
TGF-β/Smad Signalling in Neurogenesis: Implications for Neuropsychiatric Diseases


Abstract
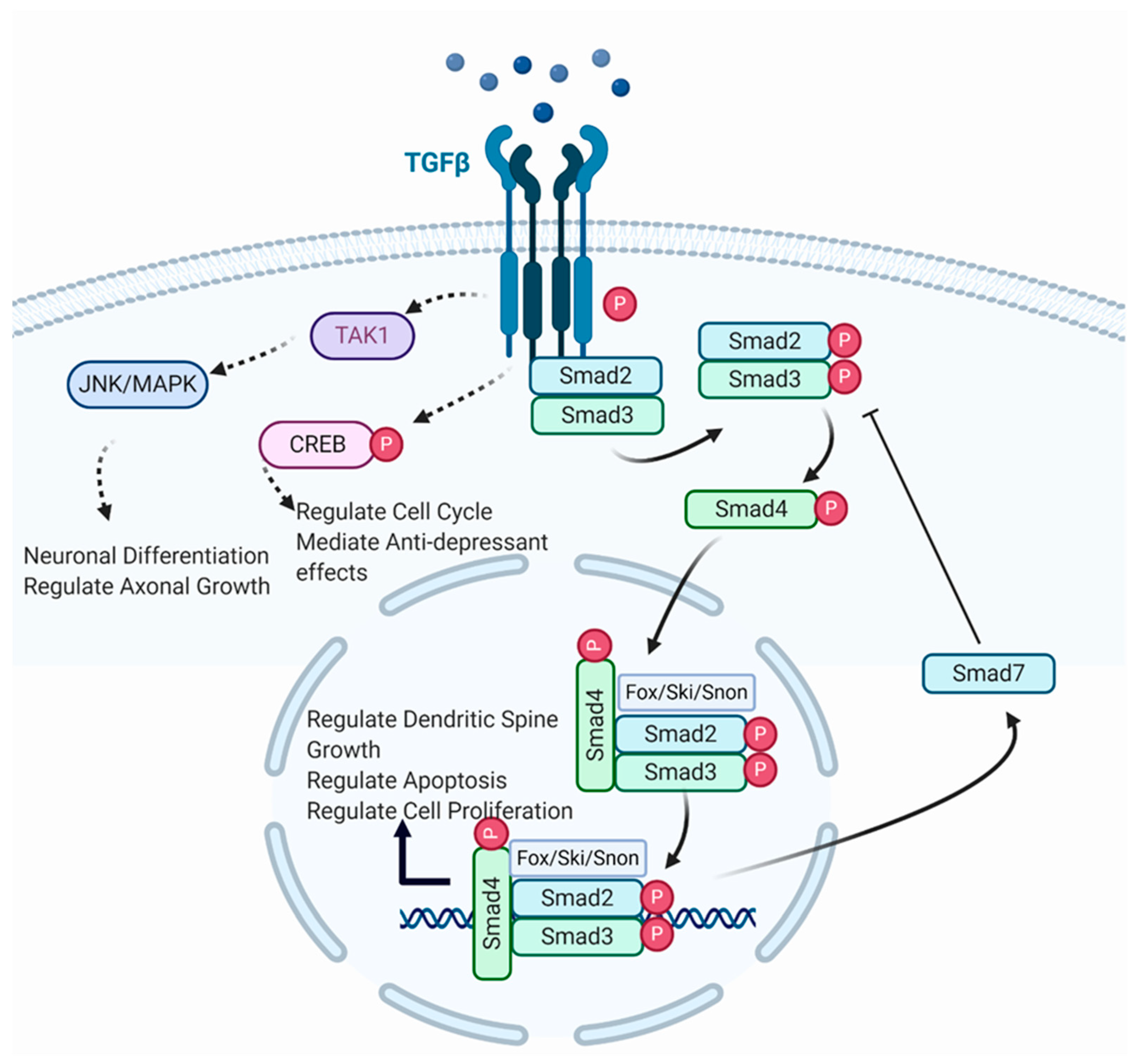
:1. Introduction
2. Non-Canonical Signalling
3. Canonical Signalling in Neurogenesis
4. TGF-β Signalling in Epigenetics
5. Neurogenesis in Neuropsychiatric Disorders
6. Effects of Stress on TGF-β Signalling and Neurogenesis
7. Discussion and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Derynck, R.; Akhurst, R.J. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat. Cell Biol. 2007, 9, 1000–1004. [Google Scholar] [CrossRef]
- Massague, J.; Blain, S.W.; Lo, R.S. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Ten Dijke, P.; Hill, C.S. New insights into TGF-beta-Smad signalling. Trends Biochem. Sci. 2004, 29, 265–273. [Google Scholar] [CrossRef]
- Nakao, A.; Afrakhte, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Lyons, R.M.; Moses, H.L. Transforming growth factors and the regulation of cell proliferation. Eur. J. Biochem. 1990, 187, 467–473. [Google Scholar] [CrossRef]
- Vogel, T.; Ahrens, S.; Buttner, N.; Krieglstein, K. Transforming growth factor beta promotes neuronal cell fate of mouse cortical and hippocampal progenitors in vitro and in vivo: Identification of Nedd9 as an essential signaling component. Cereb. Cortex 2010, 20, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Wachs, F.P.; Winner, B.; Couillard-Despres, S.; Schiller, T.; Aigner, R.; Winkler, J.; Bogdahn, U.; Aigner, L. Transforming growth factor-beta1 is a negative modulator of adult neurogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 358–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Chu, P.J.; Ishihara, A.; Saito, H. Transforming growth factor-beta 1 promotes re-elongation of injured axons of cultured rat hippocampal neurons. Brain Res. 1996, 723, 206–209. [Google Scholar] [CrossRef]
- Knoferle, J.; Ramljak, S.; Koch, J.C.; Tonges, L.; Asif, A.R.; Michel, U.; Wouters, F.S.; Heermann, S.; Krieglstein, K.; Zerr, I.; et al. TGF-beta 1 enhances neurite outgrowth via regulation of proteasome function and EFABP. Neurobiol. Dis. 2010, 38, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.J.; Barnes, A.P.; Hand, R.; Polleux, F.; Ehlers, M.D. TGF-beta signaling specifies axons during brain development. Cell 2010, 142, 144–157. [Google Scholar] [CrossRef] [Green Version]
- Beauregard, M.; Levesque, J.; Bourgouin, P. Neural correlates of conscious self-regulation of emotion. J. Neurosci. 2001, 21, RC165. [Google Scholar] [CrossRef]
- Patten, S.B. Sensitization: The sine qua non of the depressive disorders? Med. Hypotheses 2008, 71, 872–875. [Google Scholar] [CrossRef] [PubMed]
- Ekdahl, C.T.; Claasen, J.H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Jin, K. Current perspectives on the link between neuroinflammation and neurogenesis. Metab. Brain Dis. 2015, 30, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bueno, B.; Caso, J.R.; Leza, J.C. Stress as a neuroinflammatory condition in brain: Damaging and protective mechanisms. Neurosci. Biobehav. Rev. 2008, 32, 1136–1151. [Google Scholar] [CrossRef]
- Menachem-Zidon, O.B.; Goshen, I.; Kreisel, T.; Menahem, Y.B.; Reinhartz, E.; Hur, T.B.; Yirmiya, R. Intrahippocampal transplantation of transgenic neural precursor cells overexpressing interleukin-1 receptor antagonist blocks chronic isolation-induced impairment in memory and neurogenesis. Neuropsychopharmacology 2008, 33, 2251–2262. [Google Scholar] [CrossRef] [Green Version]
- Goshen, I.; Kreisel, T.; Ben-Menachem-Zidon, O.; Licht, T.; Weidenfeld, J.; Ben-Hur, T.; Yirmiya, R. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol. Psychiatry 2008, 13, 717–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, J.M.; Alekseenko, Z.; Applequist, J.M.; Ericson, J. Tgfbeta signaling regulates temporal neurogenesis and potency of neural stem cells in the CNS. Neuron 2014, 84, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandasamy, M.; Lehner, B.; Kraus, S.; Sander, P.R.; Marschallinger, J.; Rivera, F.J.; Trumbach, D.; Ueberham, U.; Reitsamer, H.A.; Strauss, O.; et al. TGF-beta signalling in the adult neurogenic niche promotes stem cell quiescence as well as generation of new neurons. J. Cell. Mol. Med. 2014, 18, 1444–1459. [Google Scholar] [CrossRef] [Green Version]
- Meyers, E.A.; Kessler, J.A. TGF-beta Family Signaling in Neural and Neuronal Differentiation, Development, and Function. Cold Spring Harb. Perspect. Biol. 2017, 9, a022244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drevets, W.C. Neuroplasticity in mood disorders. Dialogues Clin. Neurosci. 2004, 6, 199–216. [Google Scholar] [PubMed]
- Manji, H.K.; Moore, G.J.; Rajkowska, G.; Chen, G. Neuroplasticity and cellular resilience in mood disorders. Mol. Psychiatry 2000, 5, 578–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef]
