
Ion Channel Gene Mutations Causing Skeletal Muscle Disorders: Pathomechanisms and Opportunities for Therapy



Abstract
:1. Introduction
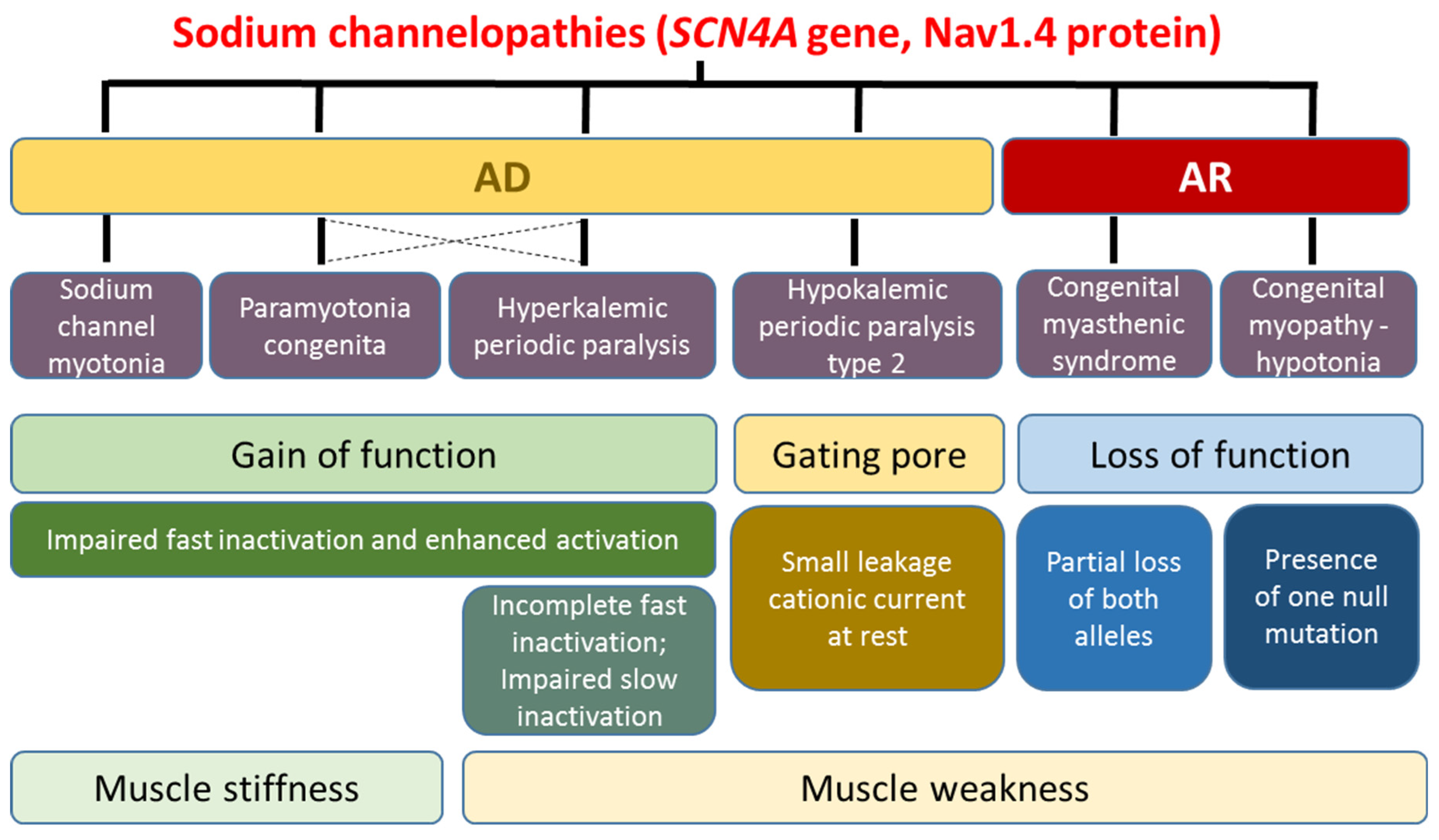
2. Skeletal Muscle Sodium Channelopathies
2.1. Hyperkalemic Periodic Paralysis, Paramyotonia Congenita, and Sodium Channel Myotonia
2.2. Hypokalemic Periodic Paralysis Type 2
2.3. Congenital Myasthenia and Congenital Myopathy Related to SCN4A
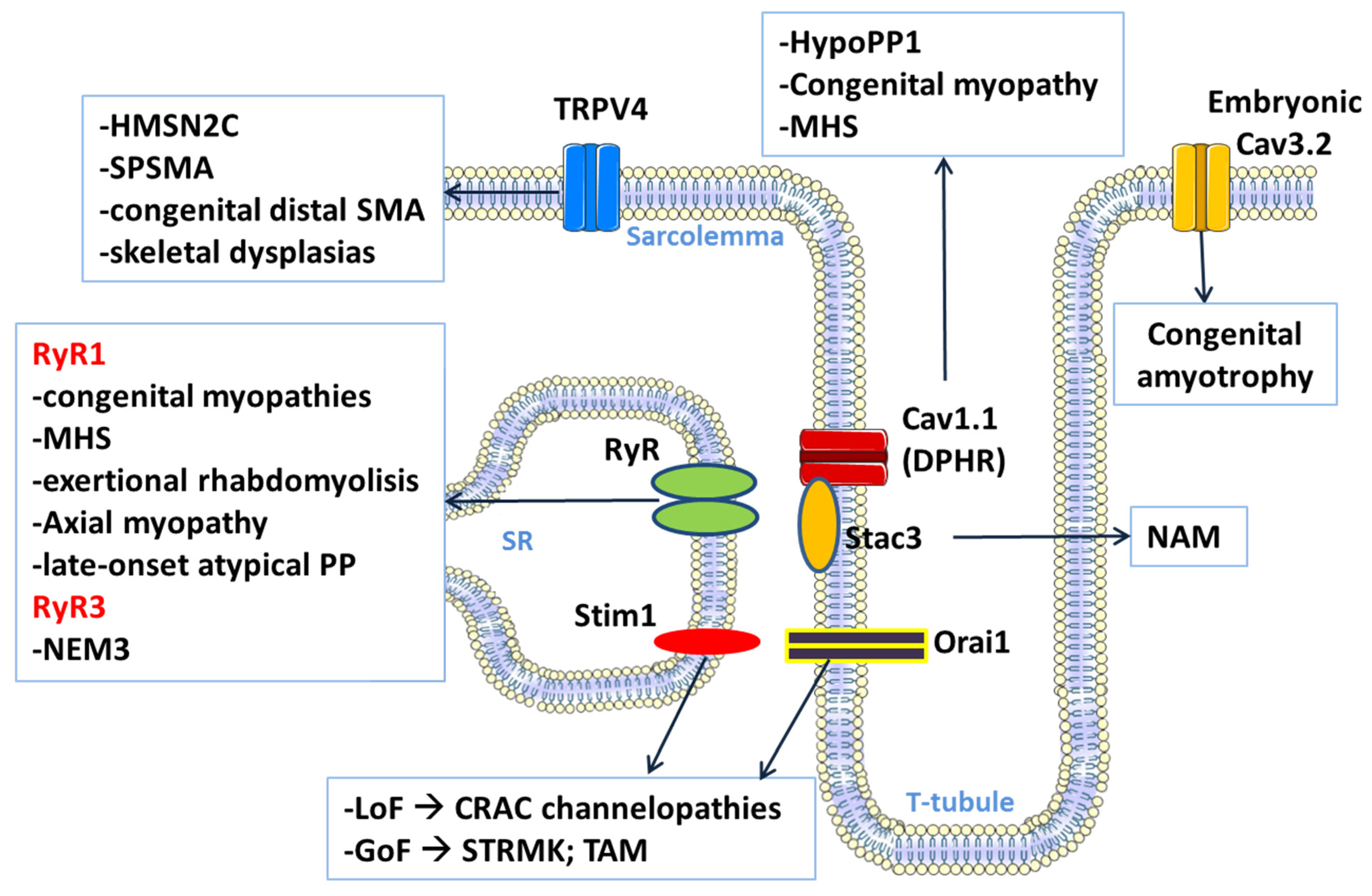
3. Calcium Channel-Related Myopathies
3.1. CACNA1S-Related Disorders
3.1.1. Hypokalemic Periodic Paralysis Type 1
3.1.2. Calcium Channel-Related Congenital Myopathy
3.2. STAC3-Related Disorder
3.3. STIM1 and ORAI1-Related Disorders
3.4. RYR1-Related Disorders
3.4.1. RYR1-Related Congenital Myopathies
3.4.2. Malignant Hyperthermia Susceptibility
3.4.3. Exertional Rhabdomyolysis
3.4.4. Other Ryr1-Related Phenotypes
3.5. RYR3-Related Myopathy with Nemaline Bodies
3.6. TRPV4 Channel Related Myopathies
3.7. Congenital Amyotrophy Related to CACNA1H (T-Type Channel)
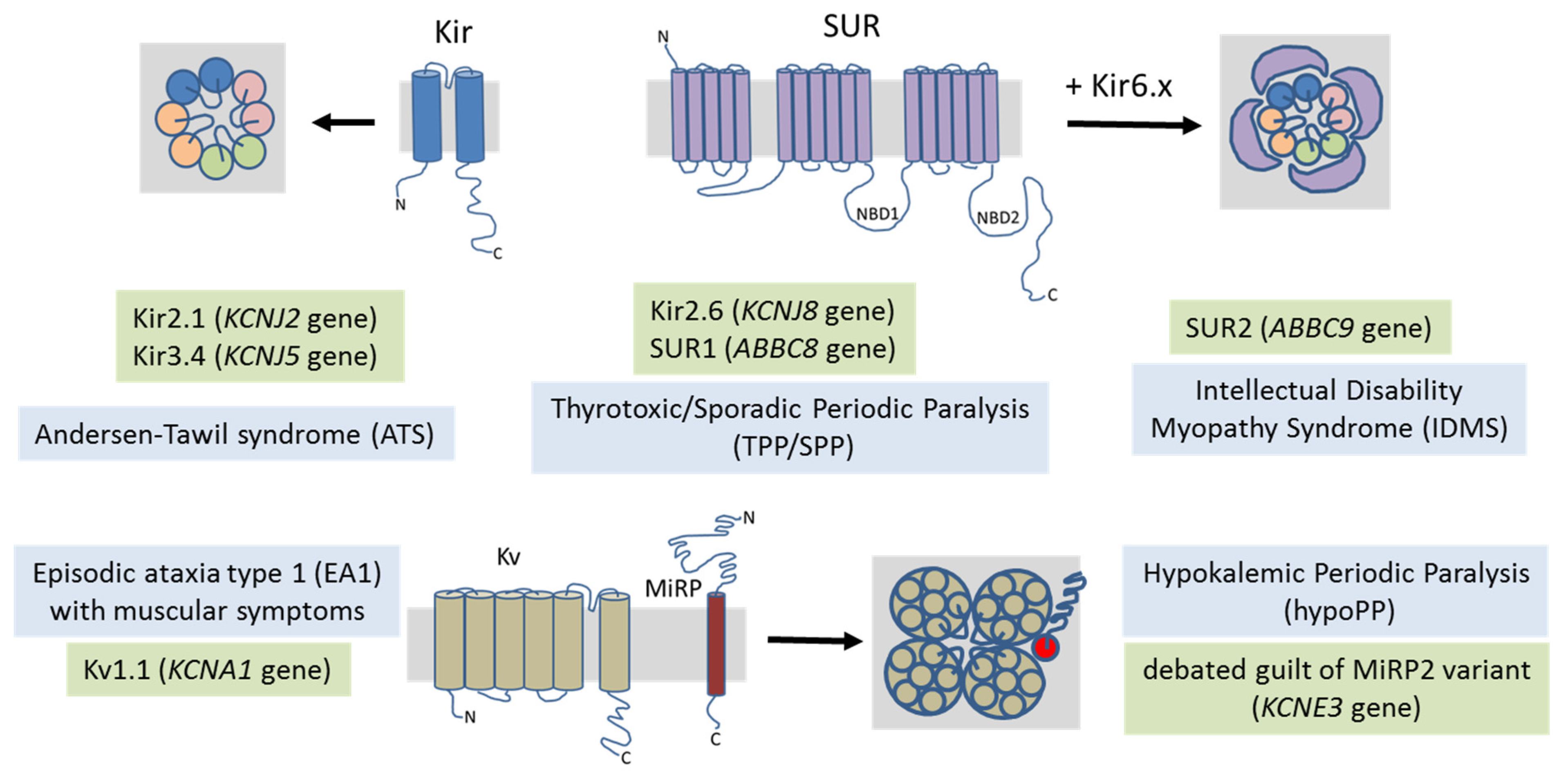
4. Potassium Channel-Related Myopathies
4.1. Andersen–Tawil Syndrome
4.2. Hypokalemic Periodic Paralysis
4.3. Thyrotoxic Periodic Paralysis
4.4. Intellectual Disability Myopathy Syndrome
4.5. Episodic Ataxia/Myokymia
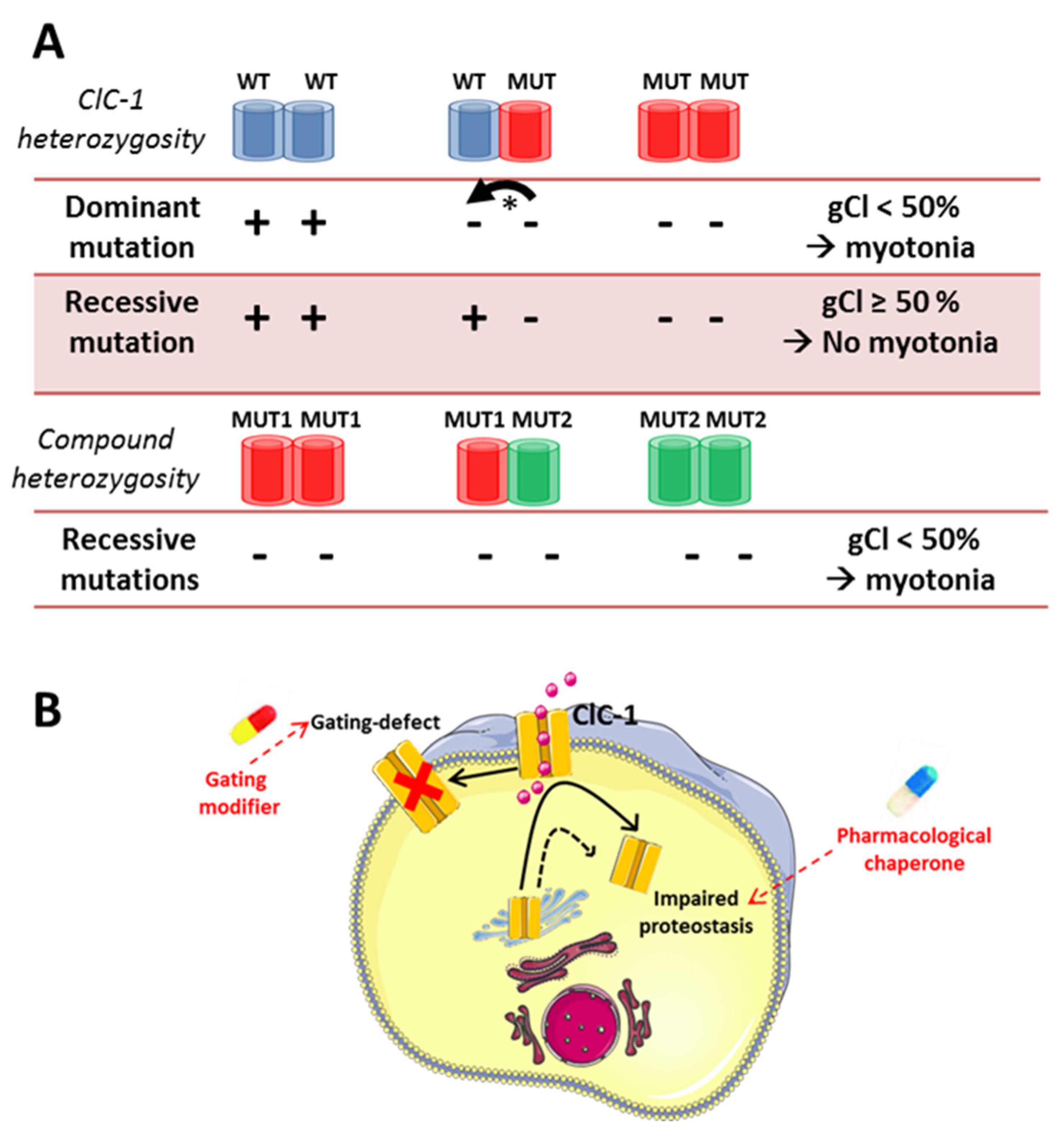
5. Chloride Channel-Related Muscle Disorders
6. Nicotinic Receptor-Channels
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fontaine, B.; Khurana, T.S.; Hoffman, E.P.; Bruns, G.A.; Haines, J.L.; Trofatter, J.A.; Hanson, M.P.; Rich, J.; McFarlane, H.; Yasek, D.M. Hyperkalemic periodic paralysis and the adult muscle sodium channel alpha-subunit gene. Science 1990, 250, 1000–1002. [Google Scholar] [CrossRef] [PubMed]
- Horga, A.; Rayan, D.L.; Matthews, E.; Sud, R.; Fialho, D.; Durran, S.C.; Burge, J.A.; Portaro, S.; Davis, M.B.; Haworth, A.; et al. Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology 2013, 80, 1472–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stunnenberg, B.C.; Raaphorst, J.; Deenen, J.C.; Links, T.P.; Wilde, A.A.; Verbove, D.J.; Kamsteeg, E.J.; van den Wijngaard, A.; Faber, C.G.; van der Wilt, G.J.; et al. Prevalence and mutation spectrum of skeletal muscle channelopathies in the Netherlands. Neuromuscul. Disord. 2018, 28, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Brugnoni, R.; Canioni, E.; Tonin, P.; Saletti, V.; Patrizia, S.; Cotti Piccinelli, S.; Colleoni, L.; Ferrigno, P.; Antonella, P.; et al. Clinical and Molecular Spectrum of Myotonia and Periodic Paralyses Associated With Mutations in SCN4A in a Large Cohort of Italian Patients. Front. Neurol. 2020, 11, 646. [Google Scholar] [CrossRef] [PubMed]
- Ptáček, L.J.; George, A.L., Jr.; Griggs, R.C.; Tawil, R.; Kallen, R.G.; Barchi, R.L.; Robertson, M.; Leppert, M.F. Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell 1991, 67, 1021–1027. [Google Scholar] [CrossRef]
- Rojas, C.V.; Wang, J.Z.; Schwartz, L.S.; Hoffman, E.P.; Powell, B.R.; Brown, R.H., Jr. A Met-to-Val mutation in the skeletal muscle Nav channel alpha-subunit in hyperkalaemic periodic paralysis. Nature 1991, 354, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.M.; Da Silva, M.D.; Miller, H.A.; Kwiecinski, H.; Mendell, J.R.; Tawil, R.; McManis, P.; Griggs, R.C.; Angelini, C.; Servidei, S.; et al. Correlating phenotype and genotype in the periodic paralyses. Neurology 2004, 63, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Ptáček, L.J.; George, A.L., Jr.; Barchi, R.L.; Griggs, R.C.; Riggs, J.E.; Robertson, M.; Leppert, M.F. Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita. Neuron 1992, 8, 891–897. [Google Scholar] [CrossRef]
- Matthews, E.; Fialho, D.; Tan, S.V.; Venance, S.L.; Cannon, S.C.; Sternberg, D.; Fontaine, B.; Amato, A.A.; Barohn, R.J.; Griggs, R.C.; et al. The non-dystrophic myotonias: Molecular pathogenesis, diagnosis and treatment. Brain 2010, 133, 9–22. [Google Scholar] [CrossRef]
- Lerche, H.; Heine, R.; Pika, U.; George Jr, A.L.; Mitrovic, N.; Browatzki, M.; Weiss, T.; Rivet-Bastide, M.; Franke, C.; Lomonaco, M. Human sodium channel myotonia: Slowed channel inactivation due to substitutions for a glycine within the III-IV linker. J. Physiol. 1993, 470, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Trip, J.; Drost, G.; Ginjaar, H.B.; Nieman, F.H.; van der Kooi, A.J.; de Visser, M.; van Engelen, B.G.; Faber, C.G. Redefining the clinical phenotypes of non-dystrophic myotonic syndromes. J. Neurol. Neurosurg. Psychiatry 2009, 80, 647–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cea, G.; Andreu, D.; Fletcher, E.; Ramdas, S.; Sud, R.; Hanna, M.G.; Matthews, E. Sodium channel myotonia may be associated with high-risk brief resolved unexplained events. Wellcome Open Res. 2020, 5, 57. [Google Scholar] [CrossRef] [PubMed]
- Fusco, C.; Frattini, D.; Salerno, G.G.; Canali, E.; Bernasconi, P.; Maggi, L. New phenotype and neonatal onset of sodium channel myotonia in a child with a novel mutation of SCN4A gene. Brain Dev. 2015, 37, 891–893. [Google Scholar] [CrossRef] [PubMed]
