
Dopamine Neuron Diversity: Recent Advances and Current Challenges in Human Stem Cell Models and Single Cell Sequencing


Abstract
:1. Introduction
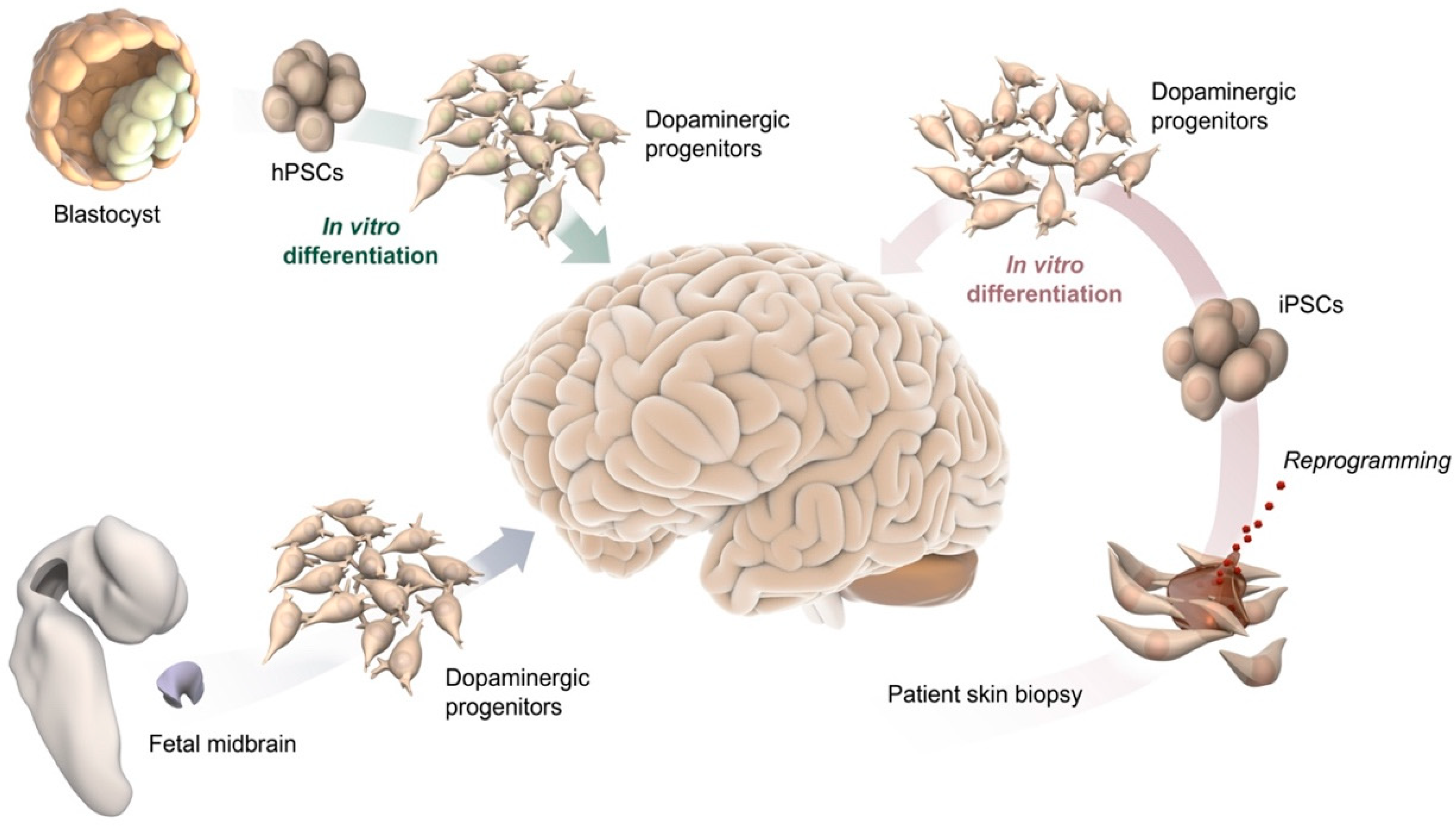
2. Cell-Based Human Models In Vitro and after Transplantation In Vivo
2.1. 2D Culture
2.2. Cell Transplantation
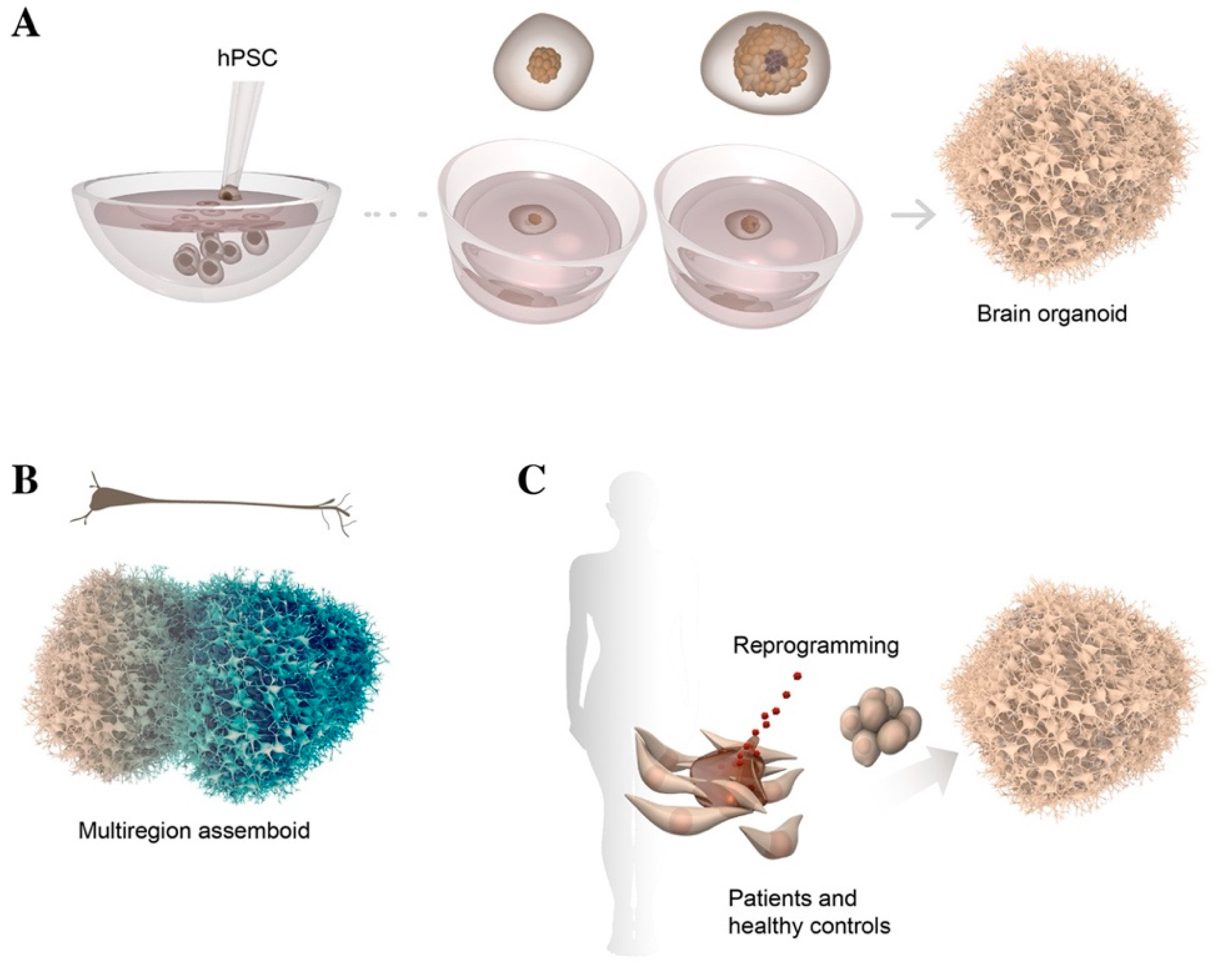
2.3. 3D Culture
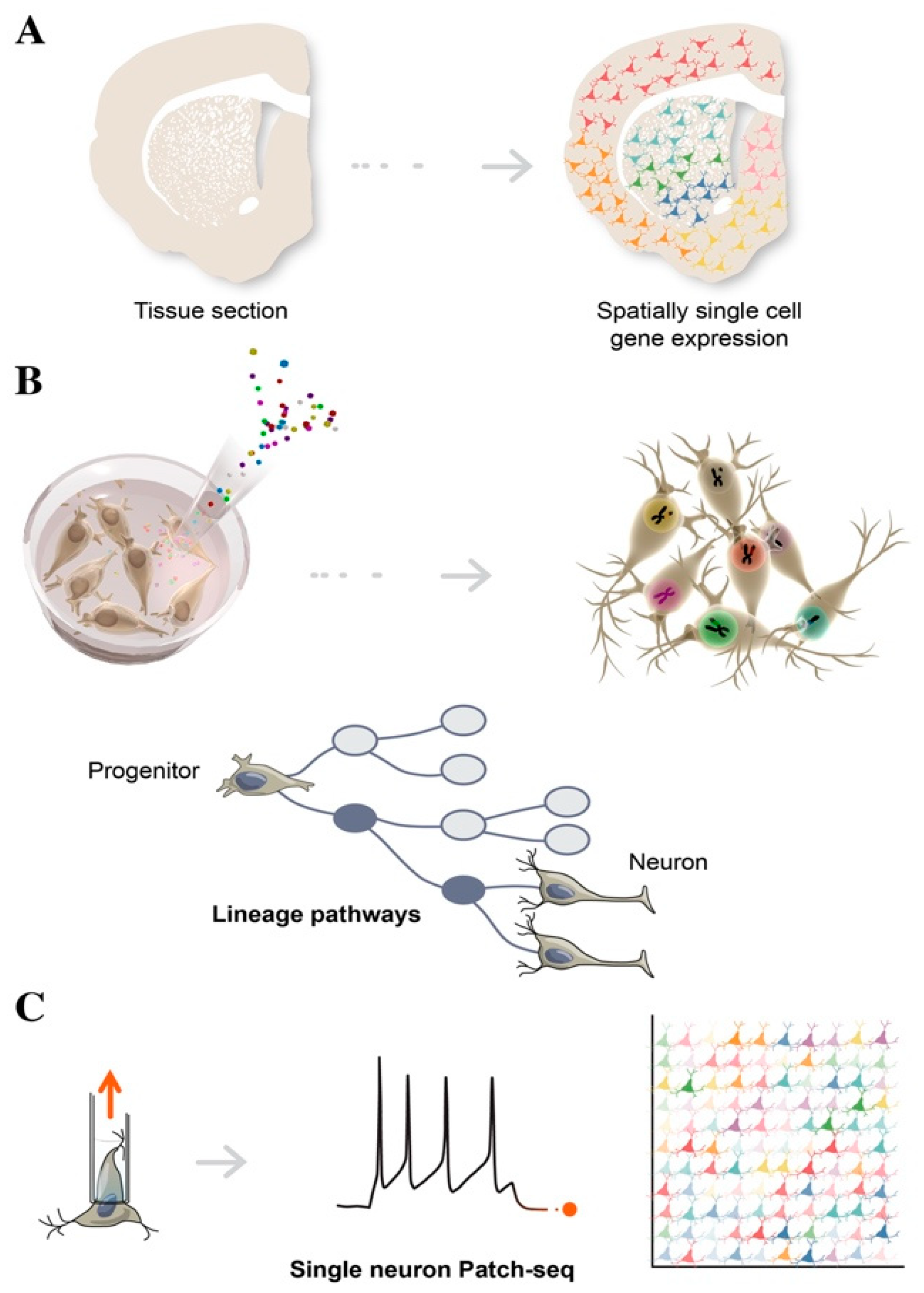
3. Single Cell Sequencing in Decoding Human Brain Complexity
4. Advances in Single Cell Transcriptomics
4.1. Spatial Transcriptomics
4.2. Cellular Barcoding
4.3. Multimodal Single Cell Data
4.4. Patch-Seq
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bjorklund, A.; Dunnett, S.B. Dopamine neuron systems in the brain: An update. Trends Neurosci. 2007, 30, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Yuan, Y.; Halliday, G.; Rusznak, Z.; Watson, C.; Paxinos, G. A cytoarchitectonic and chemoarchitectonic analysis of the dopamine cell groups in the substantia nigra, ventral tegmental area, and retrorubral field in the mouse. Brain Struct. Funct. 2012, 217, 591–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulin, J.F.; Caronia, G.; Hofer, C.; Cui, Q.; Helm, B.; Ramakrishnan, C.; Chan, C.S.; Dombeck, D.A.; Deisseroth, K.; Awatramani, R. Mapping projections of molecularly defined dopamine neuron subtypes using intersectional genetic approaches. Nat. Neurosci. 2018, 21, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Panman, L.; Papathanou, M.; Laguna, A.; Oosterveen, T.; Volakakis, N.; Acampora, D.; Kurtsdotter, I.; Yoshitake, T.; Kehr, J.; Joodmardi, E.; et al. Sox6 and Otx2 control the specification of substantia nigra and ventral tegmental area dopamine neurons. Cell Rep. 2014, 8, 1018–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brichta, L.; Greengard, P. Molecular determinants of selective dopaminergic vulnerability in Parkinson’s disease: An update. Front. Neuroanat. 2014, 8, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderegg, A.; Poulin, J.F.; Awatramani, R. Molecular heterogeneity of midbrain dopaminergic neurons—Moving toward single cell resolution. FEBS Lett. 2015, 589, 3714–3726. [Google Scholar] [CrossRef]
- Brignani, S.; Pasterkamp, R.J. Neuronal Subset-Specific Migration and Axonal Wiring Mechanisms in the Developing Midbrain Dopamine System. Front. Neuroanat. 2017, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Grimm, J.; Mueller, A.; Hefti, F.; Rosenthal, A. Molecular basis for catecholaminergic neuron diversity. Proc. Natl. Acad. Sci. USA 2004, 101, 13891–13896. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.Y.; Seo, H.; Sonntag, K.C.; Brooks, A.; Lin, L.; Isacson, O. Cell type-specific gene expression of midbrain dopaminergic neurons reveals molecules involved in their vulnerability and protection. Hum. Mol. Genet. 2005, 14, 1709–1725. [Google Scholar] [CrossRef] [Green Version]
- Greene, J.G.; Dingledine, R.; Greenamyre, J.T. Gene expression profiling of rat midbrain dopamine neurons: Implications for selective vulnerability in parkinsonism. Neurobiol. Dis. 2005, 18, 19–31. [Google Scholar] [CrossRef]
