2, 163–171. [Google Scholar]
- Keppler, O.T.; Peter, M.E.; Hinderlich, S.; Moldenhauer, G.; Stehling, P.; Schmitz, I.; Schwartz-Albiez, R.; Reutter, W.; Pawlita, M. Differential sialylation of cell surface glycoconjugates in a human B lymphoma cell line regulates susceptibility for CD95 (APO-1/Fas)-mediated apoptosis and for infection by a lymphotropic virus. Glycobiology 1999, 9, 557–569. [Google Scholar] [CrossRef] [Green Version]
- Swindall, A.F.; Bellis, S.L. Sialylation of the Fas death receptor by ST6Gal-I provides protection against Fas-mediated apoptosis in colon carcinoma cells. J. Biol. Chem. 2011, 286, 22982–22990. [Google Scholar] [CrossRef] [Green Version]
- Klíma, M.; Zájedová, J.; Doubravská, L.; Andera, L. Functional analysis of the posttranslational modifications of the death receptor 6. Biochim. Biophys. Acta 2009, 1793, 1579–1587. [Google Scholar] [CrossRef] [Green Version]
- Friedmann, E.; Hauben, E.; Maylandt, K.; Schleeger, S.; Vreugde, S.; Lichtenthaler, S.F.; Kuhn, P.H.; Stauffer, D.; Rovelli, G.; Martoglio, B. SPPL2a and SPPL2b promote intramembrane proteolysis of TNFalpha in activated dendritic cells to trigger IL-12 production. Nat. Cell Biol. 2006, 8, 843–848. [Google Scholar] [CrossRef]
- Dufour, F.; Rattier, T.; Shirley, S.; Picarda, G.; Constantinescu, A.A.; Morlé, A.; Zakaria, A.B.; Marcion, G.; Causse, S.; Szegezdi, E.; et al. N-glycosylation of mouse TRAIL-R and human TRAIL-R1 enhances TRAIL-induced death. Cell Death Differ. 2017, 24, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.W.; Punnoose, E.A.; Januario, T.; Lawrence, D.A.; Pitti, R.M.; Lancaster, K.; Lee, D.; von Goetz, M.; Yee, S.F.; Totpal, K.; et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat. Med. 2007, 13, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Meng, Y.; Sheng, X.; Guan, Y.; Zhang, F.; Han, Z.; Kang, Y.; Tai, G.; Zhou, Y.; Cheng, H. Tunicamycin enhances human colon cancer cells to TRAIL-induced apoptosis by JNK-CHOP-mediated DR5 upregulation and the inhibition of the EGFR pathway. Anticancer Drugs 2017, 28, 66–74. [Google Scholar] [CrossRef]
- Jiang, Y.; Wen, T.; Yan, R.; Kim, S.R.; Stowell, S.R.; Wang, W.; Wang, Y.; An, G.; Cummings, R.D.; Ju, T. O-glycans on death receptors in cells modulate their sensitivity to TRAIL-induced apoptosis through affecting on their stability and oligomerization. FASEB J. 2020, 34, 11786–11801. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.Y.; Seo, S.U.; Woo, S.M.; Min, K.J.; Byun, H.S.; Hur, G.M.; Kang, S.C.; Kwon, T.K. Oridonin enhances TRAIL-induced apoptosis through GALNT14-mediated DR5 glycosylation. Biochimie 2019, 165, 108–114. [Google Scholar] [CrossRef]
- Lee, H.; Oh, Y.; Jeon, Y.J.; Lee, S.Y.; Kim, H.; Lee, H.J.; Jung, Y.K. DR4-Ser424 O-GlcNAcylation Promotes Sensitization of TRAIL-Tolerant Persisters and TRAIL-Resistant Cancer Cells to Death. Cancer Res. 2019, 79, 2839–2852. [Google Scholar] [CrossRef] [Green Version]
- Holdbrooks, A.T.; Britain, C.M.; Bellis, S.L. ST6Gal-I sialyltransferase promotes tumor necrosis factor (TNF)-mediated cancer cell survival via sialylation of the TNF receptor 1 (TNFR1) death receptor. J. Biol. Chem. 2018, 293, 1610–1622. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Q.; Yang, H.; Cheng, C.; Li, C.; Wu, X.; Huan, W.; Sun, H.; Zhou, Z.; Wang, Y.; Zhao, Y.; et al. β-1,4-Galactosyltransferase I involved in Schwann cells proliferation and apoptosis induced by tumor necrosis factor-alpha via the activation of MAP kinases signal pathways. Mol. Cell Biochem. 2012, 365, 149–158. [Google Scholar] [CrossRef]