- Bednarz, M.; Stunnenberg, B.C.; Kusters, B.; Kamsteeg, E.J.; Saris, C.G.; Groome, J.; Winston, V.; Meola, G.; Jurkat-Rott, K.; Voermans, N.C. A novel Ile1455Thr variant in the skeletal muscle sodium channel alpha-subunit in a patient with a severe adult-onset proximal myopathy with electrical myotonia and a patient with mild paramyotonia phenotype. Neuromuscul. Disord. 2017, 27, 175–182. [Google Scholar] [CrossRef]
- Luo, S.; Castañeda, M.S.; Matthews, E.; Sud, R.; Hanna, M.G.; Sun, J.; Song, J.; Lu, J.; Qiao, K.; Zhao, C.; et al. Hypokalaemic periodic paralysis and myotonia in a patient with homozygous mutation p.R1451L in Nav1.4. Sci. Rep. 2018, 8, 9714. [Google Scholar] [CrossRef] [PubMed]
- Elia, N.; Nault, T.; McMillan, H.J.; Graham, G.E.; Huang, L.; Cannon, S.C. Myotonic myopathy with secondary joint and skeletal anomalies from the c.2386C>G, p.L769V mutation in SCN4A. Front. Neurol. 2020, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Poulin, H.; Gosselin-Badaroudine, P.; Vicart, S.; Habbout, K.; Sternberg, D.; Giuliano, S.; Fontaine, B.; Bendahhou, S.; Nicole, S.; Chahine, M. Substitutions of the S4DIV R2 residue (R1451) in NaV1.4 lead to complex forms of paramyotonia congenita and periodic paralyses. Sci. Rep. 2018, 8, 2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taminato, T.; Mori-Yoshimura, M.; Miki, J.; Sasaki, R.; Sato, N.; Oya, Y.; Nishino, I.; Takahashi, Y. Paramyotonia Congenita with Persistent Distal and Facial Muscle Weakness: A Case Report with Literature Review. J. Neuromuscul. Dis. 2020, 7, 193–201. [Google Scholar] [CrossRef]
- Cannon, S.C. Sodium channelopathies of skeletal muscle. Handb. Exp. Pharmacol. 2018, 246, 309–330. [Google Scholar]
- Cannon, S.C.; Brown, R.H., Jr.; Corey, D.P. Theoretical reconstruction of myotonia and paralysis caused by incomplete inactivation of sodium channels. Biophys. J. 1993, 65, 270–288. [Google Scholar] [CrossRef] [Green Version]
- Hayward, L.J.; Sandoval, G.M.; Cannon, S.C. Defective slow inactivation of sodium channels contributes to familial periodic paralysis. Neurology 1999, 52, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Statland, J.M.; Bundy, B.N.; Wang, Y.; Rayan, D.R.; Trivedi, J.R.; Sansone, V.A.; Salajegheh, M.K.; Venance, S.L.; Ciafaloni, E.; Matthews, E.; et al. Mexiletine for symptoms and signs of myotonia in nondystrophic myotonia: A randomized controlled trial. JAMA 2012, 308, 1357–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stunnenberg, B.C.; Raaphorst, J.; Groenewoud, H.M.; Statland, J.M.; Griggs, R.C.; Woertman, W.; Stegeman, D.F.; Timmermans, J.; Trivedi, J.; Matthews, E.; et al. Effect of mexiletine on muscle stiffness in patients with nondystrophic myotonia evaluated using aggregated N-of-1 trials. JAMA 2018, 320, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Suetterlin, K.J.; Bugiardini, E.; Kaski, J.P.; Morrow, J.M.; Matthews, E.; Hanna, M.G.; Fialho, D. Long-term safety and efficacy of mexiletine for patients with skeletal muscle channelopathies. JAMA Neurol. 2015, 72, 1531–1533. [Google Scholar] [CrossRef] [Green Version]
- Modoni, A.; D’Amico, A.; Primiano, G.; Capozzoli, F.; Desaphy, J.-F.; Lo Monaco, M. Long-term safety and usefulness of mexiletine in a large cohort of patients affected by non-dystrophic myotonias. Front. Neurol. 2020, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Andersen, G.; Hedermann, G.; Witting, N.; Duno, M.; Andersen, H.; Vissing, J. The antimyotonic effect of lamotrigine in non-dystrophic myotonias: A double-blind randomized study. Brain 2017, 140, 2295–2305. [Google Scholar] [CrossRef] [PubMed]
- Vereb, N.; Montagnese, F.; Gläser, D.; Schoser, B. Non-dystrophic myotonias: Clinical and mutation spectrum of 70 German patients. J. Neurol. 2021, 268, 1708–1720. [Google Scholar] [CrossRef]
- Desaphy, J.F.; Modoni, A.; Lomonaco, M.; Camerino, D.C. Dramatic improvement of myotonia permanens with flecainide: A two-case report of a possible bench-to-bedside pharmacogenetics strategy. Eur. J. Clin. Pharmacol. 2013, 69, 1037–1039. [Google Scholar] [CrossRef] [Green Version]
- Desaphy, J.F.; Carbonara, R.; D’Amico, A.; Modoni, A.; Roussel, J.; Imbrici, P.; Pagliarani, S.; Lucchiari, S.; Lo Monaco, M.; Conte Camerino, D. Translational approach to address therapy in myotonia permanens due to a new SCN4A mutation. Neurology 2016, 86, 2100–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farinato, A.; Altamura, C.; Imbrici, P.; Maggi, L.; Bernasconi, P.; Mantegazza, R.; Pasquali, L.; Siciliano, G.; Monaco, M.L.; Vial, C.; et al. Pharmacogenetics of myotonic hNav1.4 sodium channel variants situated near the fast inactivation gate. Pharmacol. Res. 2019, 141, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Desaphy, J.F.; Altamura, C.; Vicart, S.; Fontaine, B. Targeted Therapies for Skeletal Muscle Ion Channelopathies: Systematic Review and Steps Towards Precision Medicine. J. Neuromuscul. Dis. 2021, 8, 357–381. [Google Scholar] [CrossRef]
