
Neuroprotection in Glaucoma: NAD+/NADH Redox State as a Potential Biomarker and Therapeutic Target

 , and
, and
Abstract
:1. Introduction
2. Glaucoma and Mitochondrial Function
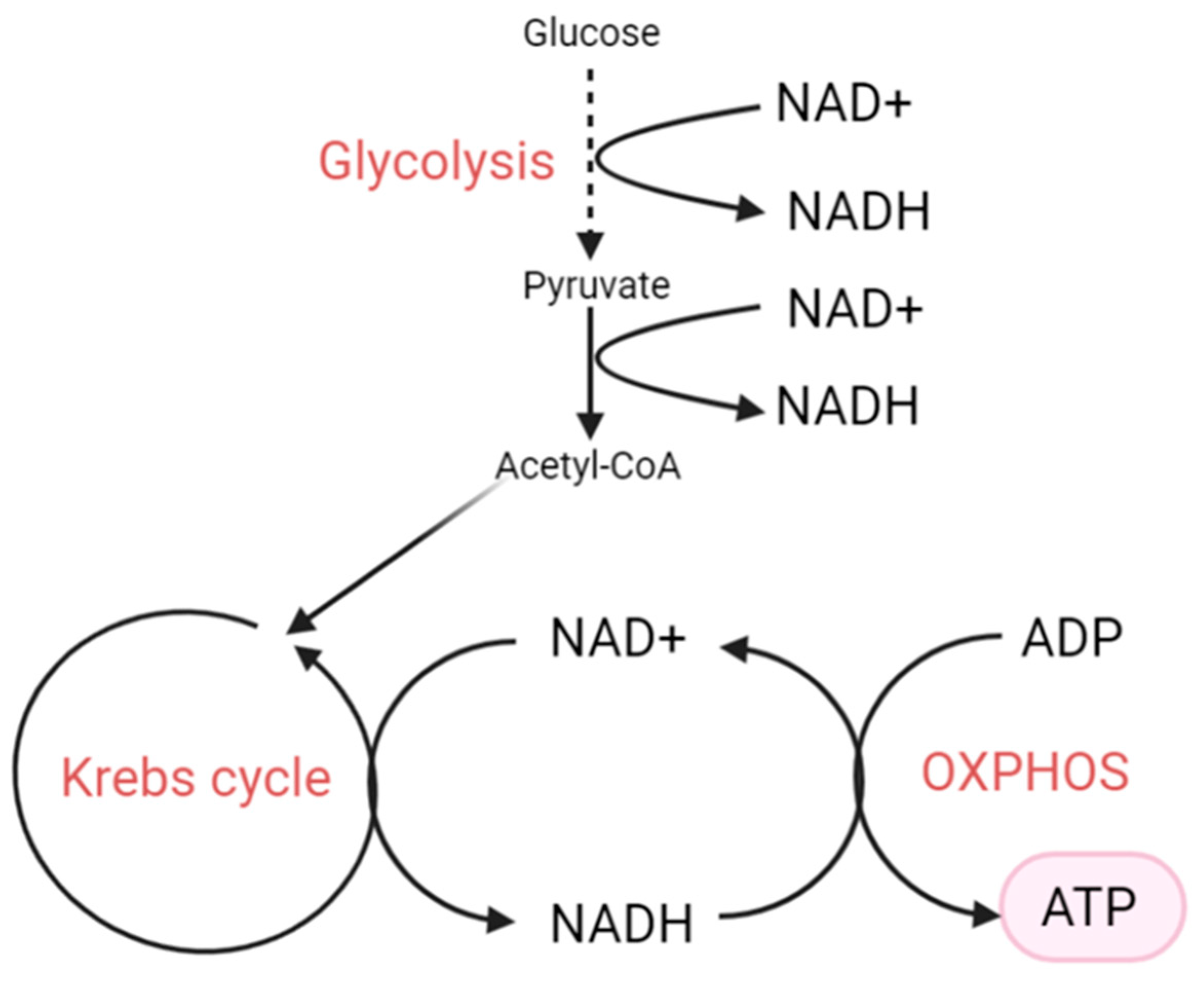
3. NAD+, NADH and Their Biological Functions
4. The NAD+/NADH Redox State
5. NAD+ Depletion
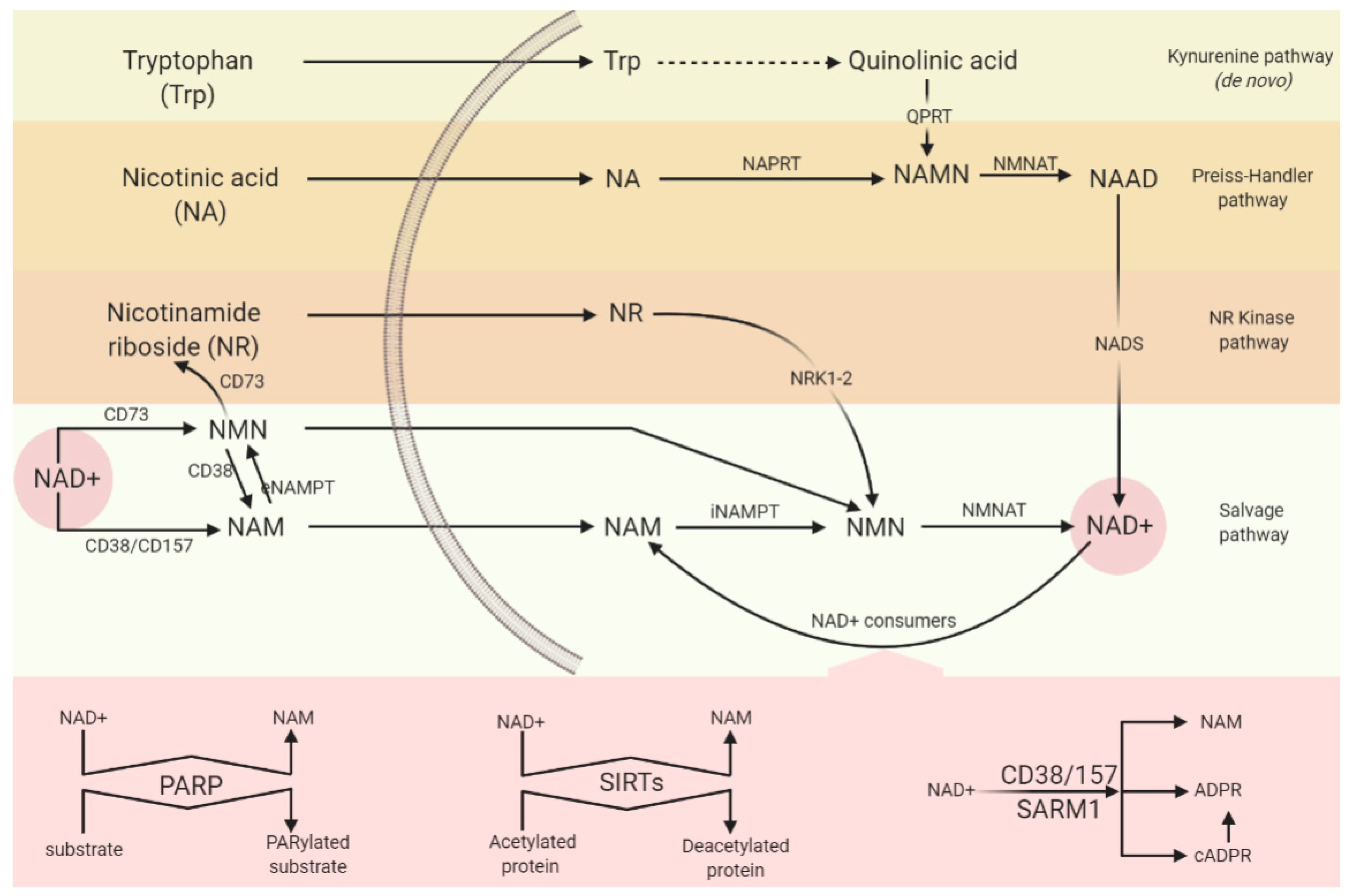
5.1. Synthesising Enzymes
5.1.1. NAM Salvage Pathway
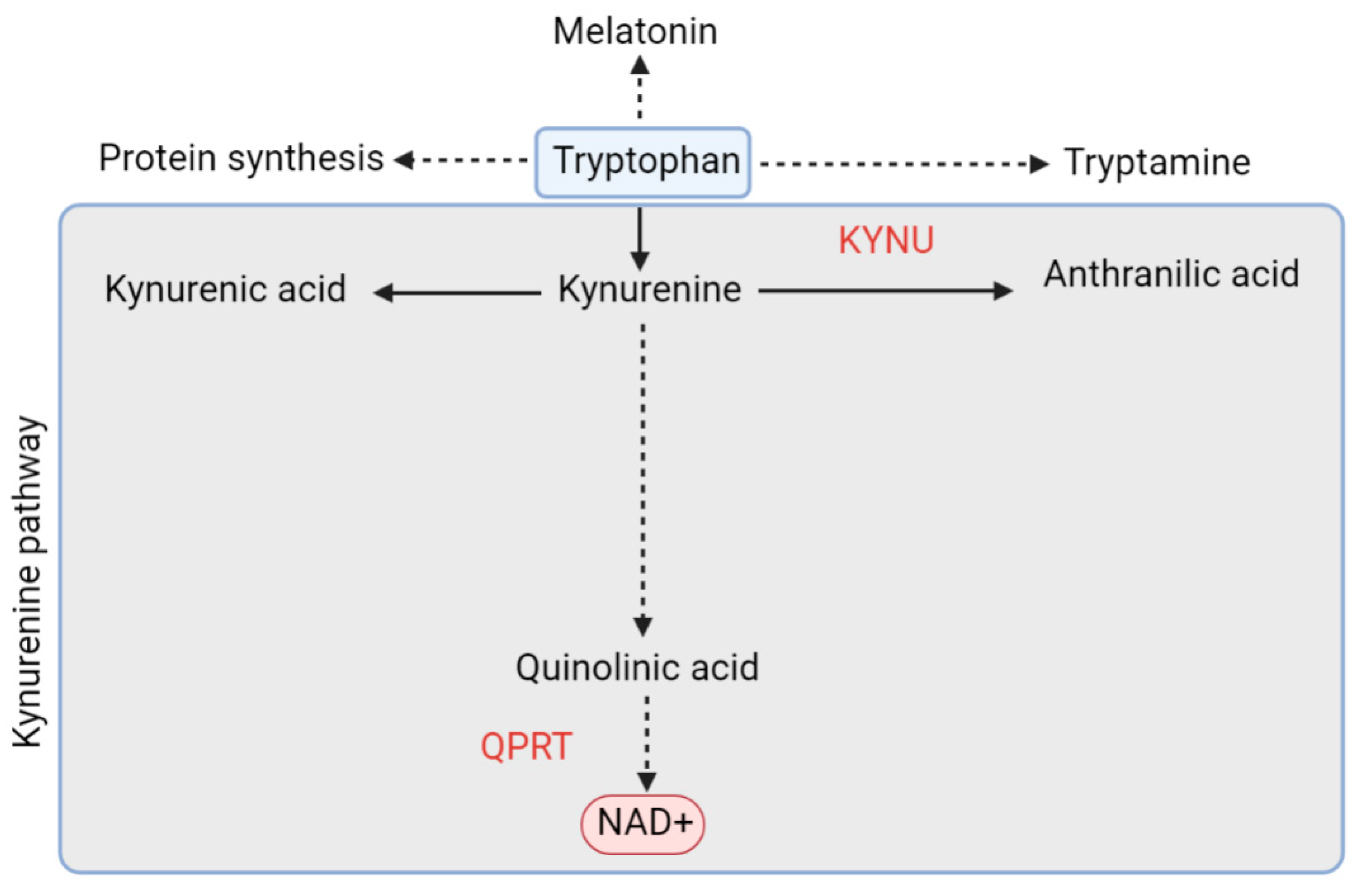
5.1.2. De Novo Pathway and QPRT
5.2. Consuming Enzymes
5.2.1. PARP
5.2.2. CD38/CD157
5.2.3. SARM1
5.2.4. Sirtuins
6. Increased NADH
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef]
- King, A.; Azuara-Blanco, A.; Tuulonen, A. Glaucoma. BMJ Br. Med. J. 2013, 346, f3518. [Google Scholar] [CrossRef]
- Agorastos, A.; Skevas, C.; Matthaei, M.; Otte, C.; Klemm, M.; Richard, G.; Huber, C.G. Depression, Anxiety, and Disturbed Sleep in Glaucoma. J. Neuropsychiatry Clin. Neurosci. 2013, 25, 205–213. [Google Scholar] [CrossRef]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Yepez, V.A.; Kremer, L.S.; Iuso, A.; Gusic, M.; Kopajtich, R.; Konarikova, E.; Nadel, A.; Wachutka, L.; Prokisch, H.; Gagneur, J. OCR-Stats: Robust estimation and statistical testing of mitochondrial respiration activities using Seahorse XF Analyzer. PLoS ONE 2018, 13, e0199938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD(+) in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal. Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Drance, S.; Anderson, D.R.; Schulzer, M.; Collaborative Normal-Tension Glaucoma Study, G. Risk factors for progression of visual field abnormalities in normal-tension glaucoma. Am. J. Ophthalmol. 2001, 131, 699–708. [Google Scholar] [CrossRef]
- Peters, D.; Bengtsson, B.; Heijl, A. Lifetime risk of blindness in open-angle glaucoma. Am. J. Ophthalmol. 2013, 156, 724–730. [Google Scholar] [CrossRef] [Green Version]
- Konieczka, K.; Choi, H.J.; Koch, S.; Fankhauser, F.; Schoetzau, A.; Kim, D.M. Relationship between normal tension glaucoma and Flammer syndrome. Epma J. 2017, 8, 111–117. [Google Scholar] [CrossRef]
- Downs, J.C. Optic nerve head biomechanics in aging and disease. Exp. Eye Res. 2015, 133, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Casson, R.J.; Chidlow, G.; Crowston, J.G.; Williams, P.A.; Wood, J.P.M. Retinal energy metabolism in health and glaucoma. Prog. Retin. Eye Res. 2020, 100881. [Google Scholar] [CrossRef]
- Wang, L.; Dong, J.; Cull, G.; Fortune, B.; Cioffi, G.A. Varicosities of intraretinal ganglion cell axons in human and nonhuman primates. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Morán, M.; Moreno-Lastres, D.; Marín-Buera, L.; Arenas, J.; Martín, M.A.; Ugalde, C. Mitochondrial respiratory chain dysfunction: Implications in neurodegeneration. Free Radic. Biol. Med. 2012, 53, 595–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genc, S.; Kurnaz, I.A.; Ozilgen, M. Astrocyte - neuron lactate shuttle may boost more ATP supply to the neuron under hypoxic conditions - in silico study supported by in vitro expression data. BMC Syst. Biol. 2011, 5, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.; Khaw, P.T.; Yin, Z.Q.; Li, D.; Raisman, G.; Li, Y. Structural basis of glaucoma: The fortified astrocytes of the optic nerve head are the target of raised intraocular pressure. Glia 2012, 60, 13–28. [Google Scholar] [CrossRef]
- Yu, D.-Y.; Cringle, S.J. Oxygen Distribution and Consumption within the Retina in Vascularised and Avascular Retinas and in Animal Models of Retinal Disease. Prog. Retin. Eye Res. 2001, 20, 175–208. [Google Scholar] [CrossRef]
- Petriti, B.; Chau, D.; Lascaratos, G.; Campbell, P.; Lazaridis, G.; Garway-Heath, D.F. Normal Tension Glaucoma patients have reduced systemic mitochondrial function compared to High Tension Glaucoma patients [ARVO abstract]. Investig. Ophthalmol. Vis. Sci. 2020, 61, 1009. [Google Scholar]
- Braidy, N.; Grant, R.; Adams, S.; Brew, B.J.; Guillemin, G.J. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox. Res. 2009, 16, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Himori, N.; Yamamoto, K.; Maruyama, K.; Ryu, M.; Taguchi, K.; Yamamoto, M.; Nakazawa, T. Critical role of Nrf2 in oxidative stress-induced retinal ganglion cell death. J. Neurochem. 2013, 127, 669–680. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, Y.; Maruyama, K.; Yamamoto, K.; Omodaka, K.; Yasuda, M.; Himori, N.; Ryu, M.; Nishiguchi, K.M.; Nakazawa, T. The role of calpain in an in vivo model of oxidative stress-induced retinal ganglion cell damage. Biochem. Biophys. Res. Commun. 2014, 451, 510–515. [Google Scholar] [CrossRef]
- Titov, D.V.; Cracan, V.; Goodman, R.P.; Peng, J.; Grabarek, Z.; Mootha, V.K. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 2016, 352, 231–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [Green Version]
- Ju, W.-K.; Kim, K.-Y.; Lindsey, J.D.; Angert, M.; Patel, A.; Scott, R.T.; Liu, Q.; Crowston, J.G.; Ellisman, M.H.; Perkins, G.A.; et al. Elevated hydrostatic pressure triggers release of OPA1 and cytochrome C, and induces apoptotic cell death in differentiated RGC-5 cells. Mol. Vis. 2009, 15, 120–134. [Google Scholar] [PubMed]
- Aung, T.; Ocaka, L.; Ebenezer, N.D.; Morris, A.G.; Krawczak, M.; Thiselton, D.L.; Alexander, C.; Votruba, M.; Brice, G.; Child, A.H.; et al. A major marker for normal tension glaucoma: Association with polymorphisms in the OPA1 gene. Hum. Genet. 2002, 110, 52–56. [Google Scholar] [CrossRef]
