
The Complexity of FGF23 Effects on Cardiomyocytes in Normal and Uremic Milieu


{kind=link}
{kind=link}
Abstract
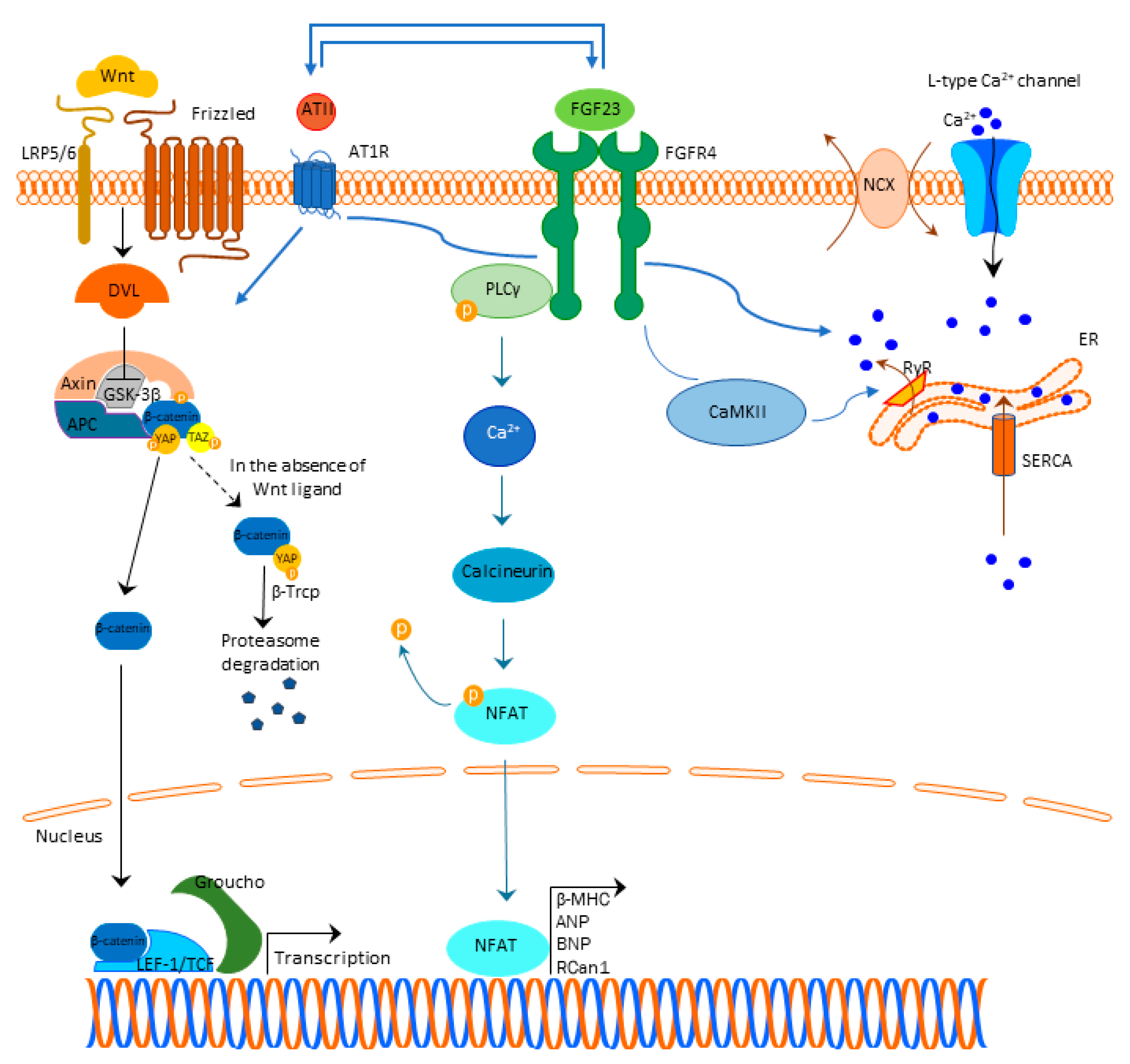
:1. FGF23 Signaling in the Physiological Milieu
2. FGF23 Signaling in the Uremic Milieu
2.1. Cardiac Hypertrophy
2.2. Cardiac Fibrosis
3. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kovesdy, C.P.; Quarles, L.D. FGF23 from Bench to Bedside Perspective. Am. J. Physiol. Ren. Physiol. 2016, 310, 1168–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacchetta, J.; Bardet, C.; Prié, D. Physiology of FGF23 and Overview of Genetic Diseases Associated with Renal Phosphate Wasting. Metab. Clin. Exp. 2020, 103, 153865. [Google Scholar] [CrossRef]
- Beck-Nielsen, S.S.; Mughal, Z.; Haffner, D.; Nilsson, O.; Levtchenko, E.; Ariceta, G.; De Lucas Collantes, C.; Schnabel, D.; Jandhyala, R.; Mäkitie, O. FGF23 and Its Role in X-Linked Hypophosphatemia-Related Morbidity. Orphanet J. Rare Dis. 2019, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Courbebaisse, M.; Lanske, B. Biology of Fibroblast Growth Factor 23: From Physiology to Pathology. Cold Spring Harb. Perspect. Med. 2018, 8, a031260. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shiizaki, K.; Kuro-O, M.; Moe, O.W. Fibroblast Growth Factor 23 and Klotho: Physiology and Pathophysiology of an Endocrine Network of Mineral Metabolism. Annu. Rev. Physiol. 2013, 75, 503–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leifheit-Nestler, M.; Haffner, D. Paracrine Effects of FGF23 on the Heart. Front. Endocrinol. 2018, 9, 278. [Google Scholar] [CrossRef] [Green Version]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.C.; Moe, O.W.; et al. Regulation of Fibroblast Growth Factor-23 Signaling by Klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef] [Green Version]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho Converts Canonical FGF Receptor into a Specific Receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef]
- Czaya, B.; Faul, C. The Role of Fibroblast Growth Factor 23 in Inflammation and Anemia. Int. J. Mol. Sci. 2019, 20, 4195. [Google Scholar] [CrossRef] [Green Version]
- Goetz, R.; Beenken, A.; Ibrahimi, O.A.; Kalinina, J.; Olsen, S.K.; Eliseenkova, A.V.; Xu, C.; Neubert, T.A.; Zhang, F.; Linhardt, R.J.; et al. Molecular Insights into the Klotho-Dependent, Endocrine Mode of Action of Fibroblast Growth Factor 19 Subfamily Members. Mol. Cell. Biol. 2007, 27, 3417–3428. [Google Scholar] [CrossRef] [Green Version]
- Tagliabracci, V.S.; Engel, J.L.; Wiley, S.E.; Xiao, J.; Gonzalez, D.J.; Appaiah, H.N.; Koller, A.; Nizet, V.; White, K.E.; Dixon, J.E. Dynamic Regulation of FGF23 by Fam20C Phosphorylation, GalNAc-T3 Glycosylation, and Furin Proteolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 5520–5525. [Google Scholar] [CrossRef] [Green Version]
- Goetz, R.; Nakada, Y.; Hu, M.C.; Kurosu, H.; Wang, L.; Nakatani, T.; Shi, M.; Eliseenkova, A.V.; Razzaque, M.S.; Moe, O.W.; et al. Isolated C-Terminal Tail of FGF23 Alleviates Hypophosphatemia by Inhibiting FGF23-FGFR-Klotho Complex Formation. Proc. Natl. Acad. Sci. USA 2010, 107, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Kuzina, E.; An, S.J.; Tome, F.; Mohanty, J.; Li, W.; Lee, S.; Liu, Y.; Lax, I.; Schlessinger, J. FGF23 Contains Two Distinct High-Affinity Binding Sites Enabling Bivalent Interactions with α-Klotho. Proc. Natl. Acad. Sci. USA 2020, 117, 31800–31807. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. α-Klotho Is a Non-Enzymatic Molecular Scaffold for FGF23 Hormone Signalling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shi, M.; Cho, H.J.; Adams-Huet, B.; Paek, J.; Hill, K.; Shelton, J.; Amaral, A.P.; Faul, C.; Taniguchi, M.; et al. Klotho and Phosphate Are Modulators of Pathologic Uremic Cardiac Remodeling. J. Am. Soc. Nephrol. 