- Barker, R.A.; Drouin-Ouellet, J.; Parmar, M. Cell-based therapies for Parkinson disease-past insights and future potential. Nat. Rev. Neurol. 2015, 11, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Parmar, M.; Studer, L.; Takahashi, J. Human Trials of Stem Cell-Derived Dopamine Neurons for Parkinson’s Disease: Dawn of a New Era. Cell Stem Cell 2017, 21, 569–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulin, J.F.; Zou, J.; Drouin-Ouellet, J.; Kim, K.Y.; Cicchetti, F.; Awatramani, R.B. Defining midbrain dopaminergic neuron diversity by single-cell gene expression profiling. Cell Rep. 2014, 9, 930–943. [Google Scholar] [CrossRef] [Green Version]
- Hook, P.W.; McClymont, S.A.; Cannon, G.H.; Law, W.D.; Morton, A.J.; Goff, L.A.; McCallion, A.S. Single-Cell RNA-Seq of Mouse Dopaminergic Neurons Informs Candidate Gene Selection for Sporadic Parkinson Disease. Am. J. Hum. Genet. 2018, 102, 427–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, D.J.; Risso, D.; Kosillo, P.; Ngai, J.; Bateup, H.S. Combinatorial Expression of Grp and Neurod6 Defines Dopamine Neuron Populations with Distinct Projection Patterns and Disease Vulnerability. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammel, S.; Steinberg, E.E.; Földy, C.; Wall, N.; Beier, K.; Luo, L.; Malenka, R.C. Diversity of transgenic mouse models for selective targeting of midbrain dopamine neurons. Neuron 2015, 85, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef]
- Kikuchi, T.; Morizane, A.; Doi, D.; Magotani, H.; Onoe, H.; Hayashi, T.; Mizuma, H.; Takara, S.; Takahashi, R.; Inoue, H.; et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 2017, 548, 592–596. [Google Scholar] [CrossRef]
- Grealish, S.; Diguet, E.; Kirkeby, A.; Mattsson, B.; Heuer, A.; Bramoulle, Y.; Van Camp, N.; Perrier, A.L.; Hantraye, P.; Bjorklund, A.; et al. Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson’s disease. Cell Stem Cell 2014, 15, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Nolbrant, S.; Heuer, A.; Parmar, M.; Kirkeby, A. Generation of high-purity human ventral midbrain dopaminergic progenitors for in vitro maturation and intracerebral transplantation. Nat. Protoc. 2017, 12, 1962–1979. [Google Scholar] [CrossRef]
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.D.; Goke, J.; Tan, Z.Y.; Saw, T.Y.; Tan, C.P.; Lokman, H.; et al. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell Stem Cell 2016, 19, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Armand, E.J.; Li, J.; Xie, F.; Luo, C.; Mukamel, E.A. Single-Cell Sequencing of Brain Cell Transcriptomes and Epigenomes. Neuron 2021, 109, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswami, S.R.; Grindberg, R.V.; Novotny, M.; Venepally, P.; Lacar, B.; Bhutani, K.; Linker, S.B.; Pham, S.; Erwin, J.A.; Miller, J.A.; et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat. Protoc. 2016, 11, 499–524. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, D.; Sandor, C.; Volpato, V.; Caffrey, T.M.; Monzon-Sandoval, J.; Bowden, R.; Alegre-Abarrategui, J.; Wade-Martins, R.; Webber, C. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat. Commun. 2020, 11, 4183. [Google Scholar] [CrossRef] [PubMed]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef] [PubMed]