- Desaphy, J.F.; Carbonara, R.; Costanza, T.; Conte Camerino, D. Preclinical evaluation of marketed sodium channel blockers in a rat model of myotonia discloses promising antimyotonic drugs. Exp. Neurol. 2014, 255, 96–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desaphy, J.F.; Farinato, A.; Altamura, C.; De Bellis, M.; Imbrici, P.; Tarantino, N.; Caccia, C.; Melloni, E.; Padoani, G.; Vailati, S.; et al. Safinamide’s potential in treating nondystrophic myotonias: Inhibition of skeletal muscle voltage-gated sodium channels and skeletal muscle hyperexcitability in vitro and in vivo. Exp. Neurol. 2020, 328, 113287. [Google Scholar] [CrossRef] [PubMed]
- Novak, K.R.; Norman, J.; Mitchell, J.R.; Pinter, M.J.; Rich, M.M. Sodium channel slow inactivation as a therapeutic target for myotonia congenita. Ann. Neurol. 2015, 77, 320–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skov, M.; de Paoli, F.V.; Nielsen, O.B.; Pedersen, T.H. The anti-convulsants lacosamide, lamotrigine, and rufinamide reduce myotonia in isolated human and rat skeletal muscle. Muscle Nerve 2017, 56, 136–142. [Google Scholar] [CrossRef] [PubMed]
- De Bellis, M.; Carbonara, R.; Roussel, J.; Farinato, A.; Massari, A.; Pierno, S.; Muraglia, M.; Corbo, F.; Franchini, C.; Carratù, M.R.; et al. Increased sodium channel use-dependent inhibition by a new potent analogue of tocainide greatly enhances in vivo antimyotonic activity. Neuropharmacology 2017, 113, 206–216. [Google Scholar] [CrossRef] [Green Version]
- De Bellis, M.; De Luca, A.; Desaphy, J.F.; Carbonara, R.; Heiny, J.A.; Kennedy, A.; Carocci, A.; Cavalluzzi, M.M.; Lentini, G.; Franchini, C.; et al. Combined modifications of mexiletine pharmacophores for new lead blockers of Na(v)1.4 channels. Biophys. J. 2013, 104, 344–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tricarico, D.; Mele, A.; Conte Camerino, D. Carbonic anhydrase inhibitors ameliorate the symptoms of hypokalaemic periodic paralysis in rats by opening the muscular Ca2+- activated-K+channels. Neuromuscul. Disord. 2006, 16, 39–45. [Google Scholar] [CrossRef]
- Altamura, C.; Fonzino, A.; Tarantino, N.; Conte, E.; Liantonio, A.; Imbrici, P.; Carratù, M.R.; Pierno, S.; Desaphy, J.F. Increased sarcolemma chloride conductance as one of the mechanisms of action of carbonic anhydrase inhibitors in muscle excitability disorders. Exp. Neurol. 2021, 342, 113758. [Google Scholar] [CrossRef]
- Sansone, V.A.; Burge, J.; McDermott, M.P.; Smith, P.C.; Herr, B.; Tawil, R.; Pandya, S.; Kissel, J.; Ciafaloni, E.; Shieh, P.; et al. Randomized, placebo-controlled trials of dichlorphenamide in periodic paralysis. Neurology 2016, 86, 1408–1416. [Google Scholar] [CrossRef] [Green Version]
- Statland, J.M.; Fontaine, B.; Hanna, M.G.; Johnson, N.E.; Kissel, J.T.; Sansone, V.A.; Shieh, P.B.; Tawil, R.N.; Trivedi, J.; Cannon, S.C.; et al. Review of the Diagnosis and Treatment of Periodic Paralysis. Muscle Nerve 2018, 57, 522–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stunnenberg, B.C.; LoRusso, S.; Arnold, W.D.; Barohn, R.J.; Cannon, S.C.; Fontaine, B.; Griggs, R.C.; Hanna, M.G.; Matthews, E.; Meola, G.; et al. Guidelines on clinical presentation and management of nondystrophic myotonias. Muscle Nerve 2020, 62, 430–444. [Google Scholar] [CrossRef]
- Bulman, D.E.; Scoggan, K.A.; van Oene, M.D.; Nicolle, M.W.; Hahn, A.F.; Tollar, L.L.; Ebers, G.C. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology 1999, 53, 1932–1936. [Google Scholar] [CrossRef]
- Jurkat-Rott, K.; Mitrovic, N.; Hang, C.; Kouzmenkine, A.; Iaizzo, P.; Herzog, J.; Lerche, H.; Nicole, S.; Vale-Santos, J.; Chauveau, D.; et al. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc. Natl. Acad. Sci. USA 2000, 97, 9549–9554. [Google Scholar] [CrossRef] [Green Version]
- Matthews, E.; Labrum, R.; Sweeney, M.G.; Sud, R.; Haworth, A.; Chinnery, P.F.; Meola, G.; Schorge, S.; Kullmann, D.M.; Davis, M.B.; et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology 2009, 72, 1544–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, E.; Portaro, S.; Ke, Q.; Sud, R.; Haworth, A.; Davis, M.B.; Griggs, R.C.; Hanna, M.G. ACZ efficacy in hypokalemic periodic paralysis and the predictive role of genotype. Neurology 2011, 77, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujino, A.; Maertens, C.; Ohno, K.; Shen, X.M.; Fukuda, T.; Harper, C.M.; Cannon, S.C.; Engel, A.G. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc. Natl. Acad. Sci. USA 2003, 100, 7377–7382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaharieva, I.T.; Thor, M.G.; Oates, E.C.; Van Karnebeek, C.; Hendson, G.; Blom, E.; Witting, N.; Rasmussen, M.; Gabbett, M.T.; Ravenscroft, G.; et al. Loss-of-function mutations in SCN4A cause severe foetal hypokinesia or classical congenital myopathy. Brain 2016, 139, 674–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, R.; Bonne, G.; Missier, P.; Lochmüller, H. Targeted therapies for congenital myasthenic syndromes: Systematic review and steps towards a treatabolome. Emerg. Top. Life Sci. 2019, 3, 19–37. [Google Scholar] [PubMed]