2015, 26, 1290–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsetti, G.; Pasini, E.; Scarabelli, T.M.; Romano, C.; Agrawal, P.R.; Chen-Scarabelli, C.; Knight, R.; Saravolatz, L.; Narula, J.; Ferrari-Vivaldi, M.; et al. Decreased Expression of Klotho in Cardiac Atria Biopsy Samples from Patients at Higher Risk of Atherosclerotic Cardiovascular Disease. J. Geriatr. Cardiol. 2016, 13, 701–711. [Google Scholar] [CrossRef]
- Richter, B.; Faul, C. FGF23 Actions on Target Tissues-with and without Klotho. Front. Endocrinol. 2018, 9, 189. [Google Scholar] [CrossRef]
- Wang, Q.; Su, W.; Shen, Z.; Wang, R. Correlation between Soluble α-Klotho and Renal Function in Patients with Chronic Kidney Disease: A Review and Meta-Analysis. BioMed Res. Int. 2018, 2018, 9481745. [Google Scholar] [CrossRef] [Green Version]
- Marthi, A.; Donovan, K.; Haynes, R.; Wheeler, D.C.; Baigent, C.; Rooney, C.M.; Landray, M.J.; Moe, S.M.; Yang, J.; Holland, L.; et al. Fibroblast Growth Factor-23 and Risks of Cardiovascular and Noncardiovascular Diseases: A Meta-Analysis. J. Am. Soc. Nephrol. 2018, 29, 2000–2013. [Google Scholar] [CrossRef] [Green Version]
- Six, I.; Okazaki, H.; Gross, P.; Cagnard, J.; Boudot, C.; Maizel, J.; Drueke, T.B.; Massy, Z.A. Direct, Acute Effects of Klotho and FGF23 on Vascular Smooth Muscle and Endothelium. PLoS ONE 2014, 9, e93423. [Google Scholar] [CrossRef]
- Semba, R.D.; Cappola, A.R.; Sun, K.; Bandinelli, S.; Dalal, M.; Crasto, C.; Guralnik, J.M.; Ferrucci, L. Plasma Klotho and Cardiovascular Disease in Adults. J. Am. Geriatr. Soc. 2011, 59, 1596–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalton, G.; An, S.W.; Al-Juboori, S.I.; Nischan, N.; Yoon, J.; Dobrinskikh, E.; Hilgemann, D.W.; Xie, J.; Luby-Phelps, K.; Kohler, J.J.; et al. Soluble Klotho Binds Monosialoganglioside to Regulate Membrane Microdomains and Growth Factor Signaling. Proc. Natl. Acad. Sci. USA 2017, 114, 752–757. [Google Scholar] [CrossRef] [Green Version]
- Navarro-García, J.A.; Rueda, A.; Romero-García, T.; Aceves-Ripoll, J.; Rodríguez-Sánchez, E.; González-Lafuente, L.; Zaragoza, C.; Fernández-Velasco, M.; Kuro-o, M.; Ruilope, L.M.; et al. Enhanced Klotho Availability Protects against Cardiac Dysfunction Induced by Uraemic Cardiomyopathy by Regulating Ca2+ Handling. Br. J. Pharmacol. 2020, 177, 4701–4719. [Google Scholar] [CrossRef] [PubMed]
- Navarro-García, J.A.; Fernández-Velasco, M.; Delgado, C.; Delgado, J.F.; Kuro-o, M.; Ruilope, L.M.; Ruiz-Hurtado, G. PTH, Vitamin D, and the FGF-23–Klotho Axis and Heart: Going beyond the Confines of Nephrology. Eur. J. Clin. Investig. 2018, 48, e12902. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Cai, C.; Xiao, Z.; Quarles, L.D. FGF23 Induced Left Ventricular Hypertrophy Mediated by FGFR4 Signaling in the Myocardium Is Attenuated by Soluble Klotho in Mice. J. Mol. Cell. Cardiol. 2020, 138, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, R.; Suhail, F.; Lerma, E.V. Cardiovascular Disease and Chronic Kidney Disease. Dis. Mon. 2015, 61, 403–413. [Google Scholar] [CrossRef]