- Powell, B.L.; Toomes, C.; Scott, S.; Yeung, A.; Marchbank, N.J.; Spry, P.G.; Lumb, R.; Inglehearn, C.F.; Churchill, A.J. Polymorphisms in OPA1 are associated with normal tension glaucoma. Mol. Vis. 2003, 9, 460–464. [Google Scholar]
- Yu-Wai-Man, P.; Stewart, J.D.; Hudson, G.; Andrews, R.M.; Griffiths, P.G.; Birch, M.K.; Chinnery, P.F. OPA1 increases the risk of normal but not high tension glaucoma. J. Med. Genet. 2010, 47, 120–125. [Google Scholar] [CrossRef] [Green Version]
- Mabuchi, F.; Tang, S.; Kashiwagi, K.; Yamagata, Z.; Iijima, H.; Tsukahara, S. The OPA1 gene polymorphism is associated with normal tension and high tension glaucoma. Am. J. Ophthalmol. 2007, 143, 125–130. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Morales, J.; Bosley, T.M. Mitochondrial abnormalities in patients with primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2533–2541. [Google Scholar] [CrossRef]
- Van Bergen, N.J.; Crowston, J.G.; Craig, J.E.; Burdon, K.P.; Kearns, L.S.; Sharma, S.; Hewitt, A.W.; Mackey, D.A.; Trounce, I.A. Measurement of Systemic Mitochondrial Function in Advanced Primary Open-Angle Glaucoma and Leber Hereditary Optic Neuropathy. PLoS ONE 2015, 10, e0140919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Sheck, L.; Crowston, J.G.; Van Bergen, N.J.; O’Neill, E.C.; O’Hare, F.; Kong, Y.X.; Chrysostomou, V.; Vincent, A.L.; Trounce, I.A. Impaired complex-I-linked respiration and ATP synthesis in primary open-angle glaucoma patient lymphoblasts. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2431–2437. [Google Scholar] [CrossRef] [Green Version]
- Wolf, C.; Gramer, E.; Muller-Myhsok, B.; Pasutto, F.; Reinthal, E.; Wissinger, B.; Weisschuh, N. Evaluation of nine candidate genes in patients with normal tension glaucoma: A case control study. BMC Med. Genet. 2009, 10, 91. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Dai, Y.; Zhang, R.; Shang, K.; Sun, X. Overexpression of Optic Atrophy Type 1 Protects Retinal Ganglion Cells and Upregulates Parkin Expression in Experimental Glaucoma. Front. Mol. Neurosci. 2018, 11, 350. [Google Scholar] [CrossRef] [PubMed]
- Khawaja, A.P.; Cooke Bailey, J.N.; Kang, J.H.; Allingham, R.R.; Hauser, M.A.; Brilliant, M.; Budenz, D.L.; Christen, W.G.; Fingert, J.; Gaasterland, D.; et al. Assessing the Association of Mitochondrial Genetic Variation With Primary Open-Angle Glaucoma Using Gene-Set Analyses. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5046–5052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, J.N.; Loomis, S.J.; Kang, J.H.; Allingham, R.R.; Gharahkhani, P.; Khor, C.C.; Burdon, K.P.; Aschard, H.; Chasman, D.I.; Igo, R.P., Jr.; et al. Genome-wide association analysis identifies TXNRD2, ATXN2 and FOXC1 as susceptibility loci for primary open-angle glaucoma. Nat. Genet. 2016, 48, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Khawaja, A.P.; Cooke Bailey, J.N.; Wareham, N.J.; Scott, R.A.; Simcoe, M.; Igo, R.P., Jr.; Song, Y.E.; Wojciechowski, R.; Cheng, C.Y.; Khaw, P.T.; et al. Genome-wide analyses identify 68 new loci associated with intraocular pressure and improve risk prediction for primary open-angle glaucoma. Nat. Genet. 2018, 50, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, P.; Simpson, D.A.; Sambare, C.; Duffy, S.; Lechner, J.; Dastane, A.; Dervan, E.W.; Vallabh, N.; Chelerkar, V.; Deshpande, M.; et al. Whole-mitochondrial genome sequencing in primary open-angle glaucoma using massively parallel sequencing identifies novel and known pathogenic variants. Genet. Med. 2015, 17, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Lascaratos, G.; Chau, K.Y.; Zhu, H.; Gkotsi, D.; King, R.; Gout, I.; Kamal, D.; Luthert, P.J.; Schapira, A.H.V.; Garway-Heath, D.F. Resistance to the most common optic neuropathy is associated with systemic mitochondrial efficiency. Neurobiol. Dis. 2015, 82, 78–85. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraenkl, S.A.; Muser, J.; Groell, R.; Reinhard, G.; Orgul, S.; Flammer, J.; Goldblum, D. Plasma citrate levels as a potential biomarker for glaucoma. J. Ocul. Pharmacol. Ther. 2011, 27, 577–580. [Google Scholar] [CrossRef]
- Goldblum, D.; Fraenkl, S.A.; Muser, J.; Groell, R.; Reinhard, G.; Flammer, J. Plasma Citrate Levels as a Biomarker for Glaucoma Diagnosis. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2669. [Google Scholar]
- Ferreira, S.M.; Lerner, S.F.; Brunzini, R.; Evelson, P.A.; Llesuy, S.F. Oxidative stress markers in aqueous humor of glaucoma patients. Am. J. Ophthalmol. 2004, 137, 62–69. [Google Scholar] [CrossRef]
- Izzotti, A.; Saccà, S.C.; Cartiglia, C.; De Flora, S. Oxidative deoxyribonucleic acid damage in the eyes of glaucoma patients. Am. J. Med. 2003, 114, 638–646. [Google Scholar] [CrossRef]
- Saccà, S.C.; Pascotto, A.; Camicione, P.; Capris, P.; Izzotti, A. Oxidative DNA damage in the human trabecular meshwork: Clinical correlation in patients with primary open-angle glaucoma. Arch. Ophthalmol. 2005, 123, 458–463. [Google Scholar] [CrossRef] [Green Version]
- Gherghel, D.; Griffiths, H.R.; Hilton, E.J.; Cunliffe, I.A.; Hosking, S.L. Systemic reduction in glutathione levels occurs in patients with primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2005, 46, 877–883. [Google Scholar] [CrossRef] [Green Version]