- Matthews, E.; Hartley, L.; Sud, R.; Hanna, M.G.; Muntoni, F.; Munot, P. Acetazolamide can improve symptoms and signs in ion channel-related congenital myopathy. J. Neurol. Neurosurg. Psychiatry 2019, 90, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Ptáček, L.J.; Tawil, R.; Griggs, R.C.; Engel, A.G.; Layzer, R.B.; Kwieciński, H.; McManis, P.G.; Santiago, L.; Moore, M.; Fouad, G.; et al. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell 1994, 77, 863–868. [Google Scholar] [CrossRef]
- Holm-Yildiz, S.; Witting, N.; Dahlqvist, J.; de Stricker Borch, J.; Solheim, T.; Fornander, F.; Eisum, A.S.; Duno, M.; Soerensen, T.; Vissing, J. Permanent muscle weakness in hypokalemic periodic paralysis. Neurology 2020, 95, e342–e352. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Lehmann-Horn, F.; Weber, M.A.; Bednarz, M.; Groome, J.R.; Jonsson, M.K. Transient compartment-like syndrome and normokalaemic periodic paralysis due to a Ca(v)1.1 mutation. Brain 2013, 136, 3775–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Mi, W.; Hernández-Ochoa, E.O.; Burns, D.K.; Fu, Y.; Gray, H.F.; Struyk, A.F.; Schneider, M.F.; Cannon, S.C. A calcium channel mutant mouse model of hypokalemic periodic paralysis. J. Clin. Investig. 2012, 122, 4580–4591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schartner, V.; Romero, N.B.; Donkervoort, S.; Treves, S.; Munot, P.; Pierson, T.M.; Dabaj, I.; Malfatti, E.; Zaharieva, I.T.; Zorzato, F.; et al. Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. 2017, 133, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Jungbluth, H.; Treves, S.; Zorzato, F.; Sarkozy, A.; Ochala, J.; Sewry, C.; Phadke, R.; Gautel, M.; Muntoni, F. Congenital myopathies: Disorders of excitation-contraction coupling and muscle contraction. Nat. Rev. Neurol. 2018, 14, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.M.; Ahearn, M.E.; Balak, C.D.; Liang, W.S.; Kurdoglu, A.; Corneveaux, J.J.; Russell, M.; Huentelman, M.J.; Craig, D.W.; Carpten, J.; et al. Novel pathogenic variants and genes for myopathies identified by whole exome sequencing. Mol. Genet. Genomic Med. 2015, 3, 283–301. [Google Scholar] [CrossRef]
- Ravenscroft, G.; Clayton, J.S.; Faiz, F.; Sivadorai, P.; Milnes, D.; Cincotta, R.; Moon, P.; Kamien, B.; Edwards, M.; Delatycki, M.; et al. Neurogenetic fetal akinesia and arthrogryposis: Genetics, expanding genotype-phenotypes and functional genomics. J. Med. Genet. 2020. [Google Scholar] [CrossRef]
- Yiş, U.; Hiz, S.; Güneş, S.; Diniz, G.; Baydan, F.; Töpf, A.; Sonmezler, E.; Lochmüller, H.; Horvath, R.; Oktay, Y. Dihydropyridine Receptor Congenital Myopathy In A Consangineous Turkish Family. J. Neuromuscul. Dis. 2019, 6, 377–384. [Google Scholar] [CrossRef]
- Flucher, B.E.; Campiglio, M. STAC proteins: The missing link in skeletal muscle EC coupling and new regulators of calcium channel function. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 1101–1110. [Google Scholar] [CrossRef]
- Nelson, B.R.; Wu, F.; Liu, Y.; Anderson, D.M.; McAnally, J.; Lin, W.; Cannon, S.C.; Bassel-Duby, R.; Olson, E.N. Skeletal muscle-specific T-tubule protein STAC3 mediates voltage-induced Ca2+ release and contractility. Proc. Natl. Acad. Sci. USA 2013, 110, 11881–11886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horstick, E.J.; Linsley, J.W.; Dowling, J.J.; Hauser, M.A.; McDonald, K.K.; Ashley-Koch, A.; Saint-Amant, L.; Satish, A.; Cui, W.W.; Zhou, W.; et al. Stac3 is a component of the excitation-contraction coupling machinery and mutated in Native American myopathy. Nat. Commun. 2013, 4, 1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, A.G.; Bloch, E.C. Malignant hyperthermia in a three-month-old American Indian infant. Anesth. Analg. 1987, 66, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Kahler, S.G.; Gilchrist, J.M. Congenital myopathy with cleft palate and increased susceptibility to malignant hyperthermia: King syndrome? Pediatr. Neurol. 1988, 4, 371–374. [Google Scholar] [CrossRef]
- Stamm, D.S.; Aylsworth, A.S.; Stajich, J.M.; Kahler, S.G.; Thorne, L.B.; Speer, M.C.; Powell, C.M. Native American myopathy: Congenital myopathy with cleft palate, skeletal anomalies, and susceptibility to malignant hyperthermia. Am. J. Med. Genet. A 2008, 146, 1832–1841. [Google Scholar] [CrossRef] [PubMed]
- Flucher, B.E. Skeletal muscle Cav1.1 channelopathies. Pflug. Arch. 2020, 472, 739–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telegrafi, A.; Webb, B.D.; Robbins, S.M.; Speck-Martins, C.E.; FitzPatrick, D.; Fleming, L.; Redett, R.; Dufke, A.; Houge, G.; van Harssel, J.J.; et al. Identification of STAC3 variants in non-Native American families with overlapping features of Carey-Fineman-Ziter syndrome and Moebius syndrome. Am. J. Med. Genet. A 2017, 173, 2763–2771. [Google Scholar] [CrossRef]