- Gutiérrez, O.M.; Mannstadt, M.; Isakova, T.; Rauh-Hain, J.A.; Tamez, H.; Shah, A.; Smith, K.; Lee, H.; Thadhani, R.; Jüppner, H.; et al. Fibroblast Growth Factor 23 and Mortality among Patients Undergoing Hemodialysis. N. Engl. J. Med. 2008, 359, 584–592. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, O.M.; Januzzi, J.L.; Isakova, T.; Laliberte, K.; Smith, K.; Collerone, G.; Sarwar, A.; Hoffmann, U.; Coglianese, E.; Christenson, R.; et al. Fibroblast Growth Factor 23 and Left Ventricular Hypertrophy in Chronic Kidney Disease. Circulation 2009, 119, 2545–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutiérrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 Induces Left Ventricular Hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Tang, W.; Fang, J.; Ren, J.; Li, H.; Xiao, Z.; Quarles, L.D. Novel Regulators of Fgf23 Expression and Mineralization in Hyp Bone. Mol. Endocrinol. 2009, 23, 1505–1518. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, J.; Tang, W.; Jiang, X.; Rowe, D.W.; Quarles, L.D. Pathogenic Role of Fgf23 in Hyp Mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Pi, M.; Ye, R.; Han, X.; Armstrong, B.; Liu, X.; Chen, Y.; Sun, Y.; Quarles, L.D. Cardiovascular Interactions between Fibroblast Growth Factor-23 and Angiotensin II. Sci. Rep. 2018, 8, 12398. [Google Scholar] [CrossRef]
- Wohlfahrt, P.; Melenovsky, V.; Kotrc, M.; Benes, J.; Jabor, A.; Franekova, J.; Lemaire, S.; Kautzner, J.; Jarolim, P. Association of Fibroblast Growth Factor-23 Levels and Angiotensin-Converting Enzyme Inhibition in Chronic Systolic Heart Failure. JACC Heart Fail. 2015, 3, 829–839. [Google Scholar] [CrossRef]
- Kuga, K.; Kusakari, Y.; Uesugi, K.; Semba, K.; Urashima, T.; Akaike, T.; Minamisawa, S. Fibrosis Growth Factor 23 Is a Promoting Factor for Cardiac Fibrosis in the Presence of Transforming Growth Factor-Β1. PLoS ONE 2020, 15, e0231905. [Google Scholar] [CrossRef] [Green Version]
- Leifheit-Nestler, M.; Kirchhoff, F.; Nespor, J.; Richter, B.; Soetje, B.; Klintschar, M.; Heineke, J.; Haffner, D. Fibroblast Growth Factor 23 Is Induced by an Activated Renin-Angiotensin-Aldosterone System in Cardiac Myocytes and Promotes the pro-Fibrotic Crosstalk between Cardiac Myocytes and Fibroblasts. Nephrol. Dial. Transplant. 2018, 33, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Kinoshita, S.; Shimba, S.; Ozono, K.; Michigami, T. Sympathetic Activation Induces Skeletal Fgf23 Expression in a Circadian Rhythm-Dependent Manner. J. Biol. Chem. 2014, 289, 1457–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fajol, A.; Chen, H.; Umbach, A.T.; Quarles, L.D.; Lang, H.; Föller, M. Enhanced FGF23 Production in Mice Expressing PI3K-insensitive GSK3 Is Normalized by Β-blocker Treatment. FASEB J. 2016, 30, 994–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrukhova, O.; Slavic, S.; Smorodchenko, A.; Zeitz, U.; Shalhoub, V.; Lanske, B.; Pohl, E.E.; Erben, R.G. FGF23 Regulates Renal Sodium Handling and Blood Pressure. EMBO Mol. Med. 2014, 6, 744–759. [Google Scholar] [CrossRef]
- Vaidya, A.; Williams, J.S. The Relationship between Vitamin D and the Renin-Angiotensin System in the Pathophysiology of Hypertension, Kidney Disease, and Diabetes. Metab. Clin. Exp. 2012, 61, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Lautze, H.-J.; Walther, T.; Braun, T.; Kostin, S.; Kubin, T. The Failing Heart Is a Major Source of Circulating FGF23 via Oncostatin M Receptor Activation. J. Heart Lung Transplant. 2015, 34, 1211–1214. [Google Scholar] [CrossRef]
- Dai, B.; David, V.; Martin, A.; Huang, J.; Li, H.; Jiao, Y.; Gu, W.; Quarles, L.D. A Comparative Transcriptome Analysis Identifying FGF23 Regulated Genes in the Kidney of a Mouse CKD Model. PLoS ONE 2012, 7, e44161. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Yoon, J.; An, S.W.; Kuro-o, M.; Huang, C.L. Soluble Klotho Protects against Uremic Cardiomyopathy Independently of Fibroblast Growth Factor 23 and Phosphate. J. Am. Soc. Nephrol. 2015, 26, 1150–1160. [Google Scholar] [CrossRef]