- Tang, M.; Wei, X.; Guo, Y.; Breslin, P.; Zhang, S.; Zhang, S.; Wei, W.; Xia, Z.; Diaz, M.; Akira, S.; et al. TAK1 is required for the survival of hematopoietic cells and hepatocytes in mice. J. Exp. Med. 2008, 205, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Shirakabe, K.; Shibuya, H.; Irie, K.; Oishi, I.; Ueno, N.; Taniguchi, T.; Nishida, E.; Matsumoto, K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 1995, 270, 2008–2011. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, F.; Wang, S.; Zhang, Y.; Fan, M.; Xu, Z. TAK1 is activated by TGF-beta signaling and controls axonal growth during brain development. J. Mol. Cell Biol. 2014, 6, 349–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Li, X.; McNamee, C.; Chen, A.P.; Baccarini, M.; Snider, W.D. Raf kinase signaling functions in sensory neuron differentiation and axon growth in vivo. Nat. Neurosci. 2007, 10, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Duman, R.S. Finding the intracellular signaling pathways affected by mood disorder treatments. Neuron 2003, 38, 157–160. [Google Scholar] [CrossRef] [Green Version]
- Duman, C.H.; Schlesinger, L.; Kodama, M.; Russell, D.S.; Duman, R.S. A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biol. Psychiatry 2007, 61, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Einat, H.; Yuan, P.; Gould, T.D.; Li, J.; Du, J.; Zhang, L.; Manji, H.K.; Chen, G. The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J. Neurosci. 2003, 23, 7311–7316. [Google Scholar] [CrossRef] [PubMed]
- Wefers, B.; Hitz, C.; Holter, S.M.; Trumbach, D.; Hansen, J.; Weber, P.; Putz, B.; Deussing, J.M.; de Angelis, M.H.; Roenneberg, T.; et al. MAPK signaling determines anxiety in the juvenile mouse brain but depression-like behavior in adults. PLoS ONE 2012, 7, e35035. [Google Scholar] [CrossRef] [PubMed]
- Lhuillier, L.; Dryer, S.E. Developmental regulation of neuronal KCa channels by TGFbeta 1: Transcriptional and posttranscriptional effects mediated by Erk MAP kinase. J Neurosci. 2000, 20, 5616–5622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, T.; Liu, R.Y.; Byrne, J.H. Transforming growth factor-beta2 modulates synaptic efficacy and plasticity and induces phosphorylation of CREB in hippocampal neurons. Hippocampus 2007, 17, 5–9. [Google Scholar] [CrossRef]
- Balschun, D.; Wolfer, D.P.; Gass, P.; Mantamadiotis, T.; Welzl, H.; Schutz, G.; Frey, J.U.; Lipp, H.P. Does cAMP response element-binding protein have a pivotal role in hippocampal synaptic plasticity and hippocampus-dependent memory? J. Neurosci. 2003, 23, 6304–6314. [Google Scholar] [CrossRef] [PubMed]
- Nibuya, M.; Nestler, E.J.; Duman, R.S. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J. Neurosci. 1996, 16, 2365–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gur, T.L.; Conti, A.C.; Holden, J.; Bechtholt, A.J.; Hill, T.E.; Lucki, I.; Malberg, J.E.; Blendy, J.A. cAMP response element-binding protein deficiency allows for increased neurogenesis and a rapid onset of antidepressant response. J. Neurosci. 2007, 27, 7860–7868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, S.; Kim, J.E.; Lee, R.; Chen, J.; Fujioka, T.; Malberg, J.; Tsuji, S.; Duman, R.S. Localization of phosphorylated cAMP response element-binding protein in immature neurons of adult hippocampus. J. Neurosci. 2002, 22, 9868–9876. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, S.; Kim, J.E.; Lee, R.; Malberg, J.E.; Chen, J.; Steffen, C.; Zhang, Y.J.; Nestler, E.J.; Duman, R.S. Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J. Neurosci. 2002, 22, 3673–3682. [Google Scholar] [CrossRef]
- Chen, A.C.; Shirayama, Y.; Shin, K.H.; Neve, R.L.; Duman, R.S. Expression of the cAMP response element binding protein (CREB) in hippocampus produces an antidepressant effect. Biol. Psychiatry 2001, 49, 753–762. [Google Scholar] [CrossRef]
- Dennler, S.; Goumans, M.J.; ten Dijke, P. Transforming growth factor beta signal transduction. J. Leukoc. Biol. 2002, 71, 731–740. [Google Scholar] [PubMed]
- Brown, K.A.; Pietenpol, J.A.; Moses, H.L. A tale of two proteins: Differential roles and regulation of Smad2 and Smad3 in TGF-beta signaling. J. Cell. Biochem. 2007, 101, 9–33. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.J. Both SMAD2 and SMAD3 mediate activin-stimulated expression of the follicle-stimulating hormone beta subunit in mouse gonadotrope cells. Mol. Endocrinol. 2004, 18, 606–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Pangas, S.A.; Jorgez, C.J.; Graff, J.M.; Weinstein, M.; Matzuk, M.M. Redundant roles of SMAD2 and SMAD3 in ovarian granulosa cells in vivo. Mol. Cell. Biol. 2008, 28, 7001–7011. [Google Scholar] [CrossRef] [Green Version]
- Brionne, T.C.; Tesseur, I.; Masliah, E.; Wyss-Coray, T. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 2003, 40, 1133–1145. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [Green Version]
- Miguez, D.G.; Gil-Guinon, E.; Pons, S.; Marti, E. Smad2 and Smad3 cooperate and antagonize simultaneously in vertebrate neurogenesis. J. Cell Sci. 2013, 126, 5335–5343. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Symes, A.J. Smad3 deficiency reduces neurogenesis in adult mice. J. Mol. Neurosci. 2010, 41, 383–396. [Google Scholar] [CrossRef]