- La Manno, G.; Gyllborg, D.; Codeluppi, S.; Nishimura, K.; Salto, C.; Zeisel, A.; Borm, L.; Stott, S.R.; Toledo, E.M.; Villaescusa, J.C.; et al. Molecular Diversity of Midbrain Development in Mouse, Human, and Stem Cells. Cell 2016, 167, 566–580 e19. [Google Scholar] [CrossRef] [Green Version]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef]
- Fisher, E.M.C.; Bannerman, D.M. Mouse models of neurodegeneration: Know your question, know your mouse. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Harschnitz, O.; Studer, L. Human stem cell models to study host-virus interactions in the central nervous system. Nat. Rev. Immunol. 2021. [Google Scholar] [CrossRef]
- Arenas, E.; Denham, M.; Villaescusa, J.C. How to make a midbrain dopaminergic neuron. Development 2015, 142, 1918–1936. [Google Scholar] [CrossRef] [Green Version]
- Kirkeby, A.; Grealish, S.; Wolf, D.A.; Nelander, J.; Wood, J.; Lundblad, M.; Lindvall, O.; Parmar, M. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 2012, 1, 703–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroof, A.M.; Keros, S.; Tyson, J.A.; Ying, S.W.; Ganat, Y.M.; Merkle, F.T.; Liu, B.; Goulburn, A.; Stanley, E.G.; Elefanty, A.G.; et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 2013, 12, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, F.; Storm, P.; Sozzi, E.; Hidalgo Gil, D.; Birtele, M.; Sharma, Y.; Parmar, M.; Fiorenzano, A. Single-Cell Profiling of Coding and Noncoding Genes in Human Dopamine Neuron Differentiation. Cells 2021, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.; Zabierowski, S.; Dubose, B.N.; Hill, E.J.; Navare, M.; Claros, N.; Rosen, S.; Ramnarine, K.; Horn, C.; Fredrickson, C.; et al. Preclinical Efficacy and Safety of a Human Embryonic Stem Cell-Derived Midbrain Dopamine Progenitor Product, MSK-DA01. Cell Stem Cell 2021, 28, 217–229 e7. [Google Scholar] [CrossRef] [PubMed]
- Readhead, B.; Hartley, B.J.; Eastwood, B.J.; Collier, D.A.; Evans, D.; Farias, R.; He, C.; Hoffman, G.; Sklar, P.; Dudley, J.T.; et al. Expression-based drug screening of neural progenitor cells from individuals with schizophrenia. Nat. Commun. 2018, 9, 4412. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Svendsen, C.N. Human stem cells and drug screening: Opportunities and challenges. Nat. Rev. Drug Discov. 2010, 9, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Minn, Y.K.; Lee, J.Y.; Choi, D.H.; Chang, M.Y.; Shim, J.W.; Ko, J.Y.; Koh, H.C.; Kang, M.J.; Kang, J.S.; et al. In vitro and in vivo analyses of human embryonic stem cell-derived dopamine neurons. J. Neurochem. 2005, 92, 1265–1276. [Google Scholar] [CrossRef]
- Perrier, A.L.; Tabar, V.; Barberi, T.; Rubio, M.E.; Bruses, J.; Topf, N.; Harrison, N.L.; Studer, L. Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2004, 101, 12543–12548. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, S.; Hall, A.C.; Pinto, L.; Attardo, A.; Gotz, M.; Huttner, W.B.; Arenas, E. Identification of midbrain floor plate radial glia-like cells as dopaminergic progenitors. Glia 2008, 56, 809–820. [Google Scholar] [CrossRef]