- Hernández-Frías, O.; Gil-Peña, H.; Pérez-Roldán, J.M.; González-Sanchez, S.; Ariceta, G.; Chocrón, S.; Loza, R.; de la Cerda Ojeda, F.; Madariaga, L.; Vergara, I.; et al. Risk of Cardiovascular Involvement in Pediatric Patients with X-Linked Hypophosphatemia. Pediatr. Nephrol. 2019, 34, 1077–1086. [Google Scholar] [CrossRef]
- Pastor-Arroyo, E.M.; Gehring, N.; Krudewig, C.; Costantino, S.; Bettoni, C.; Knöpfel, T.; Sabrautzki, S.; Lorenz-Depiereux, B.; Pastor, J.; Strom, T.M.; et al. The Elevation of Circulating Fibroblast Growth Factor 23 without Kidney Disease Does Not Increase Cardiovascular Disease Risk. Kidney Int. 2018, 94, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Mace, M.L.; Olgaard, K.; Lewin, E. New Aspects of the Kidney in the Regulation of Fibroblast Growth Factor 23 (Fgf23) and Mineral Homeostasis. Int. J. Mol. Sci. 2020, 21, 8810. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Grabner, A.; Yanucil, C.; Schramm, K.; Czaya, B.; Krick, S.; Czaja, M.J.; Bartz, R.; Abraham, R.; Di Marco, G.S.; et al. Fibroblast Growth Factor 23 Directly Targets Hepatocytes to Promote Inflammation in Chronic Kidney Disease. Kidney Int. 2016, 90, 985–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, E.A.; Han, X.; Xiao, Z.; Quarles, L.D. Role of Fibroblast Growth Factor-23 in Innate Immune Responses. Front. Endocrinol. 2018, 9, 320. [Google Scholar] [CrossRef]
- Bär, L.; Stournaras, C.; Lang, F.; Föller, M. Regulation of Fibroblast Growth Factor 23 (FGF23) in Health and Disease. FEBS Lett. 2019, 593, 1879–1900. [Google Scholar] [CrossRef] [Green Version]
- Stöhr, R.; Schuh, A.; Heine, G.H.; Brandenburg, V. FGF23 in Cardiovascular Disease: Innocent Bystander or Active Mediator? Front. Endocrinol. 2018, 9, 351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabner, A.; Schramm, K.; Silswal, N.; Hendrix, M.; Yanucil, C.; Czaya, B.; Singh, S.; Wolf, M.; Hermann, S.; Stypmann, J.; et al. FGF23/FGFR4-Mediated Left Ventricular Hypertrophy Is Reversible. Sci. Rep. 2017, 7, 1993. [Google Scholar] [CrossRef] [PubMed]
- Figurek, A.; Spasovski, G.; Popovic-Pejicic, S. FGF23 Level and Intima-Media Thickness Are Elevated From Early Stages of Chronic Kidney Disease. Ther. Apher. Dial. 2018, 22, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Larsson, T.; Nisbeth, U.; Ljunggren, Ö.; Jüppner, H.; Jonsson, K.B. Circulating Concentration of FGF-23 Increases as Renal Function Declines in Patients with Chronic Kidney Disease, but Does Not Change in Response to Variation in Phosphate Intake in Healthy Volunteers. Kidney Int. 2003, 64, 2272–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, O.; Isakova, T.; Rhee, E.; Shah, A.; Holmes, J.; Collerone, G.; Jüppner, H.; Wolf, M. Fibroblast Growth Factor-23 Mitigates Hyperphosphatemia but Accentuates Calcitriol Deficiency in Chronic Kidney Disease. J. Am. Soc. Nephrol. 2005, 16, 2205–2215. [Google Scholar] [CrossRef]
- Weber, J.R. Left Ventricular Hypertrophy: Its Prevalence, Etiology, and Significance. Clin. Cardiol. 1991, 14, 13–17. [Google Scholar] [CrossRef]
- Leifheit-Nestler, M.; Siemer, R.G.; Flasbart, K.; Richter, B.; Kirchhoff, F.; Ziegler, W.H.; Klintschar, M.; Becker, J.U.; Erbersdobler, A.; Aufricht, C.; et al. Induction of Cardiac FGF23/FGFR4 Expression Is Associated with Left Ventricular Hypertrophy in Patients with Chronic Kidney Disease. Nephrol. Dial. Transplant. 2016, 31, 1088–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touchberry, C.D.; Green, T.M.; Tchikrizov, V.; Mannix, J.E.; Mao, T.F.; Carney, B.W.; Girgis, M.; Vincent, R.J.; Wetmore, L.A.; Dawn, B.; et al. FGF23 Is a Novel Regulator of Intracellular Calcium and Cardiac Contractility in Addition to Cardiac Hypertrophy. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E863–E873. [Google Scholar] [CrossRef]