- Yildirim, O.; Ateş, N.A.; Ercan, B.; Muşlu, N.; Unlü, A.; Tamer, L.; Atik, U.; Kanik, A. Role of oxidative stress enzymes in open-angle glaucoma. Eye (Lond.) 2005, 19, 580–583. [Google Scholar] [CrossRef]
- Tanito, M.; Kaidzu, S.; Takai, Y.; Ohira, A. Status of systemic oxidative stresses in patients with primary open-angle glaucoma and pseudoexfoliation syndrome. PLoS ONE 2012, 7, e49680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorkhabi, R.; Ghorbanihaghjo, A.; Javadzadeh, A.; Rashtchizadeh, N.; Moharrery, M. Oxidative DNA damage and total antioxidant status in glaucoma patients. Mol. Vis. 2011, 17, 41–46. [Google Scholar]
- Yuki, K.; Murat, D.; Kimura, I.; Tsubota, K. Increased serum total antioxidant status and decreased urinary 8-hydroxy-2’-deoxyguanosine levels in patients with normal-tension glaucoma. Acta Ophthalmol 2010, 88, e259–e264. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhang, X.; Han, Y.Y.; Burke, N.A.; Kochanek, P.M.; Watkins, S.C.; Graham, S.H.; Carcillo, J.A.; Szabó, C.; Clark, R.S. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem. 2003, 278, 18426–18433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Chen, G. Flow injection analysis of trace amounts of NADH with inhibited chemiluminescent detection. Talanta 2002, 57, 961–967. [Google Scholar] [CrossRef]
- Braidy, N.; Guillemin, G.; Grant, R. Promotion of cellular NAD(+) anabolism: Therapeutic potential for oxidative stress in ageing and Alzheimer’s disease. Neurotox. Res. 2008, 13, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magni, G.; Amici, A.; Emanuelli, M.; Orsomando, G.; Raffaelli, N.; Ruggieri, S. Enzymology of NAD+ homeostasis in man. Cell. Mol. Life Sci. Cmls 2004, 61, 19–34. [Google Scholar] [CrossRef]
- Magni, G.; Amici, A.; Emanuelli, M.; Raffaelli, N.; Ruggieri, S. Enzymology of NAD+ synthesis. Adv. Enzymol. Relat. Areas Mol. Biol. 1999, 73, 135–182. [Google Scholar] [CrossRef]
- Wang, J.; He, Z. NAD and axon degeneration: From the Wlds gene to neurochemistry. Cell Adhes. Migr. 2009, 3, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Shats, I.; Williams, J.G.; Liu, J.; Makarov, M.V.; Wu, X.; Lih, F.B.; Deterding, L.J.; Lim, C.; Xu, X.; Randall, T.A.; et al. Bacteria Boost Mammalian Host NAD Metabolism by Engaging the Deamidated Biosynthesis Pathway. Cell Metab. 2020, 31, 564–579. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Cantó, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, F.; Hirrlinger, J. The NAD+ /NADH redox state in astrocytes: Independent control of the NAD+ and NADH content. J. Neurosci. Res. 2011, 89, 1956–1964. [Google Scholar] [CrossRef]
- Mathews, C.K.; Van Holde, K.E.; Ahern, K.G. Biochemistry, 3rd ed.; Benjamin Cummings: San Francisco, Calif, 2000. [Google Scholar]
- Lin, S.J.; Ford, E.; Haigis, M.; Liszt, G.; Guarente, L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004, 18, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.J.; Guarente, L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 2003, 15, 241–246. [Google Scholar] [CrossRef]
- Easlon, E.; Tsang, F.; Dilova, I.; Wang, C.; Lu, S.P.; Skinner, C.; Lin, S.J. The dihydrolipoamide acetyltransferase is a novel metabolic longevity factor and is required for calorie restriction-mediated life span extension. J. Biol. Chem. 2007, 282, 6161–6171. [Google Scholar] [CrossRef] [Green Version]
- Ellerby, L.M.; Ellerby, H.M.; Park, S.M.; Holleran, A.L.; Murphy, A.N.; Fiskum, G.; Kane, D.J.; Testa, M.P.; Kayalar, C.; Bredesen, D.E. Shift of the cellular oxidation-reduction potential in neural cells expressing Bcl-2. J. Neurochem. 1996, 67, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Piston, D.W.; Goodman, R.H. Regulation of Corepressor Function by Nuclear NADH. Science 2002, 295, 1895–1897. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Tarragó, M.G.; Chini, E.N. NAD and the aging process: Role in life, death and everything in between. Mol Cell Endocrinol. 2017, 455, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F. Mitophagy and NAD(+) inhibit Alzheimer disease. Autophagy 2019, 15, 1112–1114. [Google Scholar] [CrossRef] [Green Version]
- Schöndorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef]
- Yoshida, M.; Satoh, A.; Lin, J.B.; Mills, K.F.; Sasaki, Y.; Rensing, N.; Wong, M.; Apte, R.S.; Imai, S.I. Extracellular Vesicle-Contained eNAMPT Delays Aging and Extends Lifespan in Mice. Cell Metab. 2019, 30, 329–342. [Google Scholar] [CrossRef]
- Jia, H.; Li, X.; Gao, H.; Feng, Z.; Li, X.; Zhao, L.; Jia, X.; Zhang, H.; Liu, J. High doses of nicotinamide prevent oxidative mitochondrial dysfunction in a cellular model and improve motor deficit in a Drosophila model of Parkinson’s disease. J. Neurosci. Res. 2008, 86, 2083–2090. [Google Scholar] [CrossRef]
- Lehmann, S.; Loh, S.H.; Martins, L.M. Enhancing NAD(+) salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open 2017, 6, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, S.; Costa, A.C.; Celardo, I.; Loh, S.H.Y.; Martins, L.M. Parp mutations protect against mitochondrial dysfunction and neurodegeneration in a PARKIN model of Parkinson’s disease. Cell Death Dis. 2016, 7, e2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouassi Nzoughet, J.; Chao de la Barca, J.M.; Guehlouz, K.; Leruez, S.; Coulbault, L.; Allouche, S.; Bocca, C.; Muller, J.; Amati-Bonneau, P.; Gohier, P.; et al. Nicotinamide Deficiency in Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2509–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, F.; Tang, J.; Williams, P.; McGuinness, M.; Hadoux, X.; Casson, R.J.; Coote, M.; Trounce, I.; Martin, K.R.; van Wijngaarden, P.; et al. Improvement in inner retinal function in glaucoma in response to nicotinamide (Vitamin B3) supplementation: A crossover randomized clinical trial. Investig. Ophthalmol. Vis. Sci. 2020, 61, 3493. [Google Scholar]