- Ono, Y.; Nakatani, T.; Sakamoto, Y.; Mizuhara, E.; Minaki, Y.; Kumai, M.; Hamaguchi, A.; Nishimura, M.; Inoue, Y.; Hayashi, H.; et al. Differences in neurogenic potential in floor plate cells along an anteroposterior location: Midbrain dopaminergic neurons originate from mesencephalic floor plate cells. Development 2007, 134, 3213–3225. [Google Scholar] [CrossRef] [Green Version]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Drouin-Ouellet, J.; Lau, S.; Brattas, P.L.; Rylander Ottosson, D.; Pircs, K.; Grassi, D.A.; Collins, L.M.; Vuono, R.; Andersson Sjoland, A.; Westergren-Thorsson, G.; et al. REST suppression mediates neural conversion of adult human fibroblasts via microRNA-dependent and -independent pathways. EMBO Mol. Med. 2017, 9, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, U.; Kirkeby, A.; Torper, O.; Wood, J.; Nelander, J.; Dufour, A.; Bjorklund, A.; Lindvall, O.; Jakobsson, J.; Parmar, M. Direct conversion of human fibroblasts to dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2011, 108, 10343–10348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Zheng, X.; Ansari, Z.; Bunnell, M.C.; Herdy, J.R.; Traxler, L.; Lee, H.; Paquola, A.C.M.; Blithikioti, C.; Ku, M.; et al. Mitochondrial Aging Defects Emerge in Directly Reprogrammed Human Neurons due to Their Metabolic Profile. Cell Rep. 2018, 23, 2550–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, J.; Paquola, A.C.M.; Ku, M.; Hatch, E.; Bohnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victor, M.B.; Richner, M.; Olsen, H.E.; Lee, S.W.; Monteys, A.M.; Ma, C.; Huh, C.J.; Zhang, B.; Davidson, B.; Yang, X.W.; et al. Striatal neurons directly converted from Huntington’s disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat. Neurosci. 2018, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Oikonomopoulos, A.; Sayed, N.; Wu, J.C. Modeling human diseases with induced pluripotent stem cells: From 2D to 3D and beyond. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Quadrato, G.; Arlotta, P. Present and future of modeling human brain development in 3D organoids. Curr. Opin. Cell Biol. 2017, 49, 47–52. [Google Scholar] [CrossRef]
- Tiklova, K.; Nolbrant, S.; Fiorenzano, A.; Bjorklund, A.K.; Sharma, Y.; Heuer, A.; Gillberg, L.; Hoban, D.B.; Cardoso, T.; Adler, A.F.; et al. Single cell transcriptomics identifies stem cell-derived graft composition in a model of Parkinson’s disease. Nat. Commun. 2020, 11, 2434. [Google Scholar] [CrossRef]
- Lindvall, O.; Rehncrona, S.; Brundin, P.; Gustavii, B.; Astedt, B.; Widner, H.; Lindholm, T.; Bjorklund, A.; Leenders, K.L.; Rothwell, J.C.; et al. Human fetal dopamine neurons grafted into the striatum in two patients with severe Parkinson’s disease. A detailed account of methodology and a 6-month follow-up. Arch. Neurol. 1989, 46, 615–631. [Google Scholar] [CrossRef]
- Thompson, L.; Barraud, P.; Andersson, E.; Kirik, D.; Bjorklund, A. Identification of dopaminergic neurons of nigral and ventral tegmental area subtypes in grafts of fetal ventral mesencephalon based on cell morphology, protein expression, and efferent projections. J. Neurosci. 2005, 25, 6467–6477. [Google Scholar] [CrossRef] [Green Version]
- Barker, R.A.; Barrett, J.; Mason, S.L.; Bjorklund, A. Fetal dopaminergic transplantation trials and the future of neural grafting in Parkinson’s disease. Lancet Neurol. 2013, 12, 84–91. [Google Scholar] [CrossRef]