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell Metab. 2015, 22, 1020–1032. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Fujita, S.I.; Kizawa, S.; Morita, H.; Ishizaka, N. Association between FGF23, α-Klotho, and Cardiac Abnormalities among Patients with Various Chronic Kidney Disease Stages. PLoS ONE 2016, 11, e0156860. [Google Scholar] [CrossRef]
- Vainikka, S.; Joukov, V.; Wennström, S.; Bergman, M.; Peliccill, P.G.; Alitalot, K. Signal Transduction by Fibroblast Growth Factor Receptor-4 (FGFR-4): Comparison with FGFR-1. J. Biol. Chem. 1994, 269, 18320–18326. [Google Scholar] [CrossRef]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular Signaling by Fibroblast Growth Factor Receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef]
- Molkentin, J.D. Calcineurin-NFAT Signaling Regulates the Cardiac Hypertrophic Response in Coordination with the MAPKs. Cardiovasc. Res. 2004, 63, 467–475. [Google Scholar] [CrossRef]
- Zhuo, J.L.; Li, X.C. Proximal Nephron. Compr. Physiol. 2013, 3, 1079–1123. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Sánchez, S.; Poveda, J.; Navarro-García, J.A.; González-Lafuente, L.; Rodríguez-Sánchez, E.; Ruilope, L.M.; Ruiz-Hurtado, G. An Overview of FGF-23 as a Novel Candidate Biomarker of Cardiovascular Risk. Front. Physiol. 2021, 12, 268. [Google Scholar] [CrossRef]
- Gardner, D.G. Natriuretic Peptides: Markers or Modulators of Cardiac Hypertrophy? Trends Endocrinol. Metab. 2003, 14, 411–416. [Google Scholar] [CrossRef]
- Leifheit-Nestler, M.; Grabner, A.; Hermann, L.; Richter, B.; Schmitz, K.; Fischer, D.C.; Yanucil, C.; Faul, C.; Haffner, D. Vitamin D Treatment Attenuates Cardiac FGF23/FGFR4 Signaling and Hypertrophy in Uremic Rats. Nephrol. Dial. Transplant. 2017, 32, 1493–1503. [Google Scholar] [CrossRef]
- Fabiato, A.; Fabiato, F. Contractions Induced by a Calcium-Triggered Release of Calcium from the Sarcoplasmic Reticulum of Single Skinned Cardiac Cells. J. Physiol. 1975, 249, 469–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bers, D.M. Altered Cardiac Myocyte Ca Regulation in Heart Failure. Physiology 2006, 21, 380–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bovo, E.; Huke, S.; Blatter, L.A.; Zima, A.V. The Effect of PKA-Mediated Phosphorylation of Ryanodine Receptor on SR Ca2+ Leak in Ventricular Myocytes. J. Mol. Cell. Cardiol. 2017, 104, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Kao, Y.H.; Chen, Y.C.; Lin, Y.K.; Shiu, R.J.; Chao, T.F.; Chen, S.A.; Chen, Y.J. FGF-23 Dysregulates Calcium Homeostasis and Electrophysiological Properties in HL-1 Atrial Cells. Eur. J. Clin. Investig. 2014, 44, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Navarro-García, J.A.; Delgado, C.; Fernández-Velasco, M.; Val-Blasco, A.; Rodríguez-Sánchez, E.; Aceves-Ripoll, J.; Gómez-Hurtado, N.; Bada-Bosch, T.; Mérida-Herrero, E.; Hernández, E.; et al. Fibroblast Growth Factor-23 Promotes Rhythm Alterations and Contractile Dysfunction in Adult Ventricular Cardiomyocytes. Nephrol. Dial. Transplant. 2019, 34, 1864–1875. [Google Scholar] [CrossRef] [PubMed]