- Yousef, H.; Conboy, M.J.; Morgenthaler, A.; Schlesinger, C.; Bugaj, L.; Paliwal, P.; Greer, C.; Conboy, I.M.; Schaffer, D. Systemic attenuation of the TGF-beta pathway by a single drug simultaneously rejuvenates hippocampal neurogenesis and myogenesis in the same old mammal. Oncotarget 2015, 6, 11959–11978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapia-Gonzalez, S.; Munoz, M.D.; Cuartero, M.I.; Sanchez-Capelo, A. Smad3 is required for the survival of proliferative intermediate progenitor cells in the dentate gyrus of adult mice. Cell Commun. Signal. 2013, 11, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, M.D.; Antolin-Vallespin, M.; Tapia-Gonzalez, S.; Sanchez-Capelo, A. Smad3 deficiency inhibits dentate gyrus LTP by enhancing GABAA neurotransmission. J. Neurochem. 2016, 137, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Schmierer, B.; Tournier, A.L.; Bates, P.A.; Hill, C.S. Mathematical modeling identifies Smad nucleocytoplasmic shuttling as a dynamic signal-interpreting system. Proc. Natl. Acad. Sci. USA 2008, 105, 6608–6613. [Google Scholar] [CrossRef] [Green Version]
- Buckwalter, M.S.; Yamane, M.; Coleman, B.S.; Ormerod, B.K.; Chin, J.T.; Palmer, T.; Wyss-Coray, T. Chronically increased transforming growth factor-beta1 strongly inhibits hippocampal neurogenesis in aged mice. Am. J. Pathol. 2006, 169, 154–164. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zhang, H.; Yung, A.; Villeda, S.A.; Jaeger, P.A.; Olayiwola, O.; Fainberg, N.; Wyss-Coray, T. ALK5-dependent TGF-beta signaling is a major determinant of late-stage adult neurogenesis. Nat. Neurosci. 2014, 17, 943–952. [Google Scholar] [CrossRef] [Green Version]
- Ganea, K.; Menke, A.; Schmidt, M.V.; Lucae, S.; Rammes, G.; Liebl, C.; Harbich, D.; Sterlemann, V.; Storch, C.; Uhr, M.; et al. Convergent animal and human evidence suggests the activin/inhibin pathway to be involved in antidepressant response. Transl. Psychiatry 2012, 2, e177. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kasai, Y.; Ageta, H.; Hasegawa, Y.; Tsuchida, K.; Sugino, H.; Inokuchi, K. Activin increases the number of synaptic contacts and the length of dendritic spine necks by modulating spinal actin dynamics. J. Cell Sci. 2007, 120, 3830–3837. [Google Scholar] [CrossRef] [Green Version]
- Tapia-Gonzalez, S.; Giraldez-Perez, R.M.; Cuartero, M.I.; Casarejos, M.J.; Mena, M.A.; Wang, X.F.; Sanchez-Capelo, A. Dopamine and alpha-synuclein dysfunction in Smad3 null mice. Mol. Neurodegener. 2011, 6, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Villapol, S.; Wang, Y.; Adams, M.; Symes, A.J. Smad3 deficiency increases cortical and hippocampal neuronal loss following traumatic brain injury. Exp. Neurol. 2013, 250, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Moges, H.; Bharucha, Y.; Symes, A. Smad3 null mice display more rapid wound closure and reduced scar formation after a stab wound to the cerebral cortex. Exp. Neurol. 2007, 203, 168–184. [Google Scholar] [CrossRef]
- Ma, D.K.; Jang, M.H.; Guo, J.U.; Kitabatake, Y.; Chang, M.L.; Pow-Anpongkul, N.; Flavell, R.A.; Lu, B.; Ming, G.L.; Song, H. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 2009, 323, 1074–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.H.; Hong, S.J.; Salinas, R.D.; Liu, S.J.; Sun, S.W.; Sgualdino, J.; Testa, G.; Matzuk, M.M.; Iwamori, N.; Lim, D.A. Activation of neuronal gene expression by the JMJD3 demethylase is required for postnatal and adult brain neurogenesis. Cell Rep. 2014, 8, 1290–1299. [Google Scholar] [CrossRef] [Green Version]
- Estaras, C.; Akizu, N.; Garcia, A.; Beltran, S.; de la Cruz, X.; Martinez-Balbas, M.A. Genome-wide analysis reveals that Smad3 and JMJD3 HDM co-activate the neural developmental program. Development 2012, 139, 2681–2691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.Y.; Gui, W.; He, H.Y.; Wang, X.S.; Zuo, J.; Huang, L.; Zhou, N.; Wang, K.; Wang, Y. Neuronal and astroglial TGFbeta-Smad3 signaling pathways differentially regulate dendrite growth and synaptogenesis. Neuromolecular Med. 2014, 16, 457–472. [Google Scholar] [CrossRef]
- Li, Y.; Luilkart, B.W.; Birnbaum, S.; Chen, J.; Kwon, C.H.; Kernie, S.G.; Bassel-Duby, R.; Parada, L.F. TrkB Regulates Hippocampal Neurogenesis and Governs Sensitivity to Antidepressive Treatment. Neuron 2008, 59, 399–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, S.; Ren, Z.; Li, X.; Zheng, Y.; Meng, A. Smad2 and Smad3 are required for mesendoderm induction by transforming growth factor-beta/nodal signals in zebrafish. J. Biol. Chem. 2008, 283, 2418–2426. [Google Scholar] [CrossRef] [Green Version]
- Akizu, N.; Estaras, C.; Guerrero, L.; Marti, E.; Martinez-Balbas, M.A. H3K27me3 regulates BMP activity in developing spinal cord. Development 2010, 137, 2915–2925. [Google Scholar] [CrossRef] [Green Version]
- Iida, A.; Iwagawa, T.; Kuribayashi, H.; Satoh, S.; Mochizuki, Y.; Baba, Y.; Nakauchi, H.; Furukawa, T.; Koseki, H.; Murakami, A.; et al. Histone demethylase Jmjd3 is required for the development of subsets of retinal bipolar cells. Proc. Natl. Acad. Sci. USA 2014, 111, 3751–3756. [Google Scholar] [CrossRef] [Green Version]
- Grassi, D.; Franz, H.; Vezzali, R.; Bovio, P.; Heidrich, S.; Dehghanian, F.; Lagunas, N.; Belzung, C.; Krieglstein, K.; Vogel, T. Neuronal Activity, TGFbeta-Signaling and Unpredictable Chronic Stress Modulate Transcription of Gadd45 Family Members and DNA Methylation in the Hippocampus. Cereb. Cortex 2017, 27, 4166–4181. [Google Scholar] [CrossRef]
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 2000, 20, 9104–9110. [Google Scholar] [CrossRef]