- Zaharieva, I.T.; Sarkozy, A.; Munot, P.; Manzur, A.; O’Grady, G.; Rendu, J.; Malfatti, E.; Amthor, H.; Servais, L.; Urtizberea, J.A.; et al. STAC3 variants cause a congenital myopathy with distinctive dysmorphic features and malignant hyperthermia susceptibility. Hum. Mutat. 2018, 39, 1980–1994. [Google Scholar] [CrossRef] [Green Version]
- Grzybowski, M.; Schänzer, A.; Pepler, A.; Heller, C.; Neubauer, B.A.; Hahn, A. Novel STAC3 Mutations in the First Non-Amerindian Patient with Native American Myopathy. Neuropediatrics 2017, 48, 451–455. [Google Scholar]
- Wong King Yuen, S.M.; Campiglio, M.; Tung, C.C.; Flucher, B.E.; Van Petegem, F. Structural insights into binding of STAC proteins to voltage-gated calcium channels. Proc. Natl. Acad. Sci. USA 2017, 114, E9520–E9528. [Google Scholar] [CrossRef] [Green Version]
- Campiglio, M.; Kaplan, M.M.; Flucher, B.E. STAC3 incorporation into skeletal muscle triads occurs independent of the dihydropyridine receptor. J. Cell. Physiol. 2018, 233, 9045–9051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M. The molecular physiology of CRAC channels. Immunol. Rev. 2009, 231, 88–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef] [Green Version]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell. Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef] [Green Version]
- González-Sánchez, P.; Del Arco, A.; Esteban, J.A.; Satrústegui, J. Store-Operated Calcium Entry Is Required for mGluR-Dependent Long Term Depression in Cortical Neurons. Front. Cell. Neurosci. 2017, 11, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunz, V.; Romanin, C.; Frischauf, I. STIM1 activation of Orai1. Cell Calcium 2019, 77, 29–38. [Google Scholar] [CrossRef]
- Conte, E.; Pannunzio, A.; Imbrici, P.; Camerino, G.M.; Maggi, L.; Mora, M.; Gibertini, S.; Cappellari, O.; De Luca, A.; Coluccia, M.; et al. Gain-of-Function STIM1 L96V Mutation Causes Myogenesis Alteration in Muscle Cells From a Patient Affected by Tubular Aggregate Myopathy. Front. Cell. Dev. Biol. 2021, 9, 635063. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, Y.; Zhou, Y.; Hendron, E.; Mancarella, S.; Andrake, M.D.; Rothberg, B.S.; Soboloff, J.; Gill, D.L. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun. 2014, 5, 3183. [Google Scholar] [CrossRef] [PubMed]
- Schaballie, H.; Rodriguez, R.; Martin, E.; Moens, L.; Frans, G.; Lenoir, C.; Dutré, J.; Canioni, D.; Bossuyt, X.; Fischer, A.; et al. A novel hypomorphic mutation in STIM1 results in a late-onset immunodeficiency. J. Allergy Clin. Immunol. 2015, 136, 816–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stormorken, H.; Sjaastad, O.; Langslet, A.; Sulg, I.; Egge, K.; Diderichsen, J. A new syndrome: Thrombocytopathia, muscle fatigue, asplenia, miosis, migraine, dyslexia and ichthyosis. Clin. Genet. 1985, 28, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Mizobuchi, M.; Tanaka, C.; Sako, K.; Murakami, N.; Nihira, A.; Abe, T.; Tateno, Y.; Takahashi, T.; Nonaka, I. Muscle involvement of Stormorken’s syndrome. Rinsho Shinkeigaku 2000, 40, 915–920. [Google Scholar] [PubMed]
- Markello, T.; Chen, D.; Kwan, J.Y.; Horkayne-Szakaly, I.; Morrison, A.; Simakova, O.; Maric, I.; Lozier, J.; Cullinane, A.R.; Kilo, T.; et al. York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1. Mol. Genet. Metab. 2015, 114, 474–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, C.H.S.; Allen, I.V.; Patterson, V.; Avaria, M.A. Dominantly inherited tubular aggregate myopathy. J. Pathol. 1992, 168, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Engel, W.K. Mitochondrial Aggregates In Muscle Disease. J. Histochem. Cytochem. 1964, 12, 46–48. [Google Scholar] [CrossRef] [Green Version]
- Nesin, V.; Wiley, G.; Kousi, M.; Ong, E.C.; Lehmann, T.; Nicholl, D.J.; Suri, M.; Shahrizaila, N.; Katsanis, N.; Gaffney, P.M.; et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc. Nat. Acad. Sci. USA 2014, 111, 4197–4202. [Google Scholar] [CrossRef] [Green Version]
- Morin, G.; Bruechle, N.O.; Singh, A.R.; Knopp, C.; Jedraszak, G.; Elbracht, M.; Brémond-Gignac, D.; Hartmann, K.; Sevestre, H.; Deutz, P.; et al. Gain-of-function mutation in STIM1 (P.R304W) is associated with Stormorken syndrome. Hum. Mutat. 2014, 35, 1221–1232. [Google Scholar] [CrossRef]
- Guergueltcheva, V.; Müller, J.S.; Dusl, M.; Senderek, J.; Oldfors, A.; Lindbergh, C.; Maxwell, S.; Colomer, J.; Mallebrera, C.J.; Nascimento, A.; et al. Congenital myasthenic syndrome with tubular aggregates caused by GFPT1 mutations. J. Neurol. 2012, 259, 838–850. [Google Scholar] [CrossRef]
- Belaya, K.; Finlayson, S.; Slater, C.R.; Cossins, J.; Liu, W.W.; Maxwell, S.; McGowan, S.J.; Maslau, S.; Twigg, S.R.; Walls, T.J.; et al. Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates. Am. J. Hum. Genet. 2012, 91, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, D.; Maisonobe, T.; Jurkat-Rott, K.; Nicole, S.; Launay, E.; Chauveau, D.; Tabti, N.; Lehmann-Horn, F.; Hainque, B.; Fontaine, B. Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain 2001, 124, 1091–1099. [Google Scholar] [CrossRef] [Green Version]