- Laguna Goya, R.; Tyers, P.; Barker, R.A. The search for a curative cell therapy in Parkinson’s disease. J. Neurol. Sci. 2008, 265, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Doi, D.; Magotani, H.; Kikuchi, T.; Ikeda, M.; Hiramatsu, S.; Yoshida, K.; Amano, N.; Nomura, M.; Umekage, M.; Morizane, A.; et al. Pre-clinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson’s disease. Nat. Commun. 2020, 11, 3369. [Google Scholar] [CrossRef] [PubMed]
- Fiorenzano, A.; Birtele, M.; Wahlestedt, J.N.; Parmar, M. Evaluation of TH-Cre knock-in cell lines for detection and specific targeting of stem cell-derived dopaminergic neurons. Heliyon 2021, 7, e06006. [Google Scholar] [CrossRef] [PubMed]
- Corti, S.; Bonjean, R.; Legier, T.; Rattier, D.; Melon, C.; Salin, P.; Toso, E.A.; Kyba, M.; Kerkerian-Le Goff, L.; Maina, F. Enhanced differentiation of human induced pluripotent stem cells toward the midbrain dopaminergic neuron lineage through GLYPICAN-4 downregulation. Stem Cells Transl. Med. 2021, 10, 725–742. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.F.; Cardoso, T.; Nolbrant, S.; Mattsson, B.; Hoban, D.B.; Jarl, U.; Wahlestedt, J.N.; Grealish, S.; Bjorklund, A.; Parmar, M. hESC-Derived Dopaminergic Transplants Integrate into Basal Ganglia Circuitry in a Preclinical Model of Parkinson’s Disease. Cell Rep. 2019, 28, 3462–3473 e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.W.; Piao, J.; Koo, S.Y.; Kriks, S.; Chung, S.Y.; Betel, D.; Socci, N.D.; Choi, S.J.; Zabierowski, S.; Dubose, B.N.; et al. Biphasic Activation of WNT Signaling Facilitates the Derivation of Midbrain Dopamine Neurons from hESCs for Translational Use. Cell Stem Cell 2021, 28, 343–355 e5. [Google Scholar] [CrossRef] [PubMed]
- Galvan, A.; Caiola, M.J.; Albaugh, D.L. Advances in optogenetic and chemogenetic methods to study brain circuits in non-human primates. J. Neural Transm. 2018, 125, 547–563. [Google Scholar] [CrossRef]
- Jayaprakash, N.; Wang, Z.; Hoeynck, B.; Krueger, N.; Kramer, A.; Balle, E.; Wheeler, D.S.; Wheeler, R.A.; Blackmore, M.G. Optogenetic Interrogation of Functional Synapse Formation by Corticospinal Tract Axons in the Injured Spinal Cord. J. Neurosci. 2016, 36, 5877–5890. [Google Scholar] [CrossRef]
- Aldrin-Kirk, P.; Heuer, A.; Wang, G.; Mattsson, B.; Lundblad, M.; Parmar, M.; Bjorklund, T. DREADD Modulation of Transplanted DA Neurons Reveals a Novel Parkinsonian Dyskinesia Mechanism Mediated by the Serotonin 5-HT6 Receptor. Neuron 2016, 90, 955–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidsson, M.; Wang, G.; Aldrin-Kirk, P.; Cardoso, T.; Nolbrant, S.; Hartnor, M.; Mudannayake, J.; Parmar, M.; Bjorklund, T. A systematic capsid evolution approach performed in vivo for the design of AAV vectors with tailored properties and tropism. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Min Yang, S.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Maier, B.; Baillie, G.J.; Ferguson, C.; Parton, R.G.; Wolvetang, E.J.; Roost, M.S.; Lopes, S.M.; et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2016, 536, 238. [Google Scholar] [CrossRef] [Green Version]