- Jayatissa, M.N.; Henningsen, K.; Nikolajsen, G.; West, M.J.; Wiborg, O. A reduced number of hippocampal granule cells does not associate with an anhedonia-like phenotype in a rat chronic mild stress model of depression. Stress 2010, 13, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Lagace, D.C.; Donovan, M.H.; DeCarolis, N.A.; Farnbauch, L.A.; Malhotra, S.; Berton, O.; Nestler, E.J.; Krishnan, V.; Eisch, A.J. Adult hippocampal neurogenesis is functionally important for stress-induced social avoidance. Proc. Natl. Acad. Sci. USA 2010, 107, 4436–4441. [Google Scholar] [CrossRef] [Green Version]
- Eisch, A.J.; Petrik, D. Depression and hippocampal neurogenesis: A road to remission? Science 2012, 338, 72–75. [Google Scholar] [CrossRef] [Green Version]
- Lyons, D.M.; Buckmaster, P.S.; Lee, A.G.; Wu, C.; Mitra, R.; Duffey, L.M.; Buckmaster, C.L.; Her, S.; Patel, P.D.; Schatzberg, A.F. Stress coping stimulates hippocampal neurogenesis in adult monkeys. Proc. Natl. Acad. Sci. USA 2010, 107, 14823–14827. [Google Scholar] [CrossRef] [Green Version]
- Parihar, V.K.; Hattiangady, B.; Kuruba, R.; Shuai, B.; Shetty, A.K. Predictable chronic mild stress improves mood, hippocampal neurogenesis and memory. Mol. Psychiatry 2011, 16, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Petrik, D.; Lagace, D.C.; Eisch, A.J. The neurogenesis hypothesis of affective and anxiety disorders: Are we mistaking the scaffolding for the building? Neuropharmacology 2012, 62, 21–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspi, A.; Sugden, K.; Moffitt, T.E.; Taylor, A.; Craig, I.W.; Harrington, H.; McClay, J.; Mill, J.; Martin, J.; Braithwaite, A.; et al. Influence of life stress on depression: Moderation by a polymorphism in the 5-HTT gene. Science 2003, 301, 386–389. [Google Scholar] [CrossRef]
- Heuser, I. Anna-Monika-Prize paper. The hypothalamic-pituitary-adrenal system in depression. Pharmacopsychiatry 1998, 31, 10–13. [Google Scholar] [CrossRef]
- Mizoguchi, K.; Ishige, A.; Aburada, M.; Tabira, T. Chronic stress attenuates glucocorticoid negative feedback: Involvement of the prefrontal cortex and hippocampus. Neuroscience 2003, 119, 887–897. [Google Scholar] [CrossRef]
- Herman, J.P.; Spencer, R. Regulation of hippocampal glucocorticoid receptor gene transcription and protein expression in vivo. J. Neurosci. 1998, 18, 7462–7473. [Google Scholar] [CrossRef] [Green Version]
- Gould, E.; McEwen, B.S.; Tanapat, P.; Galea, L.A.; Fuchs, E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J. Neurosci. 1997, 17, 2492–2498. [Google Scholar] [CrossRef]
- Malberg, J.E.; Duman, R.S. Cell proliferation in adult hippocampus is decreased by inescapable stress: Reversal by fluoxetine treatment. Neuropsychopharmacology 2003, 28, 1562–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, K.; Nacher, J.; Hof, P.R.; McEwen, B.S. Repeated restraint stress suppresses neurogenesis and induces biphasic PSA-NCAM expression in the adult rat dentate gyrus. Eur. J. Neurosci. 2003, 17, 879–886. [Google Scholar] [CrossRef]
- You, Z.; Luo, C.; Zhang, W.; Chen, Y.; He, J.; Zhao, Q.; Zuo, R.; Wu, Y. Pro- and anti-inflammatory cytokines expression in rat’s brain and spleen exposed to chronic mild stress: Involvement in depression. Behav. Brain Res. 2011, 225, 135–141. [Google Scholar] [CrossRef]
- Anacker, C.; Cattaneo, A.; Luoni, A.; Musaelyan, K.; Zunszain, P.A.; Milanesi, E.; Rybka, J.; Berry, A.; Cirulli, F.; Thuret, S.; et al. Glucocorticoid-related molecular signaling pathways regulating hippocampal neurogenesis. Neuropsychopharmacology 2013, 38, 872–883. [Google Scholar] [CrossRef]
- Bremner, J.D.; Elzinga, B.; Schmahl, C.; Vermetten, E. Structural and functional plasticity of the human brain in posttraumatic stress disorder. Prog. Brain Res. 2008, 167, 171–186. [Google Scholar] [CrossRef] [Green Version]
- Gould, E.; Cameron, H.A.; Daniels, D.C.; Woolley, C.S.; McEwen, B.S. Adrenal hormones suppress cell division in the adult rat dentate gyrus. J. Neurosci. 1992, 12, 3642–3650. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.L.; van Praag, H.; Gage, F.H. Adult brain neurogenesis and psychiatry: A novel theory of depression. Mol. Psychiatry 2000, 5, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Nakashiba, T.; Cushman, J.D.; Pelkey, K.A.; Renaudineau, S.; Buhl, D.L.; McHugh, T.J.; Rodriguez Barrera, V.; Chittajallu, R.; Iwamoto, K.S.; McBain, C.J.; et al. Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell 2012, 149, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Anacker, C.; Pariante, C.M. Can adult neurogenesis buffer stress responses and depressive behaviour? Mol. Psychiatry 2012, 17, 9–10. [Google Scholar] [CrossRef]
- Hill, M.N.; Patel, S. Translational evidence for the involvement of the endocannabinoid system in stress-related psychiatric illnesses. Biol. Mood Anxiety Disord. 2013, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Zhou, X.; Pandey, N.R.; Vecchiarelli, H.A.; Stewart, C.A.; Zhang, X.; Lagace, D.C.; Brunel, J.M.; Beique, J.C.; Stewart, A.F.; et al. Chronic stress induces anxiety via an amygdalar intracellular cascade that impairs endocannabinoid signaling. Neuron 2015, 85, 1319–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremner, J.D.; Narayan, M.; Anderson, E.R.; Staib, L.H.; Miller, H.L.; Charney, D.S. Hippocampal volume reduction in major depression. Am. J. Psychiatry 2000, 157, 115–118. [Google Scholar] [CrossRef] [PubMed]