- Gilley, J.; Coleman, M.P. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010, 8, e1000300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Su, X.; Quinn, W.J., 3rd; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhai, Q.; Chen, Y.; Lin, E.; Gu, W.; McBurney, M.W.; He, Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J. Cell Biol. 2005, 170, 349–355. [Google Scholar] [CrossRef] [Green Version]
- Song, S.B.; Park, J.S.; Chung, G.J.; Lee, I.H.; Hwang, E.S. Diverse therapeutic efficacies and more diverse mechanisms of nicotinamide. Metabolomics 2019, 15, 137. [Google Scholar] [CrossRef]
- Nikas, I.P.; Paschou, S.A.; Ryu, H.S. The Role of Nicotinamide in Cancer Chemoprevention and Therapy. Biomolecules 2020, 10, 477. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, C.A.; Brookes, P.S. Cellular Compartmentation and the Redox/Nonredox Functions of NAD. Antioxid. Redox Signal. 2019, 31, 623–642. [Google Scholar] [CrossRef]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, C.; Niere, M.; Ziegler, M. The NMN/NaMN adenylyltransferase (NMNAT) protein family. Front Biosci (Landmark Ed.) 2009, 14, 410–431. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Nakagawa, T.; Mao, X.; DiAntonio, A.; Milbrandt, J. NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD(+) depletion. eLife 2016, 5, e19749. [Google Scholar] [CrossRef] [PubMed]
- Zhai, R.G.; Zhang, F.; Hiesinger, P.R.; Cao, Y.; Haueter, C.M.; Bellen, H.J. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature 2008, 452, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 2004, 305, 1010–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conforti, L.; Wilbrey, A.; Morreale, G.; Janeckova, L.; Beirowski, B.; Adalbert, R.; Mazzola, F.; Di Stefano, M.; Hartley, R.; Babetto, E.; et al. Wld S protein requires Nmnat activity and a short N-terminal sequence to protect axons in mice. J. Cell Biol. 2009, 184, 491–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.J.; Zhang, Q.; Nakamaru-Ogiso, E.; Kannabiran, C.; Fonseca-Kelly, Z.; Chakarova, C.; Audo, I.; Mackay, D.S.; Zeitz, C.; Borman, A.D.; et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 2012, 44, 1040–1045. [Google Scholar] [CrossRef] [Green Version]
- Koenekoop, R.K.; Wang, H.; Majewski, J.; Wang, X.; Lopez, I.; Ren, H.; Chen, Y.; Li, Y.; Fishman, G.A.; Genead, M.; et al. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet. 2012, 44, 1035–1039. [Google Scholar] [CrossRef] [Green Version]
- Greenwald, S.H.; Brown, E.E.; Scandura, M.J.; Hennessey, E.; Farmer, R.; Pawlyk, B.S.; Xiao, R.; Vandenberghe, L.H.; Pierce, E.A. Gene Therapy Preserves Retinal Structure and Function in a Mouse Model of NMNAT1-Associated Retinal Degeneration. Mol. Ther. Methods Clin. Dev. 2020, 18, 582–594. [Google Scholar] [CrossRef]
- Milde, S.; Gilley, J.; Coleman, M.P. Subcellular localization determines the stability and axon protective capacity of axon survival factor Nmnat2. PLoS Biol. 2013, 11, e1001539. [Google Scholar] [CrossRef] [Green Version]
- Fulleylove-Krause, B.K.; Sison, S.L.; Ebert, A.D. Nicotinamide mononucleotide treatment increases NAD+ levels in an iPSC Model of Parkinson’s Disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Raff, M.C.; Whitmore, A.V.; Finn, J.T. Axonal self-destruction and neurodegeneration. Science 2002, 296, 868–871. [Google Scholar] [CrossRef]
- Lunn, E.R.; Perry, V.H.; Brown, M.C.; Rosen, H.; Gordon, S. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur. J. Neurosci. 1989, 1, 27–33. [Google Scholar] [CrossRef]
- Howell, G.R.; Libby, R.T.; Jakobs, T.C.; Smith, R.S.; Phalan, F.C.; Barter, J.W.; Barbay, J.M.; Marchant, J.K.; Mahesh, N.; Porciatti, V.; et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J. Cell Biol. 2007, 179, 1523–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beirowski, B.; Babetto, E.; Coleman, M.P.; Martin, K.R. The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur. J. Neurosci. 2008, 28, 1166–1179. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, L.; Sasaki, Y.; Milbrandt, J.; Gidday, J.M. Protection of mouse retinal ganglion cell axons and soma from glaucomatous and ischemic injury by cytoplasmic overexpression of Nmnat1. Investig. Ophthalmol. Vis. Sci. 2013, 54, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Conforti, L.; Gilley, J.; Coleman, M.P. Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 2014, 15, 394–409. [Google Scholar] [CrossRef]
- Mack, T.G.A.; Reiner, M.; Beirowski, B.; Mi, W.; Emanuelli, M.; Wagner, D.; Thomson, D.; Gillingwater, T.; Court, F.; Conforti, L.; et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci. 2001, 4, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Avery, M.A.; Sheehan, A.E.; Kerr, K.S.; Wang, J.; Freeman, M.R. Wld S requires Nmnat1 enzymatic activity and N16-VCP interactions to suppress Wallerian degeneration. J. Cell Biol. 2009, 184, 501–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laser, H.; Conforti, L.; Morreale, G.; Mack, T.G.; Heyer, M.; Haley, J.E.; Wishart, T.M.; Beirowski, B.; Walker, S.A.; Haase, G.; et al. The slow Wallerian degeneration protein, WldS, binds directly to VCP/p97 and partially redistributes it within the nucleus. Mol. Biol. Cell 2006, 17, 1075–1084. [Google Scholar] [CrossRef] [Green Version]