- Curran, J.; Brown, K.H.; Santiago, D.J.; Pogwizd, S.; Bers, D.M.; Shannon, T.R. Spontaneous Ca Waves in Ventricular Myocytes from Failing Hearts Depend on Ca2+-Calmodulin-Dependent Protein Kinase II. J. Mol. Cell. Cardiol. 2010, 49, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Mhatre, K.N.; Wakula, P.; Klein, O.; Bisping, E.; Völkl, J.; Pieske, B.; Heinzel, F.R. Crosstalk between FGF23- and Angiotensin II-Mediated Ca2+ Signaling in Pathological Cardiac Hypertrophy. Cell. Mol. Life Sci. 2018, 75, 4403–4416. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, C.; Hong, X.; Miao, J.; Liao, Y.; Hou, F.F.; Zhou, L.; Liu, Y. Wnt/β-Catenin Signaling Mediates Both Heart and Kidney Injury in Type 2 Cardiorenal Syndrome. Kidney Int. 2019, 95, 815–829. [Google Scholar] [CrossRef]
- Bergmann, M.W. WNT Signaling in Adult Cardiac Hypertrophy and Remodeling: Lessons Learned from Cardiac Development. Circ. Res. 2010, 107, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niehrs, C. The Complex World of WNT Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wang, X.; Zhang, H.; Wang, Z.; Nan, G.; Li, Y.; Zhang, F.; Mohammed, M.K.; Haydon, R.C.; Luu, H.H.; et al. The Evolving Roles of Canonical WNT Signaling in Stem Cells and Tumorigenesis: Implications in Targeted Cancer Therapies. Lab. Investig. 2016, 96, 116–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ Incorporation in the β-Catenin Destruction Complex Orchestrates the Wnt Response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Castañeda, J.R.; Rodelo-Haad, C.; Pendon-Ruiz de Mier, M.V.; Martin-Malo, A.; Santamaria, R.; Rodriguez, M. Klotho/FGF23 and Wnt Signaling as Important Players in the Comorbidities Associated with Chronic Kidney Disease. Toxins 2020, 12, 185. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, V.; Bryant, H.U.; MacDougald, O.A. Regulation of Bone Mass by Wnt Signaling. J. Clin. Investig. 2006, 116, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.L.; Weis, W.I. β-Catenin Directly Displaces Groucho/TLE Repressors from Tcf/Lef in Wnt-Mediated Transcription Activation. Nat. Struct. Mol. Biol. 2005, 12, 364–371. [Google Scholar] [CrossRef]
- Evenepoel, P.; D’Haese, P.; Brandenburg, V. Sclerostin and DKK1: New Players in Renal Bone and Vascular Disease. Kidney Int. 2015, 88, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Liu, T.; Yu, C.; Yang, X.; Shao, Y.; Shi, J.; Ye, X.; Zheng, X.; Yan, J.; Xu, D.; et al. LncRNA TUG1 Alleviates Cardiac Hypertrophy by Targeting MiR-34a/DKK1/Wnt-β-catenin Signalling. J. Cell. Mol. Med. 2020, 24, 3678–3691. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Meng, W.; Ding, J.; Cheng, M. Klotho Inhibits Angiotensin II-Induced Cardiomyocyte Hypertrophy through Suppression of the AT1R/Beta Catenin Pathway. Biochem. Biophys. Res. Commun. 2016, 473, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Milovanova, L.Y.; Dobrosmyslov, I.A.; Milovanov, Y.S.; Taranova, M.V.; Kozlov, V.V.; Milovanova, S.Y.; Kozevnikova, E.I. Fibroblast Growth Factor-23 (FGF-23)/Soluble Klotho Protein (SKlotho)/Sclerostin Glycoprotein Ratio Disturbance Is a Novel Risk Factor for Cardiovascular Complications in ESRD Patients Receiving Treatment with Regular Hemodialysis or Hemodiafiltration. Ter. Arkh. 2018, 90, 48–54. [Google Scholar] [CrossRef]
- Böckmann, I.; Lischka, J.; Richter, B.; Deppe, J.; Rahn, A.; Fischer, D.C.; Heineke, J.; Haffner, D.; Leifheit-Nestler, M. FGF23-Mediated Activation of Local RAAS Promotes Cardiac Hypertrophy and Fibrosis. Int. J. Mol. Sci. 2019, 20, 4634. [Google Scholar] [CrossRef] [Green Version]
- Ames, M.K.; Atkins, C.E.; Pitt, B. The Renin-Angiotensin-Aldosterone System and Its Suppression. J. Vet. Intern. Med. 2019, 33, 363–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, H.; Li, X.; Li, Q.; Lin, H.; Chen, Z.; Xie, J.; Xuan, W.; Liao, W.; Bin, J.; Huang, X.; et al. FGF23 Promotes Myocardial Fibrosis in Mice through Activation of β-Catenin. Oncotarget 2016, 7, 64649–64664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, D.; Alampour-Rajabi, S.; Ponomariov, V.; Curaj, A.; Wu, Z.; Staudt, M.; Rusu, M.; Jankowski, V.; Marx, N.; Jankowski, J.; et al. Cardiac FGF23: New Insights into the Role and Function of FGF23 after Acute Myocardial Infarction. Cardiovasc. Pathol. 2019, 40, 47–54. [Google Scholar] [CrossRef]