- MacQueen, G.M.; Campbell, S.; McEwen, B.S.; Macdonald, K.; Amano, S.; Joffe, R.T.; Nahmias, C.; Young, L.T. Course of illness, hippocampal function, and hippocampal volume in major depression. Proc. Natl. Acad. Sci. USA 2003, 100, 1387–1392. [Google Scholar] [CrossRef] [Green Version]
- Sheline, Y.I.; Wang, P.W.; Gado, M.H.; Csernansky, J.G.; Vannier, M.W. Hippocampal atrophy in recurrent major depression. Proc. Natl. Acad. Sci. USA 1996, 93, 3908–3913. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.W.; Yu, Y.; Gu, L. Dehydroabietic acid reverses TNF-alpha-induced the activation of FOXO1 and suppression of TGF-beta1/Smad signaling in human adult dermal fibroblasts. Int. J. Clin. Exp. Pathol. 2014, 7, 8616–8626. [Google Scholar]
- Cattaneo, A.; Cattane, N.; Malpighi, C.; Czamara, D.; Suarez, A.; Mariani, N.; Kajantie, E.; Luoni, A.; Eriksson, J.G.; Lahti, J.; et al. FoxO1, A2M, and TGF-beta1: Three novel genes predicting depression in gene X environment interactions are identified using cross-species and cross-tissues transcriptomic and miRNomic analyses. Mol. Psychiatry 2018, 23, 2192–2208. [Google Scholar] [CrossRef] [PubMed]
- Etkin, A.; Prater, K.E.; Schatzberg, A.F.; Menon, V.; Greicius, M.D. Disrupted amygdalar subregion functional connectivity and evidence of a compensatory network in generalized anxiety disorder. Arch. Gen. Psychiatry 2009, 66, 1361–1372. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Loucks, R.A.; Palmer, A.L.; Brown, A.C.; Solomon, K.M.; Marchante, A.N.; Whalen, P.J. The structural and functional connectivity of the amygdala: From normal emotion to pathological anxiety. Behav. Brain Res. 2011, 223, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Roelke, C.T.; Rademacher, D.J.; Cullinan, W.E.; Hillard, C.J. Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology 2004, 145, 5431–5438. [Google Scholar] [CrossRef] [PubMed]
- Shonesy, B.C.; Bluett, R.J.; Ramikie, T.S.; Baldi, R.; Hermanson, D.J.; Kingsley, P.J.; Marnett, L.J.; Winder, D.G.; Colbran, R.J.; Patel, S. Genetic disruption of 2-arachidonoylglycerol synthesis reveals a key role for endocannabinoid signaling in anxiety modulation. Cell Rep. 2014, 9, 1644–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunduz-Cinar, O.; Hill, M.N.; McEwen, B.S.; Holmes, A. Amygdala FAAH and anandamide: Mediating protection and recovery from stress. Trends Pharmacol. Sci. 2013, 34, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambien, F.; Ricard, S.; Troesch, A.; Mallet, C.; Generenaz, L.; Evans, A.; Arveiler, D.; Luc, G.; Ruidavets, J.B.; Poirier, O. Polymorphisms of the transforming growth factor-beta 1 gene in relation to myocardial infarction and blood pressure. The Etude Cas-Temoin de l’Infarctus du Myocarde (ECTIM) Study. Hypertension 1996, 28, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Deng, Y.; Miao, R.; Hu, Z.; Zhou, Y.; Tan, Y.; Wang, J.; Hua, Z.; Ding, W.; Wang, L.; et al. TGFB1 and TGFBR2 functional polymorphisms and risk of esophageal squamous cell carcinoma: A case-control analysis in a Chinese population. J. Cancer Res. Clin. Oncol. 2008, 134, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, S.H.; Choi, Y.L.; Wang, L.H.; Park, C.K.; Shin, Y.K. Extensive alteration in the expression profiles of TGFB pathway signaling components and TP53 is observed along the gastric dysplasia-carcinoma sequence. Histol. Histopathol. 2008, 23, 1439–1452. [Google Scholar] [CrossRef]
- Yan, C.; Yang, Q.; Gong, Z. Transgenic expression of tgfb1a induces hepatic inflammation, fibrosis and metastasis in zebrafish. Biochem. Biophys. Res. Commun. 2019, 509, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.L.; Blobe, G.C. Role of transforming growth factor Beta in human cancer. J. Clin. Oncol. 2005, 23, 2078–2093. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Shi, Y.; Massague, J. TGF-beta signaling and cancer: Structural and functional consequences of mutations in Smads. Mol. Med. Today 1998, 4, 257–262. [Google Scholar] [CrossRef]
- Jahn, S.C.; Law, M.E.; Corsino, P.E.; Law, B.K. TGF-beta antiproliferative effects in tumor suppression. Front. Biosci. (Schol. Ed.) 2012, 4, 749–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.U.; Kim, H.T.; Seong, D.H.; Kim, Y.S.; Park, Y.S.; Bang, Y.J.; Yang, H.K.; Kim, S.J. Loss of the Smad3 expression increases susceptibility to tumorigenicity in human gastric cancer. Oncogene 2004, 23, 1333–1341. [Google Scholar] [CrossRef] [Green Version]
- Boulanger, C.A.; Smith, G.H. Reducing mammary cancer risk through premature stem cell senescence. Oncogene 2001, 20, 2264–2272. [Google Scholar] [CrossRef] [Green Version]
- Pierce, D.F., Jr.; Gorska, A.E.; Chytil, A.; Meise, K.S.; Page, D.L.; Coffey, R.J., Jr.; Moses, H.L. Mammary tumor suppression by transforming growth factor beta 1 transgene expression. Proc. Natl. Acad. Sci. USA 1995, 92, 4254–4258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, P.M.; Shu, W.; Cardiff, R.D.; Muller, W.J.; Massague, J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc. Natl. Acad. Sci. USA 2003, 100, 8430–8435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckwalter, M.S.; Wyss-Coray, T. Modelling neuroinflammatory phenotypes in vivo. J. Neuroinflamm. 2004, 1, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finch, C.E.; Laping, N.J.; Morgan, T.E.; Nichols, N.R.; Pasinetti, G.M. TGF-beta 1 is an organizer of responses to neurodegeneration. J. Cell. Biochem. 1993, 53, 314–322. [Google Scholar] [CrossRef]