- Kitaoka, Y.; Munemasa, Y.; Kojima, K.; Hirano, A.; Ueno, S.; Takagi, H. Axonal protection by Nmnat3 overexpression with involvement of autophagy in optic nerve degeneration. Cell Death Dis. 2013, 4, e860. [Google Scholar] [CrossRef]
- Köhler, C.; Okuno, E.; Flood, P.R.; Schwarcz, R. Quinolinic acid phosphoribosyltransferase: Preferential glial localization in the rat brain visualized by immunocytochemistry. Proc. Natl. Acad. Sci. USA 1987, 84, 3491–3495. [Google Scholar] [CrossRef] [Green Version]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Brew, B.J. Implications of the kynurenine pathway and quinolinic acid in Alzheimer’s disease. Redox Rep. 2002, 7, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Eddleston, M.; Mucke, L. Molecular profile of reactive astrocytes--implications for their role in neurologic disease. Neuroscience 1993, 54, 15–36. [Google Scholar] [CrossRef]
- Neufeld, A.H.; Hernandez, M.R.; Gonzalez, M. Nitric oxide synthase in the human glaucomatous optic nerve head. Arch. Ophthalmol. 1997, 115, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.E. Optic nerve head structure in glaucoma: Astrocytes as mediators of axonal damage. Eye 2000, 14, 437–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedorowicz, M.; Choragiewicz, T.; Turski, W.A.; Kocki, T.; Nowakowska, D.; Wertejuk, K.; Kamińska, A.; Avitabile, T.; Wełniak-Kaminska, M.; Grieb, P.; et al. Tryptophan Pathway Abnormalities in a Murine Model of Hereditary Glaucoma. Int. J. Mol. Sci. 2021, 22, 1039. [Google Scholar] [CrossRef]
- Ferreira, F.S.; Biasibetti-Brendler, H.; Pierozan, P.; Schmitz, F.; Bertó, C.G.; Prezzi, C.A.; Manfredini, V.; Wyse, A.T.S. Kynurenic Acid Restores Nrf2 Levels and Prevents Quinolinic Acid-Induced Toxicity in Rat Striatal Slices. Mol. Neurobiol. 2018, 55, 8538–8549. [Google Scholar] [CrossRef]
- Rejdak, R.; Zarnowski, T.; Turski, W.A.; Kocki, T.; Zagorski, Z.; Zrenner, E.; Schuettauf, F. Alterations of kynurenic acid content in the retina in response to retinal ganglion cell damage. Vision Res. 2003, 43, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Amé, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weise, J.; Isenmann, S.; Bähr, M. Increased expression and activation of poly(ADP-ribose) polymerase (PARP) contribute to retinal ganglion cell death following rat optic nerve transection. Cell Death Differ. 2001, 8, 801–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, S.; Barber, R.; Wilcock, G.K. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain 1999, 122, 247–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandir, A.S.; Przedborski, S.; Jackson-Lewis, V.; Wang, Z.Q.; Simbulan-Rosenthal, C.M.; Smulson, M.E.; Hoffman, B.E.; Guastella, D.B.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc. Natl. Acad. Sci. USA 1999, 96, 5774–5779. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanon-Moreno, V.; Garcia-Medina, J.J.; Moreno-Nadal, M.A.; Vinuesa-Silva, I.; Pinazo-Duran, M.D. Cell Death Markers in Primary Open-Angle Glaucoma [ARVO abstract]. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3630. [Google Scholar]
- Salech, F.; Ponce, D.P.; Paula-Lima, A.C.; SanMartin, C.D.; Behrens, M.I. Nicotinamide, a Poly [ADP-Ribose] Polymerase 1 (PARP-1) Inhibitor, as an Adjunctive Therapy for the Treatment of Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12. [Google Scholar] [CrossRef]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.T.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef]
- Jin, D.; Liu, H.X.; Hirai, H.; Torashima, T.; Nagai, T.; Lopatina, O.; Shnayder, N.A.; Yamada, K.; Noda, M.; Seike, T.; et al. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 2007, 446, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, M.; Otsuka, N.; Sato, M.; Ishii, Y.; Kon, S.; Yamada, M.; Nishina, H.; Katada, T.; Ikeda, K. Neuronal localization of CD38 antigen in the human brain. Brain Res. 1995, 697, 235–240. [Google Scholar] [CrossRef]
- Verderio, C.; Bruzzone, S.; Zocchi, E.; Fedele, E.; Schenk, U.; De Flora, A.; Matteoli, M. Evidence of a role for cyclic ADP-ribose in calcium signalling and neurotransmitter release in cultured astrocytes. J. Neurochem. 2001, 78, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Moutin, M.J.; Lund, F.E.; Stein, R. Dual role of CD38 in microglial activation and activation-induced cell death. J. Immunol. 2008, 181, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Wang, H.; Wang, Q.; Deng, H. Overexpression of CD38 decreases cellular NAD levels and alters the expression of proteins involved in energy metabolism and antioxidant defense. J. Proteome Res. 2014, 13, 786–795. [Google Scholar] [CrossRef]
- Braidy, N.; Poljak, A.; Grant, R.; Jayasena, T.; Mansour, H.; Chan-Ling, T.; Guillemin, G.J.; Smythe, G.; Sachdev, P. Mapping NAD(+) metabolism in the brain of ageing Wistar rats: Potential targets for influencing brain senescence. Biogerontology 2014, 15, 177–198. [Google Scholar] [CrossRef]