- Böhm, J.; Chevessier, F.; De Paula, A.M.; Koch, C.; Attarian, S.; Feger, C.; Hantaï, D.; Laforêt, P.; Ghorab, K.; Vallat, J.M.; et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am. J. Hum. Genet. 2013, 92, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böhm, J.; Chevessier, F.; Koch, C.; Peche, G.A.; Mora, M.; Morandi, L.; Pasanisi, B.; Moroni, I.; Tasca, G.; Fattori, F.; et al. Clinical, histological and genetic characterisation of patients with tubular aggregate myopathy caused by mutations in STIM1. J. Med. Genet. 2014, 51, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Hedberg, C.; Niceta, M.; Fattori, F.; Lindvall, B.; Ciolfi, A.; D’Amico, A.; Tasca, G.; Petrini, S.; Tulinius, M.; Tartaglia, M.; et al. Childhood onset tubular aggregate myopathy associated with de novo STIM1 mutations. J. Neurol. 2014, 261, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Claeys, T.; Goosens, V.; Racé, V.; Theys, T.; Thal, D.R.; Depuydt, C.E.; Claeys, K.G. Clinical and muscle MRI features in a family with tubular aggregate myopathy and novel STIM1 mutation. Neuromuscul. Disord. 2020, 30, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Silva-Rojas, R.; Laporte, J.; Böhm, J. STIM1/ORAI1 Loss-of-Function and Gain-of-Function Mutations Inversely Impact on SOCE and Calcium Homeostasis and Cause Multi-Systemic Mirror Diseases. Front. Physiol. 2020, 11, 604941. [Google Scholar] [CrossRef] [PubMed]
- Misceo, D.; Holmgren, A.; Louch, W.E.; Holme, P.A.; Mizobuchi, M.; Morales, R.J.; De Paula, A.M.; Stray-Pedersen, A.; Lyle, R.; Dalhus, B.; et al. A dominant STIM1 mutation causes Stormorken syndrome. Hum. Mutat. 2014, 35, 556–564. [Google Scholar] [CrossRef]
- Endo, Y.; Noguchi, S.; Hara, Y.; Hayashi, Y.K.; Motomura, K.; Miyatake, S.; Murakami, N.; Tanaka, S.; Yamashita, S.; Kizu, R.; et al. Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca(2)(+) channels. Hum. Mol. Genet. 2015, 24, 637–648. [Google Scholar] [CrossRef] [Green Version]
- Böhm, J.; Bulla, M.; Urquhart, J.E.; Malfatti, E.; Williams, S.G.; O’Sullivan, J.; Szlauer, A.; Koch, C.; Baranello, G.; Mora, M.; et al. ORAI1 Mutations with Distinct Channel Gating Defects in Tubular Aggregate Myopathy. Hum. Mutat. 2017, 38, 426–438. [Google Scholar] [CrossRef] [Green Version]
- Garibaldi, M.; Fattori, F.; Riva, B.; Labasse, C.; Brochier, G.; Ottaviani, P.; Sacconi, S.; Vizzaccaro, E.; Laschena, F.; Romero, N.B.; et al. A novel gain-of-function mutation in ORAI1 causes late-onset tubular aggregate myopathy and congenital miosis. Clin. Genet. 2017, 91, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Stauderman, K.A. CRAC channels as targets for drug discovery and development. Cell. Calcium. 2018, 74, 147–159. [Google Scholar] [CrossRef]
- Phillips, M.S.; Fujii, J.; Khanna, V.K.; DeLeon, S.; Yokobata, K.; de Jong, P.J.; MacLennan, D.H. The structural organization of the human skeletal muscle ryanodine receptor (RYR1) gene. Genomics 1996, 34, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.J.D.; Gonorazky, H.; Cohn, R.D.; Campbell, C. Treating pediatric neuromuscular disorders: The future is now. Am. J. Med. Genet. A 2018, 176, 804–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.; Lillis, S.; Munteanu, I.; Scoto, M.; Zhou, H.; Quinlivan, R.; Straub, V.; Manzur, A.Y.; Roper, H.; Jeannet, P.Y.; et al. Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene-associated myopathies. Hum. Mutat. 2012, 33, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Dlamini, N.; Voermans, N.C.; Lillis, S.; Stewart, K.; Kamsteeg, E.J.; Drost, G.; Quinlivan, R.; Snoeck, M.; Norwood, F.; Radunovic, A.; et al. Mutations in RYR1 are a common cause of exertional myalgia and rhabdomyolysis. Neuromuscul. Disord. 2013, 23, 540–548. [Google Scholar] [CrossRef]
- Løseth, S.; Voermans, N.C.; Torbergsen, T.; Lillis, S.; Jonsrud, C.; Lindal, S.; Kamsteeg, E.J.; Lammens, M.; Broman, M.; Dekomien, G.; et al. A novel late-onset axial myopathy associated with mutations in the skeletal muscle ryanodine receptor (RYR1) gene. J. Neurol. 2013, 260, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Liang, Y.; Gong, H.; Zhou, N.; Ma, H.; Guan, A.; Sun, A.; Wang, P.; Niu, Y.; Jiang, H.; et al. Ryanodine receptor type 2 is required for the development of pressure overload-induced cardiac hypertrophy. Hypertension 2011, 58, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Rokach, O.; Feng, L.; Munteanu, I.; Mamchaoui, K.; Wilmshurst, J.M.; Sewry, C.; Manzur, A.Y.; Pillay, K.; Mouly, V.; et al. RyR1 deficiency in congenital myopathies disrupts excitation-contraction coupling. Hum. Mutat. 2013, 34, 986–996. [Google Scholar] [CrossRef] [Green Version]