- O’Brien, M.F.; Lenke, L.G.; Lou, J.; Bridwell, K.H.; Joyce, M.E. Astrocyte response and transforming growth factor-beta localization in acute spinal cord injury. Spine 1994, 19, 2321–2329. [Google Scholar] [CrossRef]
- Buss, A.; Pech, K.; Kakulas, B.A.; Martin, D.; Schoenen, J.; Noth, J.; Brook, G.A. TGF-beta1 and TGF-beta2 expression after traumatic human spinal cord injury. Spinal Cord 2008, 46, 364–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoletti, F.; Di Marco, R.; Patti, F.; Reggio, E.; Nicoletti, A.; Zaccone, P.; Stivala, F.; Meroni, P.L.; Reggio, A. Blood levels of transforming growth factor-beta 1 (TGF-beta1) are elevated in both relapsing remitting and chronic progressive multiple sclerosis (MS) patients and are further augmented by treatment with interferon-beta 1b (IFN-beta1b). Clin. Exp. Immunol. 1998, 113, 96–99. [Google Scholar] [CrossRef]
- Estrada, L.D.; Oliveira-Cruz, L.; Cabrera, D. Transforming Growth Factor Beta Type I Role in Neurodegeneration: Implications for Alzheimer s Disease. Curr. Protein Pept. Sci. 2018, 19, 1180–1188. [Google Scholar] [CrossRef]
- Chao, C.C.; Hu, S.; Frey, W.H., 2nd; Ala, T.A.; Tourtellotte, W.W.; Peterson, P.K. Transforming growth factor beta in Alzheimer’s disease. Clin. Diagn. Lab. Immunol. 1994, 1, 109–110. [Google Scholar] [CrossRef]
- Vawter, M.P.; Dillon-Carter, O.; Tourtellotte, W.W.; Carvey, P.; Freed, W.J. TGFbeta1 and TGFbeta2 concentrations are elevated in Parkinson’s disease in ventricular cerebrospinal fluid. Exp. Neurol. 1996, 142, 313–322. [Google Scholar] [CrossRef]
- Saunier, E.F.; Akhurst, R.J. TGF beta inhibition for cancer therapy. Curr. Cancer Drug Targets 2006, 6, 565–578. [Google Scholar] [CrossRef]
- Prud’homme, G.J. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab. Investig. 2007, 87, 1077–1091. [Google Scholar] [CrossRef] [Green Version]
- Ruzek, M.C.; Hawes, M.; Pratt, B.; McPherson, J.; Ledbetter, S.; Richards, S.M.; Garman, R.D. Minimal effects on immune parameters following chronic anti-TGF-beta monoclonal antibody administration to normal mice. Immunopharmacol. Immunotoxicol. 2003, 25, 235–257. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.A.; Dukhanina, O.; Tang, B.; Mamura, M.; Letterio, J.J.; MacGregor, J.; Patel, S.C.; Khozin, S.; Liu, Z.Y.; Green, J.; et al. Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. J. Clin. Investig. 2002, 109, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Forrester, E.; Chytil, A.; Bierie, B.; Aakre, M.; Gorska, A.E.; Sharif-Afshar, A.R.; Muller, W.J.; Moses, H.L. Effect of conditional knockout of the type II TGF-beta receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res. 2005, 65, 2296–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss-Coray, T. Tgf-Beta pathway as a potential target in neurodegeneration and Alzheimer’s. Curr. Alzheimer Res. 2006, 3, 191–195. [Google Scholar] [CrossRef]
- Sahay, A.; Drew, M.R.; Hen, R. Dentate gyrus neurogenesis and depression. Prog. Brain Res. 2007, 163, 697–722. [Google Scholar] [CrossRef]
- Sahay, A.; Hen, R. Adult hippocampal neurogenesis in depression. Nat. Neurosci. 2007, 10, 1110–1115. [Google Scholar] [CrossRef]
- Vollmayr, B.; Mahlstedt, M.M.; Henn, F.A. Neurogenesis and depression: What animal models tell us about the link. Eur. Arch. Psychiatry Clin. Neurosci. 2007, 257, 300–303. [Google Scholar] [CrossRef]
- Gold, P.W.; Goodwin, F.K.; Chrousos, G.P. Clinical and biochemical manifestations of depression. Relation to the neurobiology of stress (2). N. Engl. J. Med. 1988, 319, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Sapolsky, R.M. The possibility of neurotoxicity in the hippocampus in major depression: A primer on neuron death. Biol. Psychiatry 2000, 48, 755–765. [Google Scholar] [CrossRef]
- Davidson, R.J.; Lewis, D.A.; Alloy, L.B.; Amaral, D.G.; Bush, G.; Cohen, J.D.; Drevets, W.C.; Farah, M.J.; Kagan, J.; McClelland, J.L.; et al. Neural and behavioral substrates of mood and mood regulation. Biol. Psychiatry 2002, 52, 478–502. [Google Scholar] [CrossRef]
- Czeh, B.; Michaelis, T.; Watanabe, T.; Frahm, J.; de Biurrun, G.; van Kampen, M.; Bartolomucci, A.; Fuchs, E. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc. Natl. Acad. Sci. USA 2001, 98, 12796–12801. [Google Scholar] [CrossRef] [Green Version]
- Anacker, C.; Zunszain, P.A.; Cattaneo, A.; Carvalho, L.A.; Garabedian, M.J.; Thuret, S.; Price, J.; Pariante, C.M. Antidepressants increase human hippocampal neurogenesis by activating the glucocorticoid receptor. Mol. Psychiatry 2011, 16, 738–750. [Google Scholar] [CrossRef] [Green Version]
- Perera, T.D.; Coplan, J.D.; Lisanby, S.H.; Lipira, C.M.; Arif, M.; Carpio, C.; Spitzer, G.; Santarelli, L.; Scharf, B.; Hen, R.; et al. Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J. Neurosci. 2007, 27, 4894–4901. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003, 301, 805–809. [Google Scholar] [CrossRef] [Green Version]