- Guerreiro, S.; Privat, A.-L.; Bressac, L.; Toulorge, D. CD38 in Neurodegeneration and Neuroinflammation. Cells 2020, 9, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, K.; Hirano, T. BST-1/CD157 regulates the humoral immune responses in vivo. Chem. Immunol. 2000, 75, 235–255. [Google Scholar] [CrossRef] [PubMed]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Loring, H.S.; Icso, J.D.; Nemmara, V.V.; Thompson, P.R. Initial Kinetic Characterization of Sterile Alpha and Toll/Interleukin Receptor Motif-Containing Protein 1. Biochemistry 2020, 59, 933–942. [Google Scholar] [CrossRef]
- Gerdts, J.; Brace, E.J.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 activation triggers axon degeneration locally via NAD⁺ destruction. Science 2015, 348, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Loreto, A.; Di Stefano, M.; Gering, M.; Conforti, L. Wallerian Degeneration Is Executed by an NMN-SARM1-Dependent Late Ca(2+) Influx but Only Modestly Influenced by Mitochondria. Cell Rep. 2015, 13, 2539–2552. [Google Scholar] [CrossRef] [Green Version]
- Loreto, A.; Hill, C.S.; Hewitt, V.L.; Orsomando, G.; Angeletti, C.; Gilley, J.; Lucci, C.; Sanchez-Martinez, A.; Whitworth, A.J.; Conforti, L.; et al. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. bioRxiv 2019. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Zhou, P.; Qian, L.; Chuang, J.Z.; Lee, J.; Li, C.; Iadecola, C.; Nathan, C.; Ding, A. MyD88-5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. J. Exp. Med. 2007, 204, 2063–2074. [Google Scholar] [CrossRef] [Green Version]
- Ko, K.W.; Milbrandt, J.; DiAntonio, A. SARM1 acts downstream of neuroinflammatory and necroptotic signaling to induce axon degeneration. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, E.; Gibbons, L.; Neto, N.G.; Kenna, P.; Carty, M.; Humphries, M.; Humphries, P.; Campbell, M.; Monaghan, M.; Bowie, A.; et al. SARM1 deficiency promotes rod and cone photoreceptor cell survival in a model of retinal degeneration. Life Sci. Alliance 2020, 3. [Google Scholar] [CrossRef]
- Uccellini, M.B.; Bardina, S.V.; Sánchez-Aparicio, M.T.; White, K.M.; Hou, Y.J.; Lim, J.K.; García-Sastre, A. Passenger Mutations Confound Phenotypes of SARM1-Deficient Mice. Cell Rep. 2020, 31, 107498. [Google Scholar] [CrossRef]
- Summers, D.W.; DiAntonio, A.; Milbrandt, J. Mitochondrial dysfunction induces Sarm1-dependent cell death in sensory neurons. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 9338–9350. [Google Scholar] [CrossRef] [Green Version]
- Massoll, C.; Mando, W.; Chintala, S.K. Excitotoxicity upregulates SARM1 protein expression and promotes Wallerian-like degeneration of retinal ganglion cells and their axons. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2771–2780. [Google Scholar] [CrossRef]
- Fernandes, K.A.; Mitchell, K.L.; Patel, A.; Marola, O.J.; Shrager, P.; Zack, D.J.; Libby, R.T.; Welsbie, D.S. Role of SARM1 and DR6 in retinal ganglion cell axonal and somal degeneration following axonal injury. Exp. Eye Res. 2018, 171, 54–61. [Google Scholar] [CrossRef]
- Osterloh, J.M.; Yang, J.; Rooney, T.M.; Fox, A.N.; Adalbert, R.; Powell, E.H.; Sheehan, A.E.; Avery, M.A.; Hackett, R.; Logan, M.A.; et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012, 337, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Khanna, A.; Acharjee, P.; Acharjee, A.; Trigun, S.K. Mitochondrial SIRT3 and neurodegenerative brain disorders. J. Chem. Neuroanat. 2019, 95, 43–53. [Google Scholar] [CrossRef]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Herranz, D.; Serrano, M. SIRT1: Recent lessons from mouse models. Nature reviews. Cancer 2010, 10, 819–823. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Jing, E.; Emanuelli, B.; Hirschey, M.D.; Boucher, J.; Lee, K.Y.; Lombard, D.; Verdin, E.M.; Kahn, C.R. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2011, 108, 14608–14613. [Google Scholar] [CrossRef] [Green Version]
- Zaganjor, E.; Vyas, S.; Haigis, M.C. SIRT4 Is a Regulator of Insulin Secretion. Cell Chem. Biol. 2017, 24, 656–658. [Google Scholar] [CrossRef] [Green Version]
- Nasrin, N.; Wu, X.; Fortier, E.; Feng, Y.; Bare, O.C.; Chen, S.; Ren, X.; Wu, Z.; Streeper, R.S.; Bordone, L. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J. Biol. Chem. 2010, 285, 31995–32002. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Jiang, M.; Wu, X.; Diao, F.; Qiu, D.; Hou, X.; Wang, H.; Li, L.; Li, C.; Ge, J.; et al. SIRT4 is essential for metabolic control and meiotic structure during mouse oocyte maturation. Aging Cell 2018, 17, e12789. [Google Scholar] [CrossRef]
- Nakagawa, T.; Lomb, D.J.; Haigis, M.C.; Guarente, L. SIRT5 Deacetylates Carbamoyl Phosphate Synthetase 1 and Regulates the Urea Cycle. Cell 2009, 137, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Bharathi, S.S.; Rardin, M.J.; Lu, J.; Maringer, K.V.; Sims-Lucas, S.; Prochownik, E.V.; Gibson, B.W.; Goetzman, E.S. Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J. Biol. Chem. 2017, 292, 10239–10249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, Y.; Rardin, M.J.; Carrico, C.; He, W.; Sahu, A.K.; Gut, P.; Najjar, R.; Fitch, M.; Hellerstein, M.; Gibson, B.W.; et al. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol. Cell 2015, 59, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Kanfi, Y.; Naiman, S.; Amir, G.; Peshti, V.; Zinman, G.; Nahum, L.; Bar-Joseph, Z.; Cohen, H.Y. The sirtuin SIRT6 regulates lifespan in male mice. Nature 2012, 483, 218–221. [Google Scholar] [CrossRef]