- Guo, Y.; Xiao, L.; Sun, L.; Liu, F. Wnt/β-Catenin Signaling: A Promising New Target for Fibrosis Diseases. Physiol. Res. 2012, 61, 337–346. [Google Scholar] [CrossRef]
- Działo, E.; Tkacz, K.; Błyszczuk, P. Crosstalk between the TGF-β and WNT Signalling Pathways during Cardiac Fibrogenesis. Acta Biochim. Pol. 2018, 65, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-β, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhu, L.J.; Waaga-Gasser, A.M.; Ding, Y.; Cao, M.; Jadhav, S.J.; Kirollos, S.; Shekar, P.S.; Padera, R.F.; Chang, Y.C.; et al. The Axis of Local Cardiac Endogenous Klotho-TGF-Β1-Wnt Signaling Mediates Cardiac Fibrosis in Human. J. Mol. Cell. Cardiol. 2019, 136, 113–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, S.; Zou, Y.; Togao, O.; Pastor, J.V.; John, G.B.; Wang, L.; Shiizaki, K.; Gotschall, R.; Schiavi, S.; Yorioka, N.; et al. Klotho Inhibits Transforming Growth Factor-Β1 (TGF-Β1) Signaling and Suppresses Renal Fibrosis and Cancer Metastasis in Mice. J. Biol. Chem. 2011, 286, 8655–8665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalhoub, V.; Shatzen, E.M.; Ward, S.C.; Davis, J.; Stevens, J.; Bi, V.; Renshaw, L.; Hawkins, N.; Wang, W.; Chen, C.; et al. FGF23 Neutralization Improves Chronic Kidney Disease-Associated Hyperparathyroidism yet Increases Mortality. J. Clin. Investig. 2012, 122, 2543–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanochko, G.M.; Vitsky, A.; Heyen, J.R.; Hirakawa, B.; Lam, J.L.; May, J.; Nichols, T.; Sace, F.; Trajkovic, D.; Blasi, E. Pan-FGFR Inhibition Leads to Blockade of FGF23 Signaling, Soft Tissue Mineralization, and Cardiovascular Dysfunction. Toxicol. Sci. 2013, 135, 451–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leask, A. TGFβ, Cardiac Fibroblasts, and the Fibrotic Response. Cardiovasc. Res. 2007, 74, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Eyries, M.; Agrapart, M.; Alonso, A.; Soubrier, F. Phorbol Ester Induction of Angiotensin-Converting Enzyme Transcription Is Mediated by Egr-1 and AP-1 in Human Endothelial Cells via ERK1/2 Pathway. Circ. Res. 2002, 91, 899–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.; Li, A.-Y.; Meng, F.; Dong, L.-H.; Zheng, B.; Hu, H.-J.; Nie, L.; Wen, J.-K. Synergistic Co-Operation of Signal Transducer and Activator of Transcription 5B with Activator Protein 1 in Angiotensin II-Induced Angiotensinogen Gene Activation in Vascular Smooth Muscle Cells. FEBS J. 2009, 276, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Siragy, H.M. Regulation of (pro)Renin Receptor Expression by Glucose-Induced Mitogen-Activated Protein Kinase, Nuclear Factor-ΚB, and Activator Protein-1 Signaling Pathways. Endocrinology 2010, 151, 3317–3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Gao, L.; Roy, S.K.; Cornish, K.G.; Zucker, I.H. Neuronal Angiotensin II Type 1 Receptor Upregulation in Heart Failure: Activation of Activator Protein 1 and Jun N-Terminal Kinase. Circ. Res. 2006, 99, 1004–1011. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Jain, S.; Puri, N.; Kaw, M.; Sirianni, N.; Eren, D.; Mopidevi, B.R.; Kumar, A. The Transcriptional Regulation of the Human Angiotensinogen Gene after High-Fat Diet Is Haplotype-Dependent: Novel Insights into the Gene-Regulatory Networks and Implications for Human Hypertension. PLoS ONE 2017, 12, e0176373. [Google Scholar] [CrossRef]