- Han, L.; Zhang, D.; Tao, T.; Sun, X.; Liu, X.; Zhu, G.; Xu, Z.; Zhu, L.; Zhang, Y.; Liu, W.; et al. The role of N-glycan modification of TNFR1 in inflammatory microglia activation. Glycoconj. J. 2015, 32, 685–693. [Google Scholar] [CrossRef]
- Yang, H.; Yuan, Q.; Chen, Q.; Li, C.; Wu, X.; Peng, C.; Kang, L.; Lu, X.; Sun, H.; Zhou, Z.; et al. β-1,4-galactosyltransferase I promotes tumor necrosis factor-α autocrine via the activation of MAP kinase signal pathways in Schwann cells. J. Mol. Neurosci. 2011, 45, 269–276. [Google Scholar] [CrossRef]
- Haltiwanger, R.S. Fucose is on the TRAIL of colon cancer. Gastroenterology 2009, 137, 36–39. [Google Scholar] [CrossRef] [Green Version]
- Moriwaki, K.; Shinzaki, S.; Miyoshi, E. GDP-mannose-4,6-dehydratase (GMDS) deficiency renders colon cancer cells resistant to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor- and CD95-mediated apoptosis by inhibiting complex II formation. J. Biol. Chem. 2011, 286, 43123–43133. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; van Roosmalen, I.A.M.; Reis, C.R.; Setroikromo, R.; Quax, W.J. Death receptor 5 is activated by fucosylation in colon cancer cells. FEBS J. 2019, 286, 555–571. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Swindall, A.F.; Kesterson, R.A.; Schoeb, T.R.; Bullard, D.C.; Bellis, S.L. ST6Gal-I regulates macrophage apoptosis via α2-6 sialylation of the TNFR1 death receptor. J. Biol. Chem. 2011, 286, 39654–39662. [Google Scholar] [CrossRef] [Green Version]
- Alexander, K.L.; Serrano, C.A.; Chakraborty, A.; Nearing, M.; Council, L.N.; Riquelme, A.; Garrido, M.; Bellis, S.L.; Smythies, L.E.; Smith, P.D. Modulation of glycosyltransferase ST6Gal-I in gastric cancer-derived organoids disrupts homeostatic epithelial cell turnover. J. Biol. Chem. 2020, 295, 14153–14163. [Google Scholar] [CrossRef]