- Vazquez, B.N.; Thackray, J.K.; Simonet, N.G.; Kane-Goldsmith, N.; Martinez-Redondo, P.; Nguyen, T.; Bunting, S.; Vaquero, A.; Tischfield, J.A.; Serrano, L. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 2016, 35, 1488–1503. [Google Scholar] [CrossRef]
- Zuo, L.; Khan, R.S.; Lee, V.; Dine, K.; Wu, W.; Shindler, K.S. SIRT1 Promotes RGC Survival and Delays Loss of Function Following Optic Nerve Crush. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5097–5102. [Google Scholar] [CrossRef]
- Verdin, E.; Hirschey, M.D.; Finley, L.W.; Haigis, M.C. Sirtuin regulation of mitochondria: Energy production, apoptosis, and signaling. Trends Biochem. Sci. 2010, 35, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Jaliffa, C.; Ameqrane, I.; Dansault, A.; Leemput, J.; Vieira, V.r.; Lacassagne, E.; Provost, A.; Bigot, K.; Masson, C.; Menasche, M.; et al. Sirt1 Involvement in rd10 Mouse Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3562–3572. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.H.; Chang, Y.L.; Kao, C.L.; Tseng, L.M.; Wu, C.C.; Chen, Y.C.; Tsai, C.Y.; Woung, L.C.; Liu, J.H.; Chiou, S.H.; et al. SirT1--A sensor for monitoring self-renewal and aging process in retinal stem cells. Sensors (Basel) 2010, 10, 6172–6194. [Google Scholar] [CrossRef] [PubMed]
- Mimura, T.; Kaji, Y.; Noma, H.; Funatsu, H.; Okamoto, S. The role of SIRT1 in ocular aging. Exp. Eye Res. 2013, 116, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cai, J.; Powell, D.W.; Paladugu, H.; Kuehn, M.H.; Tezel, G. Up-regulation of sirtuins in the glaucomatous human retina. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2398. [Google Scholar]
- Balaiya, S.; Ferguson, L.R.; Chalam, K.V. Evaluation of Sirtuin Role in Neuroprotection of Retinal Ganglion Cells in Hypoxia. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4315–4322. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Loscalzo, J. Metabolic Responses to Reductive Stress. Antioxid. Redox Signal. 2020, 32, 1330–1347. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, K.D.; Williams, C.H., Jr. NADH inhibition and NAD activation of Escherichia coli lipoamide dehydrogenase catalyzing the NADH-lipoamide reaction. J. Biol. Chem. 1981, 256, 2307–2314. [Google Scholar] [CrossRef]
- Ussher, J.R.; Jaswal, J.S.; Lopaschuk, G.D. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circ. Res. 2012, 111, 628–641. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Reinstadler, B.; Engelstad, K.; Skinner, O.S.; Stackowitz, E.; Haller, R.G.; Clish, C.B.; Pierce, K.; Walker, M.A.; Fryer, R.; et al. Circulating markers of NADH-reductive stress correlate with mitochondrial disease severity. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Annesley, S.J.; Fisher, P.R. Lymphoblastoid Cell Lines as Models to Study Mitochondrial Function in Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 4536. [Google Scholar] [CrossRef]
- Friedman, D.S.; Wilson, M.R.; Liebmann, J.M.; Fechtner, R.D.; Weinreb, R.N. An evidence-based assessment of risk factors for the progression of ocular hypertension and glaucoma. Am. J. Ophthalmol. 2004, 138, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Burr, J.M.; Mowatt, G.; Hernández, R.; Siddiqui, M.A.; Cook, J.; Lourenco, T.; Ramsay, C.; Vale, L.; Fraser, C.; Azuara-Blanco, A.; et al. The clinical effectiveness and cost-effectiveness of screening for open angle glaucoma: A systematic review and economic evaluation. Health Technol. Assess. 2007, 11, iii. [Google Scholar] [CrossRef] [Green Version]
- Burr, J.M.; Campbell, M.K.; Campbell, S.E.; Francis, J.J.; Greene, A.; Hernández, R.; Hopkins, D.; McCann, S.K.; Vale, L.D. Developing the clinical components of a complex intervention for a glaucoma screening trial: A mixed methods study. BMC Med. Res. Methodol. 2011, 11, 54. [Google Scholar] [CrossRef] [Green Version]
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Group, B.D.W. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar]
- VanderWall, K.B.; Huang, K.-C.; Pan, Y.; Lavekar, S.S.; Fligor, C.M.; Allsop, A.R.; Lentsch, K.A.; Dang, P.; Zhang, C.; Tseng, H.C.; et al. Retinal Ganglion Cells With a Glaucoma OPTN(E50K) Mutation Exhibit Neurodegenerative Phenotypes when Derived from Three-Dimensional Retinal Organoids. Stem Cell Rep. 2020, 15, 52–66. [Google Scholar] [CrossRef]
- Alonso-Lavin, A.J.; Bajić, D.; Poyatos, J.F. Tolerance to NADH/NAD+ imbalance anticipates aging and anti-aging interventions. bioRxiv 2019. [Google Scholar] [CrossRef]
- Zhu, X.; Shen, W.; Wang, Y.; Jaiswal, A.; Ju, Z.; Sheng, Q. Nicotinamide adenine dinucleotide replenishment rescues colon degeneration in aged mice. Signal. Transduct. Target. Ther. 2017, 2, 17017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.U.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar]
- Wu, J.; Jin, Z.; Zheng, H.; Yan, L.-J. Sources and implications of NADH/NAD(+) redox imbalance in diabetes and its complications. DiabetesMetab. Syndr. Obes. Targets Ther. 2016, 9, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Akie, T.E.; Liu, L.; Nam, M.; Lei, S.; Cooper, M.P. OXPHOS-Mediated Induction of NAD+ Promotes Complete Oxidation of Fatty Acids and Interdicts Non-Alcoholic Fatty Liver Disease. PLoS ONE 2015, 10, e0125617. [Google Scholar] [CrossRef] [Green Version]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C., Jr.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, G.R.; Pride, P.M.; Babbey, C.M.; Payne, R.M. Friedreich’s ataxia reveals a mechanism for coordinate regulation of oxidative metabolism via feedback inhibition of the SIRT3 deacetylase. Hum. Mol. Genet. 2012, 21, 2688–2697. [Google Scholar] [CrossRef] [Green Version]