- Garibaldi, M.; Rendu, J.; Brocard, J.; Lacene, E.; Fauré, J.; Brochier, G.; Beuvin, M.; Labasse, C.; Madelaine, A.; Malfatti, E.; et al. Dusty core disease (DuCD): Expanding morphological spectrum of RYR1 recessive myopathies. Acta Neuropathol. Commun. 2019, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Neuwirth, C.; Jaffer, F.; Scalco, R.S.; Fialho, D.; Parton, M.; Rayan, D.R.; Suetterlin, K.; Sud, R.; Spiegel, R.; et al. Atypical periodic paralysis and myalgia: A novel RYR1 phenotype. Neurology 2018, 90, e412–e418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawal, T.A.; Todd, J.J.; Witherspoon, J.W.; Bönnemann, C.G.; Dowling, J.J.; Hamilton, S.L.; Meilleur, K.G.; Dirksen, R.T. Ryanodine receptor 1-related disorders: An historical perspective and proposal for a unified nomenclature. Skelet. Muscle 2020, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.R.; Haan, E.; Jungbluth, H.; Sewry, C.; North, K.; Muntoni, F.; Kuntzer, T.; Lamont, P.; Bankier, A.; Tomlinson, P.; et al. Principal mutation hotspot for central core disease and related myopathies in the C-terminal transmembrane region of the RYR1 gene. Neuromuscul. Disord. 2003, 13, 151–157. [Google Scholar] [CrossRef]
- Levano, S.; Vukcevic, M.; Singer, M.; Matter, A.; Treves, S.; Urwyler, A.; Girard, T. Increasing the number of diagnostic mutations in malignant hyperthermia. Hum. Mutat. 2009, 30, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.; Carpenter, D.; Shaw, M.A.; Halsall, J.; Hopkins, P. Mutations in RYR1 in malignant hyperthermia and central core disease. Hum. Mutat. 2006, 27, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Ibarra, M.C.; Malicdan, M.C.; Murayama, K.; Ichihara, Y.; Kikuchi, H.; Nonaka, I.; Noguchi, S.; Hayashi, Y.K.; Nishino, I. Central core disease is due to RYR1 mutations in more than 90% of patients. Brain 2006, 129, 1470–1480. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Jungbluth, H.; Sewry, C.A.; Feng, L.; Bertini, E.; Bushby, K.; Straub, V.; Roper, H.; Rose, M.R.; Brockington, M.; et al. Molecular mechanisms and phenotypic variation in RYR1-related congenital myopathies. Brain 2007, 130, 2024–2036. [Google Scholar] [CrossRef] [Green Version]
- Maggi, L.; Scoto, M.; Cirak, S.; Robb, S.A.; Klein, A.; Lillis, S.; Cullup, T.; Feng, L.; Manzur, A.Y.; Sewry, C.A.; et al. Congenital myopathies—clinical features and frequency of individual subtypes diagnosed over a 5-year period in the United Kingdom. Neuromuscul. Disord. 2013, 23, 195–205. [Google Scholar] [CrossRef]
- Colombo, I.; Scoto, M.; Manzur, A.Y.; Robb, S.A.; Maggi, L.; Gowda, V.; Cullup, T.; Yau, M.; Phadke, R.; Sewry, C.; et al. Congenital myopathies: Natural history of a large pediatric cohort. Neurology 2015, 84, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungbluth, H. Central core disease. Orphanet J. Rare Dis. 2007, 2, 25. [Google Scholar] [CrossRef] [Green Version]
- Jungbluth, H. Multi-minicore disease. Orphanet J. Rare Dis. 2007, 2, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, N.F.; Waddell, L.B.; Cooper, S.T.; Perry, M.; Smith, R.L.; Kornberg, A.J.; Muntoni, F.; Lillis, S.; Straub, V.; Bushby, K.; et al. Recessive mutations in RYR1 are a common cause of congenital fibre type disproportion. Hum. Mutat. 2010, 31, e1544–e1550. [Google Scholar] [CrossRef] [PubMed]
- Monnier, N.; Marty, I.; Faure, J.; Castiglioni, C.; Desnuelle, C.; Sacconi, S.; Estournet, B.; Ferreiro, A.; Romero, N.; Laquerriere, A.; et al. Null mutations causing depletion of the type 1 ryanodine receptor (RYR1) are commonly associated with recessive structural congenital myopathies with cores. Hum. Mutat. 2008, 29, 670–678. [Google Scholar] [CrossRef]
- Monnier, N.; Romero, N.B.; Lerale, J.; Nivoche, Y.; Qi, D.; MacLennan, D.H.; Fardeau, M.; Lunardi, J. An autosomal dominant congenital myopathy with cores and rods is associated with a neomutation in the RYR1 gene encoding the skeletal muscle ryanodine receptor. Hum. Mol. Genet. 2000, 9, 2599–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungbluth, H.; Dowling, J.J.; Ferreiro, A.; Muntoni, F. 182nd ENMC International Workshop: RYR1-related myopathies, 15-17th April 2011, Naarden, The Netherlands. Neuromuscul. Disord. 2012, 22, 453–462. [Google Scholar] [CrossRef]
- Wilmshurst, J.M.; Lillis, S.; Zhou, H.; Pillay, K.; Henderson, H.; Kress, W.; Müller, C.R.; Ndondo, A.; Cloke, V.; Cullup, T.; et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann. Neurol. 2010, 68, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Ferreiro, A.; Monnier, N.; Romero, N.B.; Leroy, J.P.; Bönnemann, C.; Haenggeli, C.A.; Straub, V.; Voss, W.D.; Nivoche, Y.; Jungbluth, H.; et al. A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann. Neurol. 2002, 51, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.B.; Monnier, N.; Viollet, L.; Cortey, A.; Chevallay, M.; Leroy, J.P.; Lunardi, J.; Fardeau, M. Dominant and recessive central core disease associated with RYR1 mutations and fetal akinesia. Brain 2003, 126, 2341–2349. [Google Scholar] [CrossRef] [PubMed]