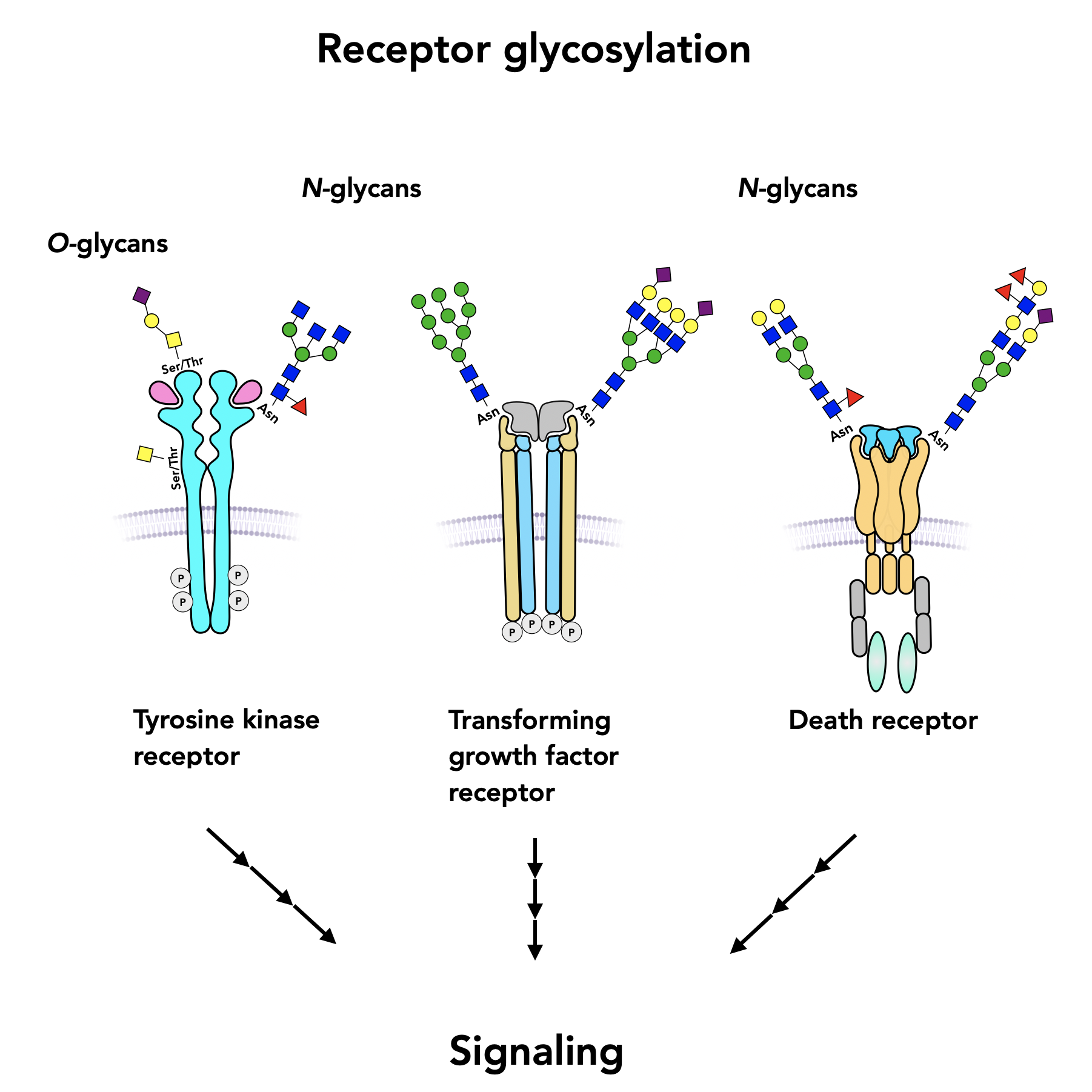{kind=link}
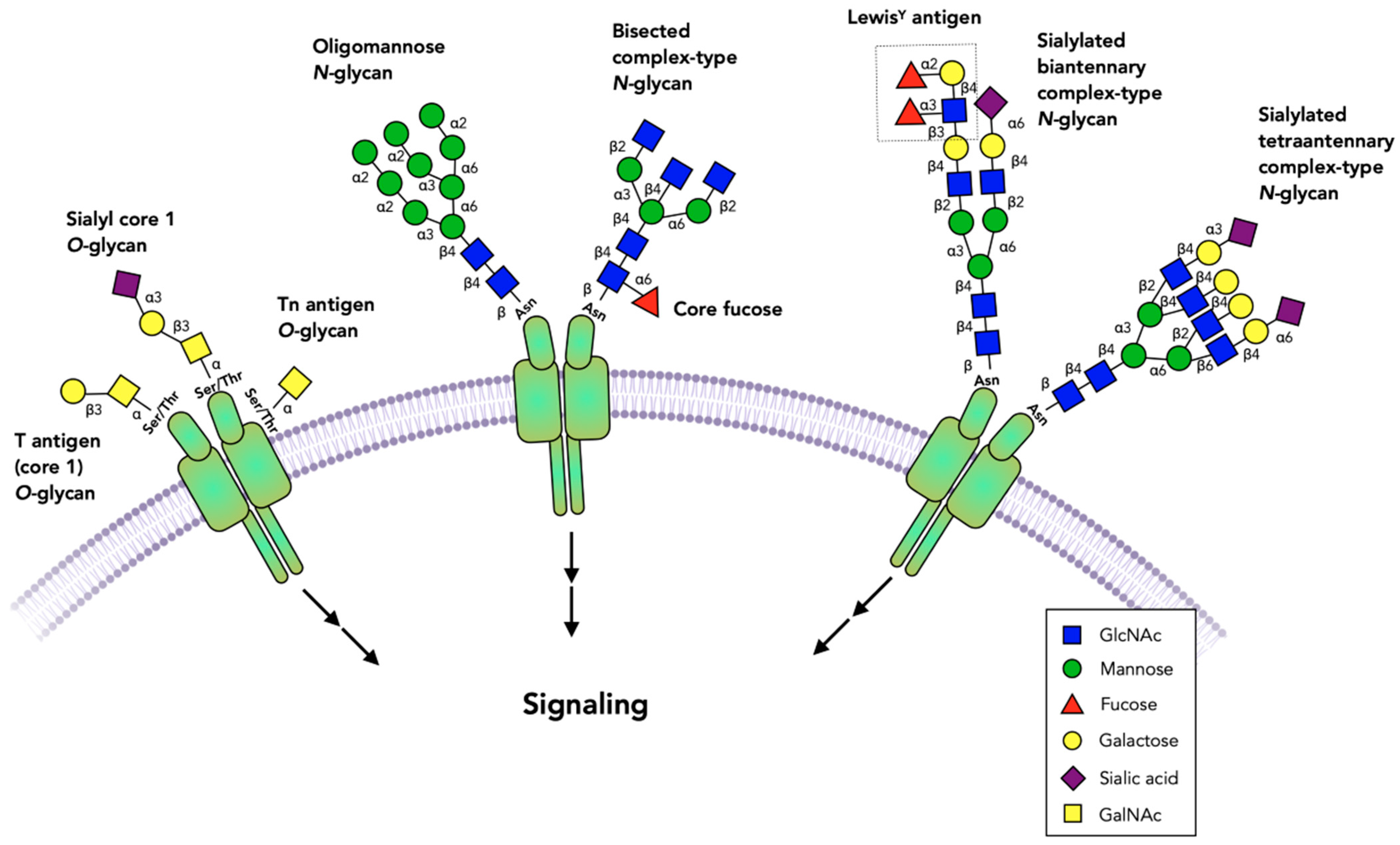{kind=link}
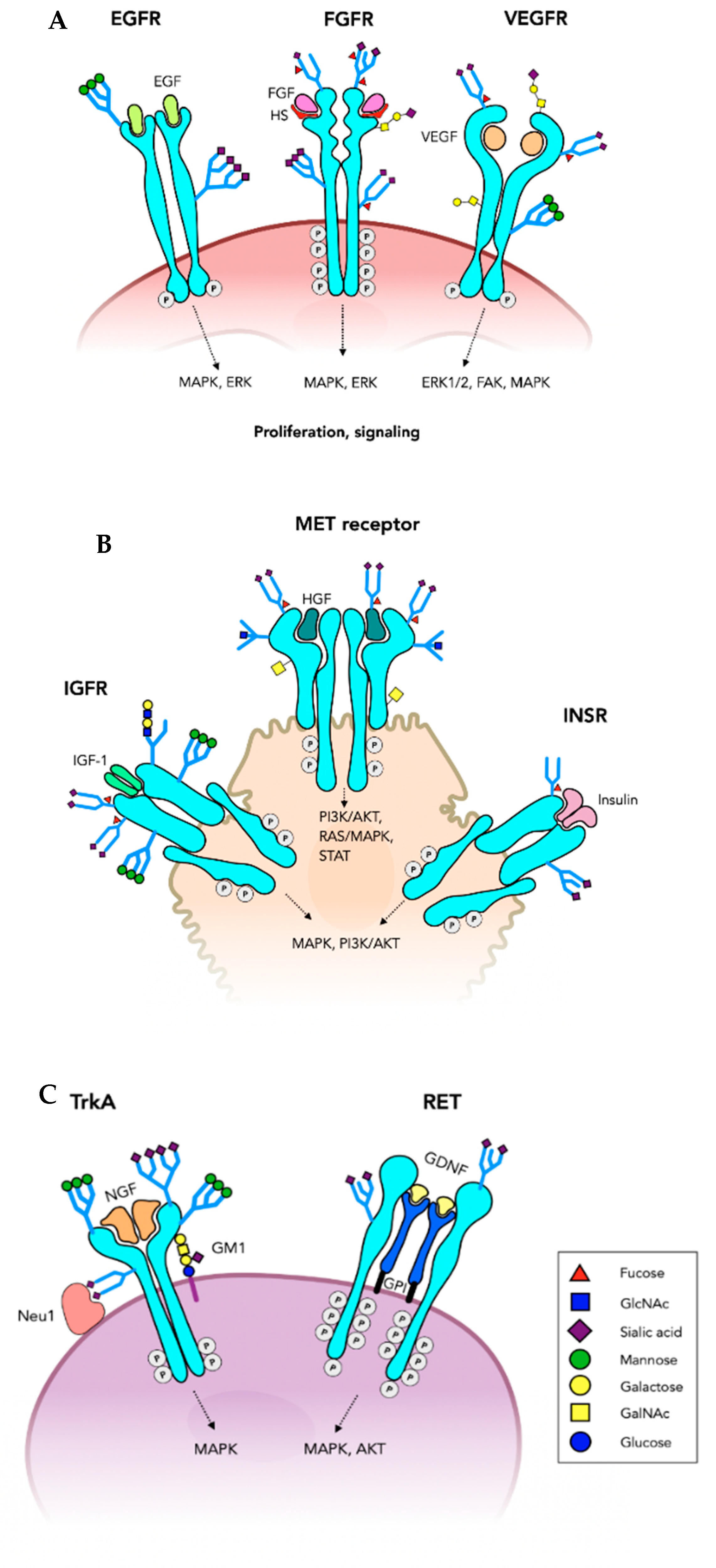{kind=link}
{kind=link}
{kind=link}
| Protein Uniprot No. | Extracellular Domain | N-glycosylation Sites | Cytoplasmic Domain | Function |
|---|---|---|---|---|
| EGFR P00533 | binds EGF, TGFα, MUC1, Neu3, at least 4 signaling cascades | 13 | Tyr-kinase | cell migration, proliferation |
| ErbB2 P04626 | 7 | Tyr-kinase | proliferation | |
| ErbB3 P21860 | 10 | Tyr-kinase | proliferation | |
| Erb4 Q15303 | 11 | Tyr-kinase | proliferation | |
| Hepatocyte GFR,MET P08581 | Siaα6, O-glycans, binds MUC20, HGF | 11 | Tyr-kinase | proliferation |
| FGFR1 P11362 | oligoMan, complex N-glycans, binds FGF | 8 | Tyr-kinase | proliferation |
| FGFR2 P21802 | poly-LacNAc, O-glycans, binds FGF | 8 | Tyr-kinase | proliferation |
| FGFR3 P22607 | binds FGF | 6 | Tyr-kinase | proliferation |
| FGFR4 P22455 | binds FGF | 5 | Tyr-kinase | proliferation |
| VEGFR1 P17948 | binds VEGF | 13 | Tyr-kinase | proliferation, angiogenesis |
| VEGFR2 P35968 | oligoMan, Siaα6, O-glycans, binds VEGF | 18 | Tyr-kinase | proliferation, angiogenesis |
| VEGFR3 P35916 | binds VEGF | 12 | Tyr-kinase | proliferation, angiogenesis |
| IGFR1 P08069 | biantennary, hybrid, oligoMan, N-glycans, core Fuc, binds insulin, IGF1, IGF2 | 16 | Tyr-kinase | cell growth, survival |
| INSR P06213 | binds insulin, IGF1, IGF2 | 18 | Tyr-kinase | cell growth, survival |
| Glycosylation | Cell Type | EGFR Function | Reference |
|---|---|---|---|
| ↓ N-glycans | cancer | ↑ activation | [29] |
| ↓ Asn418 | CHO | ↑ proliferation | [30] |
| Mutation at Asn420 | A431 epidermoid | ↑ EGF-independent proliferation | [32] |