- Vollmayr, B.; Simonis, C.; Weber, S.; Gass, P.; Henn, F. Reduced cell proliferation in the dentate gyrus is not correlated with the development of learned helplessness. Biol. Psychiatry 2003, 54, 1035–1040. [Google Scholar] [CrossRef]
- Czeh, B.; Welt, T.; Fischer, A.K.; Erhardt, A.; Schmitt, W.; Muller, M.B.; Toschi, N.; Fuchs, E.; Keck, M.E. Chronic psychosocial stress and concomitant repetitive transcranial magnetic stimulation: Effects on stress hormone levels and adult hippocampal neurogenesis. Biol. Psychiatry 2002, 52, 1057–1065. [Google Scholar] [CrossRef]
- Lee, K.J.; Kim, S.J.; Kim, S.W.; Choi, S.H.; Shin, Y.C.; Park, S.H.; Moon, B.H.; Cho, E.; Lee, M.S.; Choi, S.H.; et al. Chronic mild stress decreases survival, but not proliferation, of new-born cells in adult rat hippocampus. Exp. Mol. Med. 2006, 38, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Bennett, A.J.; Lesch, K.P.; Heils, A.; Long, J.C.; Lorenz, J.G.; Shoaf, S.E.; Champoux, M.; Suomi, S.J.; Linnoila, M.V.; Higley, J.D. Early experience and serotonin transporter gene variation interact to influence primate CNS function. Mol. Psychiatry 2002, 7, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Coe, C.L.; Kramer, M.; Czeh, B.; Gould, E.; Reeves, A.J.; Kirschbaum, C.; Fuchs, E. Prenatal stress diminishes neurogenesis in the dentate gyrus of juvenile rhesus monkeys. Biol. Psychiatry 2003, 54, 1025–1034. [Google Scholar] [CrossRef]
- Kaufman, J.; Yang, B.Z.; Douglas-Palumberi, H.; Grasso, D.; Lipschitz, D.; Houshyar, S.; Krystal, J.H.; Gelernter, J. Brain-derived neurotrophic factor-5-HTTLPR gene interactions and environmental modifiers of depression in children. Biol. Psychiatry 2006, 59, 673–680. [Google Scholar] [CrossRef]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Bostrom, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. Dynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldrini, M.; Fulmore, C.A.; Tartt, A.N.; Simeon, L.R.; Pavlova, I.; Poposka, V.; Rosoklija, G.B.; Stankov, A.; Arango, V.; Dwork, A.J.; et al. Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell 2018, 22, 589–599.e585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toda, T.; Parylak, S.L.; Linker, S.B.; Gage, F.H. The role of adult hippocampal neurogenesis in brain health and disease. Mol. Psychiatry 2019, 24, 67–87. [Google Scholar] [CrossRef]
- Snyder, J.S.; Soumier, A.; Brewer, M.; Pickel, J.; Cameron, H.A. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 2011, 476, 458–461. [Google Scholar] [CrossRef]
- Jung, S.; Choe, S.; Woo, H.; Jeong, H.; An, H.K.; Moon, H.; Ryu, H.Y.; Yeo, B.K.; Lee, Y.W.; Choi, H.; et al. Autophagic death of neural stem cells mediates chronic stress-induced decline of adult hippocampal neurogenesis and cognitive deficits. Autophagy 2020, 16, 512–530. [Google Scholar] [CrossRef] [Green Version]
- Neumeister, A.; Wood, S.; Bonne, O.; Nugent, A.C.; Luckenbaugh, D.A.; Young, T.; Bain, E.E.; Charney, D.S.; Drevets, W.C. Reduced hippocampal volume in unmedicated, remitted patients with major depression versus control subjects. Biol. Psychiatry 2005, 57, 935–937. [Google Scholar] [CrossRef]
- Sivakumar, P.T.; Kalmady, S.V.; Venkatasubramanian, G.; Bharath, S.; Reddy, N.N.; Rao, N.P.; Kovoor, J.M.; Jain, S.; Varghese, M. Volumetric analysis of hippocampal sub-regions in late onset depression: A 3 tesla magnetic resonance imaging study. Asian J. Psychiatry 2015, 13, 38–43. [Google Scholar] [CrossRef]
- Pinto, V.; Costa, J.C.; Morgado, P.; Mota, C.; Miranda, A.; Bravo, F.V.; Oliveira, T.G.; Cerqueira, J.J.; Sousa, N. Differential impact of chronic stress along the hippocampal dorsal-ventral axis. Brain Struct. Funct. 2015, 220, 1205–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahima, R.S.; Harlan, R.E. Charting of type II glucocorticoid receptor-like immunoreactivity in the rat central nervous system. Neuroscience 1990, 39, 579–604. [Google Scholar] [CrossRef]
- Aggleton, J.P. A description of the amygdalo-hippocampal interconnections in the macaque monkey. Exp. Brain Res. 1986, 64, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Pikkarainen, M.; Ronkko, S.; Savander, V.; Insausti, R.; Pitkanen, A. Projections from the lateral, basal, and accessory basal nuclei of the amygdala to the hippocampal formation in rat. J. Comp. Neurol. 1999, 403, 229–260. [Google Scholar] [CrossRef]
- Czeh, B.; Simon, M.; Schmelting, B.; Hiemke, C.; Fuchs, E. Astroglial plasticity in the hippocampus is affected by chronic psychosocial stress and concomitant fluoxetine treatment. Neuropsychopharmacology 2006, 31, 1616–1626. [Google Scholar] [CrossRef] [Green Version]
- Czeh, B.; Varga, Z.K.; Henningsen, K.; Kovacs, G.L.; Miseta, A.; Wiborg, O. Chronic stress reduces the number of GABAergic interneurons in the adult rat hippocampus, dorsal-ventral and region-specific differences. Hippocampus 2015, 25, 393–405. [Google Scholar] [CrossRef]
- Cobb, J.A.; O’Neill, K.; Milner, J.; Mahajan, G.J.; Lawrence, T.J.; May, W.L.; Miguel-Hidalgo, J.; Rajkowska, G.; Stockmeier, C.A. Density of GFAP-immunoreactive astrocytes is decreased in left hippocampi in major depressive disorder. Neuroscience 2016, 316, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Czeh, B.; Abumaria, N.; Rygula, R.; Fuchs, E. Quantitative changes in hippocampal microvasculature of chronically stressed rats: No effect of fluoxetine treatment. Hippocampus 2010, 20, 174–185. [Google Scholar] [CrossRef]