- Huch, M.; Koo, B.K. Modeling mouse and human development using organoid cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Buth, J.E.; Vishlaghi, N.; de la Torre-Ubieta, L.; Taxidis, J.; Khakh, B.S.; Coppola, G.; Pearson, C.A.; Yamauchi, K.; Gong, D.; et al. Self-Organized Cerebral Organoids with Human-Specific Features Predict Effective Drugs to Combat Zika Virus Infection. Cell Rep. 2017, 21, 517–532. [Google Scholar] [CrossRef] [Green Version]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Kanton, S.; Boyle, M.J.; He, Z.; Santel, M.; Weigert, A.; Sanchis-Calleja, F.; Guijarro, P.; Sidow, L.; Fleck, J.S.; Han, D.; et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 2019, 574, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, H.J.; Choi, H.; Chang, Y.; Park, H.; Shin, J.; Kim, J.; Lengner, C.J.; Lee, Y.K.; Kim, J. Modeling G2019S-LRRK2 Sporadic Parkinson’s Disease in 3D Midbrain Organoids. Stem Cell Rep. 2019, 12, 518–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, L.M.; Reinhardt, L.; Reinhardt, P.; Glatza, M.; Monzel, A.S.; Stanslowsky, N.; Rosato-Siri, M.D.; Zanon, A.; Antony, P.M.; Bellmann, J.; et al. Modeling Parkinson’s disease in midbrain-like organoids. NPJ Parkinsons Dis. 2019, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rifes, P.; Isaksson, M.; Rathore, G.S.; Aldrin-Kirk, P.; Moller, O.K.; Barzaghi, G.; Lee, J.; Egerod, K.L.; Rausch, D.M.; Parmar, M.; et al. Publisher Correction: Modeling neural tube development by differentiation of human embryonic stem cells in a microfluidic WNT gradient. Nat. Biotechnol. 2020, 38, 1357. [Google Scholar] [CrossRef] [PubMed]
- Demers, C.J.; Soundararajan, P.; Chennampally, P.; Cox, G.A.; Briscoe, J.; Collins, S.D.; Smith, R.L. Development-on-chip: In vitro neural tube patterning with a microfluidic device. Development 2016, 143, 1884–1892. [Google Scholar] [CrossRef] [Green Version]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Sloan, S.A.; Andersen, J.; Pasca, A.M.; Birey, F.; Pasca, S.P. Generation and assembly of human brain region-specific three-dimensional cultures. Nat. Protoc. 2018, 13, 2062–2085. [Google Scholar] [CrossRef]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Huch, M. Disease modelling in human organoids. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodusek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef]
- Galet, B.; Cheval, H.; Ravassard, P. Patient-Derived Midbrain Organoids to Explore the Molecular Basis of Parkinson’s Disease. Front. Neurol. 2020, 11, 1005. [Google Scholar] [CrossRef] [PubMed]
- Hoban, D.B.; Shrigley, S.; Mattsson, B.; Breger, L.S.; Jarl, U.; Cardoso, T.; Wahlestedt, J.N.; Luk, K.C.; Björklund, A.; Parmar, M. Impact of alpha-synuclein pathology on transplanted hESC-derived dopaminergic neurons in a humanized alpha-synuclein rat model of PD. Proc. Natl. Acad. Sci. USA 2020, 117, 15209–15220. [Google Scholar] [CrossRef] [PubMed]
- Pasca, A.M.; Park, J.Y.; Shin, H.W.; Qi, Q.; Revah, O.; Krasnoff, R.; O’Hara, R.; Willsey, A.J.; Palmer, T.D.; Pasca, S.P. Human 3D cellular model of hypoxic brain injury of prematurity. Nat. Med. 2019, 25, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Corsini, N.S.; Wolfinger, S.; Gustafson, E.H.; Phillips, A.W.; Burkard, T.R.; Otani, T.; Livesey, F.J.; Knoblich, J.A. Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol. 2017, 35, 659–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Su, Y.; Adam, C.D.; Deutschmann, A.U.; Pather, S.R.; Goldberg, E.M.; Su, K.; Li, S.; Lu, L.; Jacob, F.; et al. Sliced Human Cortical Organoids for Modeling Distinct Cortical Layer Formation. Cell Stem Cell 2020, 26, 766–781 e9. [Google Scholar] [CrossRef]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef]