| Mutation at Asn579 | A431 epidermoid | ↑ dimerization | [33] |
| ↑ Gn-T III | HeLa | ↑ phosphorylation | [34] |
| ↑ Gn-T III | glioma | ↓ phosphorylation ↑ proliferation ↓ ligand binding | [35] |
| ↑ Gn-T III | rat pheochromocytoma PC12 | ↓ neurite outgrowth ↓ activation | [36] |
| ↓ Gn-T V | breast cancer | ↓ activation | [37] |
| ↓ FUT8 | mouse embryonic fibroblasts | ↓ activation ↓ phosphorylation | [38] |
| ↑ FUT8 | HEK293 | ↑ signaling | [39] |
| ↓ FUT1 | oral squamous carcinoma | ↑ cell migration | [40,41] |
| ↓ FUT1 | gastric cancer NCI-N87 | ↓ cell migration, ↑ degradation, ↓ expression ↓ phosphorylation | [42] |
| ↑ FUT1 | ovarian cancer RMG-I | ↑ phosphorylation ↑ proliferation | [43] |
| ↓ FUT4 | epidermoid cancer A431 | ↓ phosphorylation ↓ tumor growth | [44] |
| ↓ FUT4 | melanoma | ↓ phosphorylation ↓ proliferation | [45] |
| ↓ FUT4 | bronchial epithelial | ↓ phosphorylation | [46] |
| ↑ FUT4, ↑ FUT6 | A549 lung cancer | ↓ phosphorylation ↓ dimerization | [47] |
| Fucosidase, sialidase | A549 lung cancer | ↑ dimerization ↑ proliferation | [47] |
| ↑ Sialylation | A549 lung cancer | ↓ invasion | [47] |
| ↓ FUT1, ↓ FUT4 | epidermoid cancer A431 | ↓ activation ↓ phosphorylation ↓ tumor growth | [44] |
| ↓ Sialylation | lung cancer | ↑ phosphorylation ↑ TKI sensitivity | [48] |
| Sialidase | A549 lung cancer | ↑ invasion | [47] |
| Sialidase | A549 lung cancer | ↑ activation | [49,50] |
| Sialidase | A549 lung cancer | ↓ activation | [51] |
| ↑ ST6Gal I | ovarian cancer | ↑ activation | [52] |
| ↑ ST6Gal I | pancreatic cancer | ↑ activation ↑ EMT | [53] |
| ↓ ST6Gal I | colon cancer | ↑ activation | [54] |
| ↑ GALNT2 | gastric adenocarcinoma | ↓ activation ↓ tumorigenesis | [55] |
| ↓ GALNT2 | gastric adenocarcinoma | ↑ activation ↑ phosphorylation | [55] |