- Rajkowska, G.; Stockmeier, C.A. Astrocyte pathology in major depressive disorder: Insights from human postmortem brain tissue. Curr. Drug Targets 2013, 14, 1225–1236. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Signalling Molecule | Models | Behavioural and Physiological Changes | Mechanisms | References |
|---|---|---|---|---|
| TGF-β | Receptor inhibition in vivo (C57BL6 Mouse) | Increased neurogenesis | Reduction of inflammatory response mediated by B2M through attenuation of pSmad3 activity | [50] |
| Knockout in vivo (C57BL6 Mouse) | Increased neuronal degeneration and microgliosis | TGF-β-related decrease in laminin-reduced survivability and increased susceptibility to apoptosis | [46] | |
| Chronic upregulation of TGF-β1 in vivo (C57BL6 Mouse) | Decreased immature hippocampal neurones and neurogenesis | Induced early cell cycle exit of neural progenitor cells | [54] | |
| Exogenous upregulation of TGF-β2 in vitro (Sprague-Dawley Rat Hippocampus) | Induction of evoked post-synaptic currents and inhibition of miniature post-synaptic currents | TGF-β-related upregulation of CREB in hippocampal neurones | [35] | |
| Receptor knockout in vivo (C57BL6 Mouse) | Reduction of immature neurones and neurogenesis | Increased expression of pro-apoptotic effectors; decreased expression of anti-apoptotic effectors | [55] | |
| Smad3 | Knockout in vivo (C57BL6 Mouse) | Reduction of Neurogenesis | Disruption of neuronal proliferation and migration | [49] |
| Inhibition of long-term potentiation | Impairment of NMDA activity by Smad3-related increase in GABAergic signalling | [52]) | ||
| Rostral increase of proliferative cells; caudal decrease in neurogenesis | Potential compensatory mechanism in rostral DG to maintain cell numbers; increased apoptosis at intermediate cell stage reduces neurogenesis | [58] | ||
| Decreased neuronal viability following injury | Disruption of Smad3 signalling in astrocytes | [59] | ||
| Accelerated wound closure and decreased activation of microglia | Reduced expression of MCP-1 and reduced leukocyte activity | [60] | ||
| Transient knockdown in vivo (Chick Embryo) | Decreased neurogenesis | Preferential activation of Smad2 targets due to loss of Smad3 activity | [48] | |
| Smad2 | Transient knockdown in vivo (Chick Embryo) | Increased neurogenesis | Preferential activation of Smad3 targets due to loss of Smad2 activity | [48] |
| Braf | Knockout in vivo (129S1/Sv + C57BL6 Mouse) | Increase of depressive-like behaviour in adults, decrease of anxiety in juveniles; reduction of dendritic spine growth | Disturbance of Erk/MAP signalling and alteration of serotonergic transmission | [33] |
| CREB | Inhibition by dominant negative mutant in vivo (129SvEv + C57BL6 Mouse) | Anti-depressant effects mediated by increase in neurogenesis | Potential mCREB interaction with non-CREB targets | [38] |
| Decreased granule cell proliferation | Disruption of cAMP-CREB signalling | [39] | ||
| Conditional knockout in vivo (129SvEv + C57BL6 Mouse) | Impairment of performance in spatial retention | Upregulation of CREB | [36] | |
| Transient overexpression of CREB in vivo (Sprague-Dawley Rat) | Reduction of depressive-like behaviours | Improved adaptation due to CREB-related regulation of granule cells | [41] | |
| Gadd45b | Knockout in vivo (Mouse) | Reduction of the effectiveness of ECT in inducing neurogenesis and dendritic spine growth | Attenuation of Gadd45b-mediated demethylation in regulatory regions of BDNF and FGF1 | [61] |
| JMJD3 | Knockout in vivo (Mouse) | Disruption of neuronal migration and reduction of neurogenesis | Reduction of Dlx2 activation and H3K27me3 demethylation | [62] |
| Disruption of TGF-β/Smad signalling cascade | [63] | |||
| MAPK | Transient inhibition in vivo (Mouse) | Increased behavioural despair and reduced effectiveness of anti-depressant treatment | Disruption of MEK-ERK signalling | [31] |
| TAK1 | Knockdown in vivo (Mouse) | Reduction of axonal length | Impairment of JNK activity mediated by TAK1 | [64] |
| TrkB | Conditional knockout in vivo (Mouse) | Impairment of neurogenesis; resistance to anti-depressant treatment | Blockade of BDNF signalling | [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hiew, L.-F.; Poon, C.-H.; You, H.-Z.; Lim, L.-W. TGF-β/Smad Signalling in Neurogenesis: Implications for Neuropsychiatric Diseases. Cells 2021, 10, 1382. https://doi.org/10.3390/cells10061382
Hiew L-F, Poon C-H, You H-Z, Lim L-W. TGF-β/Smad Signalling in Neurogenesis: Implications for Neuropsychiatric Diseases. Cells. 2021; 10(6):1382. https://doi.org/10.3390/cells10061382
Chicago/Turabian StyleHiew, Lih-Fhung, Chi-Him Poon, Heng-Ze You, and Lee-Wei Lim. 2021. "TGF-β/Smad Signalling in Neurogenesis: Implications for Neuropsychiatric Diseases" Cells 10, no. 6: 1382. https://doi.org/10.3390/cells10061382
APA StyleHiew, L. -F., Poon, C. -H., You, H. -Z., & Lim, L. -W. (2021). TGF-β/Smad Signalling in Neurogenesis: Implications for Neuropsychiatric Diseases. Cells, 10(6), 1382. https://doi.org/10.3390/cells10061382