- Shi, Y.; Sun, L.; Wang, M.; Liu, J.; Zhong, S.; Li, R.; Li, P.; Guo, L.; Fang, A.; Chen, R.; et al. Vascularized human cortical organoids (vOrganoids) model cortical development in vivo. PLoS Biol. 2020, 18, e3000705. [Google Scholar] [CrossRef]
- Adriani, G.; Ma, D.; Pavesi, A.; Kamm, R.D.; Goh, E.L. A 3D neurovascular microfluidic model consisting of neurons, astrocytes and cerebral endothelial cells as a blood-brain barrier. Lab. Chip 2017, 17, 448–459. [Google Scholar] [CrossRef]
- Osaki, T.; Sivathanu, V.; Kamm, R.D. Engineered 3D vascular and neuronal networks in a microfluidic platform. Sci. Rep. 2018, 8, 5168. [Google Scholar] [CrossRef] [Green Version]
- Poulin, J.F.; Gaertner, Z.; Moreno-Ramos, O.A.; Awatramani, R. Classification of Midbrain Dopamine Neurons Using Single-Cell Gene Expression Profiling Approaches. Trends Neurosci. 2020, 43, 155–169. [Google Scholar] [CrossRef]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030 e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashima, Y.; Suzuki, A.; Suzuki, Y. An Informative Approach to Single-Cell Sequencing Analysis. Adv. Exp. Med. Biol. 2019, 1129, 81–96. [Google Scholar] [PubMed]
- Cao, J.; Packer, J.S.; Ramani, V.; Cusanovich, D.A.; Huynh, C.; Daza, R.; Qiu, X.; Lee, C.; Furlan, S.N.; Steemers, F.J.; et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 2017, 357, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picelli, S.; Bjorklund, A.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 173, 1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiklova, K.; Bjorklund, A.K.; Lahti, L.; Fiorenzano, A.; Nolbrant, S.; Gillberg, L.; Volakakis, N.; Yokota, C.; Hilscher, M.M.; Hauling, T.; et al. Single-cell RNA sequencing reveals midbrain dopamine neuron diversity emerging during mouse brain development. Nat. Commun. 2019, 10, 581. [Google Scholar] [CrossRef] [Green Version]
- Cahan, P.; Cacchiarelli, D.; Dunn, S.J.; Hemberg, M.; de Sousa Lopes, S.M.C.; Morris, S.A.; Rackham, O.J.L.; Del Sol, A.; Wells, C.A. Computational Stem Cell Biology: Open Questions and Guiding Principles. Cell Stem Cell 2021, 28, 20–32. [Google Scholar] [CrossRef]
- Stahl, P.L.; Salmen, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Moncada, R.; Barkley, D.; Wagner, F.; Chiodin, M.; Devlin, J.C.; Baron, M.; Hajdu, C.H.; Simeone, D.M.; Yanai, I. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 2020, 38, 333–342. [Google Scholar] [CrossRef]
- Wang, C.; Balch, W.E. Bridging Genomics to Phenomics at Atomic Resolution through Variation Spatial Profiling. Cell Rep. 2018, 24, 2013–2028 e6. [Google Scholar] [CrossRef] [Green Version]
- Weinreb, C.; Rodriguez-Fraticelli, A.; Camargo, F.D.; Klein, A.M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 2020, 367. [Google Scholar] [CrossRef]