- Figurek, A.; Spasovski, G. Is Serum Sclerostin a Marker of Atherosclerosis in Patients with Chronic Kidney Disease–Mineral and Bone Disorder? Int. Urol. Nephrol. 2018, 50, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Figurek, A.; Rroji, M.; Spasovski, G. Sclerostin: A New Biomarker of CKD–MBD. Int. Urol. Nephrol. 2020, 52, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Udell, J.A.; Morrow, D.A.; Jarolim, P.; Sloan, S.; Hoffman, E.B.; O’Donnell, T.F.; Vora, A.N.; Omland, T.; Solomon, S.D.; Pfeffer, M.A.; et al. Fibroblast Growth Factor-23, Cardiovascular Prognosis, and Benefit of Angiotensin-Converting Enzyme Inhibition in Stable Ischemic Heart Disease. J. Am. Coll. Cardiol. 2014, 63, 2421–2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moe, S.M.; Chertow, G.M.; Parfrey, P.S.; Kubo, Y.; Block, G.A.; Correa-Rotter, R.; Drüeke, T.B.; Herzog, C.A.; London, G.M.; Mahaffey, K.W.; et al. Cinacalcet, Fibroblast Growth Factor-23, and Cardiovascular Disease in Hemodialysis: The Evaluation of Cinacalcet HCl Therapy to Lower Cardiovascular Events (EVOLVE) Trial. Circulation 2015, 132, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Rodelo-Haad, C.; Rodríguez-Ortiz, M.E.; Martin-Malo, A.; Pendon-Ruiz de Mier, M.V.; Agüera, M.L.; Muñoz-Castañeda, J.R.; Soriano, S.; Caravaca, F.; Alvarez-Lara, M.A.; Felsenfeld, A.; et al. Phosphate Control in Reducing FGF23 Levels in Hemodialysis Patients. PLoS ONE 2018, 13, e0201537. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, J.B.; Liu, S.; Krebill, R.; Menard, R.; Quarles, L.D. Effects of Cinacalcet and Concurrent Low-Dose Vitamin D on FGF23 Levels in ESRD. Clin. J. Am. Soc. Nephrol. 2010, 5, 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodyak, N.; Ayus, J.C.; Achinger, S.; Shivalingappa, V.; Ke, Q.; Chen, Y.S.; Rigor, D.L.; Stillman, I.; Tamez, H.; Kroeger, P.E.; et al. Activated Vitamin D Attenuates Left Ventricular Abnormalities Induced by Dietary Sodium in Dahl Salt-Sensitive Animals. Proc. Natl. Acad. Sci. USA 2007, 104, 16810–16815. [Google Scholar] [CrossRef] [Green Version]
- Hyung, W.K.; Cheol, W.P.; Young, S.S.; Young, S.K.; Seok, J.S.; Kim, Y.S.; Euy, J.C.; Yoon, S.C.; Byung, K.B. Calcitriol Regresses Cardiac Hypertrophy and QT Dispersion in Secondary Hyperparathyroidism on Hemodialysis. Nephron Clin. Pract. 2006, 102, c21–c29. [Google Scholar] [CrossRef]
- D’Arrigo, G.; Pizzini, P.; Cutrupi, S.; Tripepi, R.; Tripepi, G.; Mallamaci, F.; Zoccali, C. FGF23 and the PTH Response to Paricalcitol in Chronic Kidney Disease. Eur. J. Clin. Investig. 2020, 50, e13196. [Google Scholar] [CrossRef] [PubMed]
- Lekawanvijit, S. Cardiotoxicity of Uremic Toxins: A Driver of Cardiorenal Syndrome. Toxins 2018, 10, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figurek, A.; Rroji, M.; Spasovski, G. The Complexity of FGF23 Effects on Cardiomyocytes in Normal and Uremic Milieu. Cells 2021, 10, 1266. https://doi.org/10.3390/cells10051266
Figurek A, Rroji M, Spasovski G. The Complexity of FGF23 Effects on Cardiomyocytes in Normal and Uremic Milieu. Cells. 2021; 10(5):1266. https://doi.org/10.3390/cells10051266
Chicago/Turabian StyleFigurek, Andreja, Merita Rroji, and Goce Spasovski. 2021. "The Complexity of FGF23 Effects on Cardiomyocytes in Normal and Uremic Milieu" Cells 10, no. 5: 1266. https://doi.org/10.3390/cells10051266
APA StyleFigurek, A., Rroji, M., & Spasovski, G. (2021). The Complexity of FGF23 Effects on Cardiomyocytes in Normal and Uremic Milieu. Cells, 10(5), 1266. https://doi.org/10.3390/cells10051266