| GALNT2 | oral squamous cellular carcinoma | migration, invasion | [56] |
| ↓ GALNT2 | hepatocellular carcinoma | ↓ phosphorylation ↓ activation | [56] |
| GALNT2 | glioma | ↑ activation | [57] |
| ↓ GALNT2 | glioma | ↓ tumor growth ↓ phosphorylation | [57] |
| Protein Uniprot No. | Extracellular Domain | N-glycosylation Sites | Cytoplasmic Domain | Function |
|---|---|---|---|---|
| RET P07949 | Complex N-glycans | 12 | Tyr-kinase | survival |
| NTRK1, TrkA P04629 | oligoMan, Sia binds NGF | 13 | Tyr-kinase | proliferation differentiation |
| NTRK2, TrkB Q16620 | binds BDNF, NGF4 | 11 | Tyr-kinase | neuronal development proliferation |
| NTRK3, TrkC Q16288 | binds NGF3 | 13 | Tyr-kinase | survival, differentiation |
| NGFR, p75 P08138 | O-glycans, binds NGF | 1 | protein-protein interaction | circadian rhythm, apoptosis, differentiation, survival |
| GFRα1 P56159 | binds GDNF | 3 | GPI anchor | |
| GFRα2 O00451 | binds NRTN binds RET | 4 | GPI anchor | |
| GFRα3 O60609 | binds RET binds GDNF | 4 | GPI anchor | |
| GFRα4 Q9GZZ7 | binds RET binds GDNF | 1 | GPI anchor |
| Protein Uniprot No. | Extracellular Domain | N-glycosylation Sites | Role | Cytoplasmic Domain |
|---|---|---|---|---|
| TGFR1 P36897 | Tetraantennary N-glycan core Fuc, Fucα1-3 GlcNAcβ1-6 bisected N-glycan binds TGFβ | 1 | promotes signaling promotes signaling promotes signaling promotes EMT inhibits EMT | Ser/Thr kinase, proliferation binds SMAD2 |
| TGFR II, P37173 | oligoMan tetraantennary N-glycans, core Fuc, Fucα1-3, Sia-Lewisx, Sia-Lewisa Ser31-O-glycan Binds TGFβ3 | 3 | promotes signaling blocks signaling | Ser/Thr kinase proliferation binds SMAD4 |
| TGFR III Q03167 | 2 Xyl-Ser GAGs binds TGFβ | 5 | truncated |
| Protein Uniprot No. | Extracellular Domain | N-glycosylation Sites | Cytoplasmic Domain | Function |