- Kebschull, J.M.; Zador, A.M. Cellular barcoding: Lineage tracing, screening and beyond. Nat. Methods 2018, 15, 871–879. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Gagnon, J.A. Recording development with single cell dynamic lineage tracing. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, J.D.; Kozareva, V.; Ferreira, A.; Vanderburg, C.; Martin, C.; Macosko, E.Z. Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell 2019, 177, 1873–1887 e17. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.B.; Chen, S.; Sos, B.C.; Fan, J.; Kaeser, G.E.; Yung, Y.C.; Duong, T.E.; Gao, D.; Chun, J.; Kharchenko, P.V.; et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018, 36, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Preissl, S.; Ren, B. Single-cell multimodal omics: The power of many. Nat. Methods 2020, 17, 11–14. [Google Scholar] [CrossRef]
- Rooijers, K.; Markodimitraki, C.M.; Rang, F.J.; de Vries, S.S.; Chialastri, A.; de Luca, K.L.; Mooijman, D.; Dey, S.S.; Kind, J. Simultaneous quantification of protein-DNA contacts and transcriptomes in single cells. Nat. Biotechnol. 2019, 37, 766–772. [Google Scholar] [CrossRef]
- Cadwell, C.R.; Palasantza, A.; Jiang, X.; Berens, P.; Deng, Q.; Yilmaz, M.; Reimer, J.; Shen, S.; Bethge, M.; Tolias, K.F.; et al. Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch-seq. Nat. Biotechnol. 2016, 34, 199–203. [Google Scholar] [CrossRef] [Green Version]
- Cadwell, C.R.; Scala, F.; Li, S.; Livrizzi, G.; Shen, S.; Sandberg, R.; Jiang, X.; Tolias, A.S. Multimodal profiling of single-cell morphology, electrophysiology, and gene expression using Patch-seq. Nat. Protoc. 2017, 12, 2531–2553. [Google Scholar] [CrossRef]
- van den Hurk, M.; Erwin, J.A.; Yeo, G.W.; Gage, F.H.; Bardy, C. Patch-Seq Protocol to Analyze the Electrophysiology, Morphology and Transcriptome of Whole Single Neurons Derived From Human Pluripotent Stem Cells. Front. Mol. Neurosci. 2018, 11, 261. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| 2D Cell Culture | 3D Cell Culture | Assembloid Culture | Cell Transplantation | |
| Ease of experimental set-up | ✓ | ✓ | ✓ | ✓ |
| Generation of functionally mature neurons | ✓ | ✓ | ✓ | ✓ |
| Reproducibility | ✓ | ✓ | ✓ | ✓ |
| Genetic manipulation | ✓ | ✓ | ✓ | ✓ |
| Single cell dissociation | ✓ | ✓ | ✓ | ✓ |
| Cell diversity | ✓ | ✓ | ✓ | ✓ |
| Cell-cell interaction | ✓ | ✓ | ✓ | ✓ |
| Tissue viability | ✓ | ✓ | ✓ | ✓ |
| Amendable for HTS | ✓ | ✓ | ✓ | |
| Fully humanized | ✓ | ✓ | ✓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorenzano, A.; Sozzi, E.; Parmar, M.; Storm, P. Dopamine Neuron Diversity: Recent Advances and Current Challenges in Human Stem Cell Models and Single Cell Sequencing. Cells 2021, 10, 1366. https://doi.org/10.3390/cells10061366
Fiorenzano A, Sozzi E, Parmar M, Storm P. Dopamine Neuron Diversity: Recent Advances and Current Challenges in Human Stem Cell Models and Single Cell Sequencing. Cells. 2021; 10(6):1366. https://doi.org/10.3390/cells10061366
Chicago/Turabian StyleFiorenzano, Alessandro, Edoardo Sozzi, Malin Parmar, and Petter Storm. 2021. "Dopamine Neuron Diversity: Recent Advances and Current Challenges in Human Stem Cell Models and Single Cell Sequencing" Cells 10, no. 6: 1366. https://doi.org/10.3390/cells10061366
APA StyleFiorenzano, A., Sozzi, E., Parmar, M., & Storm, P. (2021). Dopamine Neuron Diversity: Recent Advances and Current Challenges in Human Stem Cell Models and Single Cell Sequencing. Cells, 10(6), 1366. https://doi.org/10.3390/cells10061366