- Tribble, J.R.; Otmani, A.; Sun, S.; Ellis, S.A.; Cimaglia, G.; Vohra, R.; Jöe, M.; Lardner, E.; Venkataraman, A.P.; Domínguez-Vicent, A.; et al. Nicotinamide provides neuroprotection in glaucoma by protecting against mitochondrial and metabolic dysfunction. bioRxiv 2020. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Study | Finding |
|---|---|
| Ju et al. 2009 [26] | Elevated hydrostatic pressure triggered mitochondrial changes and altered OPA1 gene expression before the onset of apoptosis in differentiated RGC–5 cells |
| Aung et al. 2002; Powell et al. 2003; Yu–Wai–Man et al. 2010 [27,28,29,30] | Polymorphisms in the OPA1 gene are associated with NTG, and they also influence the phenotypic feature in patients with HTG. |
| Abu–Amero et al. 2006 [31] | Reduced mitochondrial respiratory activity in lymphocytes of POAG patients compared controls |
| N. J. Van Bergen et al. 2015a [32] | Reduced Complex–I enzyme specific activity and ATP synthesis in POAG lymphoblasts |
| [33] | Complex I defect in POAG lymphoblasts, leading to decreased rates of respiration and ATP production |
| Wolf et al. 2009 [34] | Association of NTG with common sequence variants of OPTN, MFN1, MFN2 and PARL |
| X. Hu et al. 2018 [35] | OPA1 overexpression may protect RGCs by ways of enhancing mitochondria fusion and parkin mediated mitophagy |
| Bailey et al. 2016; Khawaja et al. 2016; Khawaja et al. 2018; Sundaresan et al. 2015 [36,37,38,39] | Various genes encoding for mitochondrial proteins have been found to be associated with POAG, and in particular NTG, including TXNRD2, ME3, VPS13C, GCAT, PTCD2, ND5 |
| Lascaratos et al. 2015 [40] | Resistance to developing glaucoma is associated with systemic mitochondrial efficiency |
| Williams et al. 2017 [41] Fraenkl et al. 2011; Goldblum et al. 2010 [42,43] | Metabolic dysfunction and mitochondrial abnormalities occur prior to glaucomatous neurodegenerationReduced plasma citrate levels in patients with glaucoma compared to controls—citrate is a major component in mitochondrial metabolism |
| Study | Finding |
|---|---|
| Ferreira et al. 2004 [44] | Reduced levels of water–soluble antioxidants (glutathione, ascorbate, tyrosine) in aqueous humour of POAG compared to controls |
| Izzotti et al. 2003; Saccà et al. 2005 [45,46] | Oxidative DNA damage is exaggerated in the trabecular meshwork of POAG patients |
| Gherghel et al. 2005 [47] | Glaucoma patients have lower serum GSH and total glutathoine (t–GSH) levels as compared with age–matched controls |
| Yildirim et al. 2005 [48] | Malonyldialdehyde (marker of oxidative stress) levels were more than 2–fold greater in the serum of POAG patients as compared with healthy controls |
| Tanito et al. 2012 [49] | Biological antioxidant potential level, a measure of total antioxidative stress activity, was lower in plasma in the POAG and pseudo–exfoliation syndrome groups compared with the control groups |
| Sorkhabi et al. 2011 [50] | Increased oxidative DNA damage in the serum and aqueous humour of glaucoma patients |
| Yuki et al. 2010 [51] | Increased serum total antioxidant and decreased 8–hydroxy–2′–deoxyguanosine in response to increased systemic oxidative stress in patients with normal–tension glaucoma |
| Sirtuin | Activity | Location | Biological Function | References |
|---|---|---|---|---|
| SIRT1 | Deacetylation | Nucleus | Regulation of DNA damage, stress response, mitochondrial biogenesis, glucose and lipid metabolism | [147,148,149,150] |
| SIRT2 | Deacetylation | Cytosol | Lipid and glucose metabolism, control of cell cycle | [151,152] |
| SIRT3 | Deacetylation | Mitochondria | Regulation of ATP production, metabolism, apoptosis, cell signalling | [153,154,155] |
| SIRT4 | ADP–ribosylation | Mitochondria | Inhibition of insulin secretion, repression of fatty acid oxidation, tumour suppressor | [156,157,158] |
| SIRT5 | Deacetylation | Mitochondria, cytosol | Urea cycle, ATP production, glycolysis | [159,160,161] |
| SIRT6 | Deacetylation, ADP–ribosylation | Nucleus | Genomic stability and repair, metabolism and aging | [162,163,164] |
| SIRT7 | Deacetylation, ADP–ribosylation | Nucleolus | DNA repair, ageing | [165] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petriti, B.; Williams, P.A.; Lascaratos, G.; Chau, K.-Y.; Garway-Heath, D.F. Neuroprotection in Glaucoma: NAD+/NADH Redox State as a Potential Biomarker and Therapeutic Target. Cells 2021, 10, 1402. https://doi.org/10.3390/cells10061402
Petriti B, Williams PA, Lascaratos G, Chau K-Y, Garway-Heath DF. Neuroprotection in Glaucoma: NAD+/NADH Redox State as a Potential Biomarker and Therapeutic Target. Cells. 2021; 10(6):1402. https://doi.org/10.3390/cells10061402
Chicago/Turabian StylePetriti, Bledi, Pete A. Williams, Gerassimos Lascaratos, Kai-Yin Chau, and David F. Garway-Heath. 2021. "Neuroprotection in Glaucoma: NAD+/NADH Redox State as a Potential Biomarker and Therapeutic Target" Cells 10, no. 6: 1402. https://doi.org/10.3390/cells10061402
APA StylePetriti, B., Williams, P. A., Lascaratos, G., Chau, K. -Y., & Garway-Heath, D. F. (2021). Neuroprotection in Glaucoma: NAD+/NADH Redox State as a Potential Biomarker and Therapeutic Target. Cells, 10(6), 1402. https://doi.org/10.3390/cells10061402