|---|---|---|---|---|
| Fas/CD95 P25445 | Siaα6 binds FasL, TNF | 2 | death domain, DISC binds FADD, caspase-8 | apoptosis |
| TNFRSF25 DR3 Q93038 | binds TNF | 2 | death domain | apoptosis |
| TNFRSF10A DR4 O00220 | O-glycans, Fuc binds TNF, TRAIL | 1 | death domain, DISC, binds FADD, caspase-8, O-GlcNAc-phosphate at Ser 424 | apoptosis |
| TNFRSF10B DR5 O14763 | O-glycans,T, ST antigens, binds TRAIL, TNF | - | death domain, DISC, binds FADD, caspase-8 | apoptosis |
| TNFRSF21 DR6 O75509 | O-glycans, binds TRAIL, TNF | 6 | death domain, DISC, binds FADD, caspase-8 | apoptosis |
| TNFRSF1A TNFR1 P19438 | binds TNF | 3 | death domain, DISC, binds TRADD, FADD | apoptosis, survival |
| TNFRSF10D Q9UBN6 | N-glycans at Asn127,182 binds TRAIL | 2 | truncated, secreted | - |
| TNFRSF10C O14798 | binds TRAIL | 3 | truncated, GPI anchored | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Luan, X.; Melamed, J.; Brockhausen, I. Role of Glycans on Key Cell Surface Receptors That Regulate Cell Proliferation and Cell Death. Cells 2021, 10, 1252. https://doi.org/10.3390/cells10051252
Gao Y, Luan X, Melamed J, Brockhausen I. Role of Glycans on Key Cell Surface Receptors That Regulate Cell Proliferation and Cell Death. Cells. 2021; 10(5):1252. https://doi.org/10.3390/cells10051252
Chicago/Turabian StyleGao, Yin, Xue Luan, Jacob Melamed, and Inka Brockhausen. 2021. "Role of Glycans on Key Cell Surface Receptors That Regulate Cell Proliferation and Cell Death" Cells 10, no. 5: 1252. https://doi.org/10.3390/cells10051252
APA StyleGao, Y., Luan, X., Melamed, J., & Brockhausen, I. (2021). Role of Glycans on Key Cell Surface Receptors That Regulate Cell Proliferation and Cell Death. Cells, 10(5), 1252. https://doi.org/10.3390/cells10051252