- De Wel, B.; Claeys, K.G. Malignant hyperthermia: Still an issue for neuromuscular diseases? Curr. Opin. Neurol. 2018, 31, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, D.; Robinson, R.L.; Quinnell, R.J.; Ringrose, C.; Hogg, M.; Casson, F.; Booms, P.; Iles, D.E.; Halsall, P.J.; Steele, D.S.; et al. Genetic variation in RYR1 and malignant hyperthermia phenotypes. Br. J. Anaesth. 2009, 103, 538–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, H.; Pollock, N.; Schiemann, A.; Bulger, T.; Stowell, K. Malignant hyperthermia: A review. Orphanet J. Rare Dis. 2015, 10, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann-Horn, F.; Iaizzo, P.A. Are myotonias and periodic paralyses associated with susceptibility to malignant hyper—thermia? Br. J. Anaesth. 1990, 65, 692–697. [Google Scholar] [CrossRef]
- Dowling, J.J.; Lillis, S.; Amburgey, K.; Zhou, H.; Al-Sarraj, S.; Buk, S.J.; Wraige, E.; Chow, G.; Abbs, S.; Leber, S.; et al. King-Denborough syndrome with and without mutations in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul. Disord. 2011, 21, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Jungbluth, H.; Lillis, S.; Zhou, H.; Abbs, S.; Sewry, C.; Swash, M.; Muntoni, F. Late-onset axial myopathy with cores due to a novel heterozygous dominant mutation in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul. Disord. 2009, 19, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lillis, S.; Loy, R.E.; Ghassemi, F.; Rose, M.R.; Norwood, F.; Mills, K.; Al-Sarraj, S.; Lane, R.J.; Feng, L.; et al. Multi-minicore disease and atypical periodic paralysis associated with novel mutations in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul. Disord. 2010, 20, 166–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertocchini, F.; Ovitt, C.E.; Conti, A.; Barone, V.; Schöler, H.R.; Bottinelli, R.; Reggiani, C.; Sorrentino, V. Requirement for the ryanodine receptor type 3 for efficient contraction in neonatal skeletal muscles. EMBO J. 1997, 16, 6956–6963. [Google Scholar] [CrossRef]
- Perez, C.F.; López, J.R.; Allen, P.D. Expression levels of RyR1 and RyR3 control resting free Ca2+ in skeletal muscle. Am. J. Physiol. Cell. Physiol. 2005, 88, C640–C649. [Google Scholar] [CrossRef] [PubMed]
- Nilipour, Y.; Nafissi, S.; Tjust, A.E.; Ravenscroft, G.; Hossein Nejad Nedai, H.; Taylor, R.L.; Varasteh, V.; Pedrosa Domellöf, F.; Zangi, M.; Tonekaboni, S.H.; et al. Ryanodine receptor type 3 (RYR3) as a novel gene associated with a myopathy with nemaline bodies. Eur. J. Neurol. 2018, 25, 841–847. [Google Scholar] [CrossRef]
- Krüger, J.; Kunert-Keil, C.; Bisping, F.; Brinkmeier, H. Transient receptor potential cation channels in normal and dystrophic mdx muscle. Neuromuscul. Disord. 2008, 18, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.; Horn, N.A.; Huynh, T.; Kelava, L.; Lansman, J.B. Evidence TRPV4 contributes to mechanosensitive ion channels in mouse skeletal muscle fibers. Channels 2012, 6, 246–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritschow, B.W.; Lange, T.; Kasch, J.; Kunert-Keil, C.; Liedtke, W.; Brinkmeier, H. Functional TRPV4 channels are expressed in mouse skeletal muscle and can modulate resting Ca2+ influx and muscle fatigue. Pflug. Arch. 2011, 46, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Auer-Grumbach, M.; Olschewski, A.; Papić, L.; Kremer, H.; McEntagart, M.E.; Uhrig, S.; Fischer, C.; Fröhlich, E.; Bálint, Z.; Tang, B.; et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat. Genet. 2010, 42, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Klein, C.J.; Yan, J.; Shi, Y.; Wu, Y.; Fecto, F.; Yau, H.J.; Yang, Y.; Zhai, H.; Siddique, N.; et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat. Genet. 2010, 42, 165–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landouré, G.; Zdebik, A.A.; Martinez, T.L.; Burnett, B.G.; Stanescu, H.C.; Inada, H.; Shi, Y.; Taye, A.A.; Kong, L.; Munns, C.H.; et al. Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C. Nat. Genet. 2010, 42, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Zimoń, M.; Baets, J.; Auer-Grumbach, M.; Berciano, J.; Garcia, A.; Lopez-Laso, E.; Merlini, L.; Hilton-Jones, D.; McEntagart, M.; Crosby, A.H.; et al. Dominant mutations in the cation channel gene transient receptor potential vanilloid 4 cause an unusual spectrum of neuropathies. Brain 2010, 133, 1798–1809. [Google Scholar] [CrossRef] [Green Version]
- Rock, M.J.; Prenen, J.; Funari, V.A.; Funari, T.L.; Merriman, B.; Nelson, S.F.; Lachman, R.S.; Wilcox, W.R.; Reyno, S.; Quadrelli, R.; et al. Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat. Genet. 2008, 40, 999–1003. [Google Scholar] [CrossRef] [Green Version]
- Krakow, D.; Vriens, J.; Camacho, N.; Luong, P.; Deixler, H.; Funari, T.L.; Bacino, C.A.; Irons, M.B.; Holm, I.A.; Sadler, L.; et al. Mutations in the gene encoding the calcium-permeable ion channel TRPV4 produce spondylometaphyseal dysplasia, Kozlowski type and metatropic dysplasia. Am. J. Hum. Genet. 2009, 84, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, T.J.; Matsumoto, K.; Fano, V.; Dai, J.; Kim, O.H.; Chae, J.H.; Yoo, W.J.; Tanaka, Y.; Matsui, Y.; Takigami, I.; et al. TRPV4-pathy manifesting both skeletal dysplasia and peripheral neuropathy: A report of three patients. Am. J. Med. Genet. A 2012, 158A, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Dubourg, O.; Carlier, P.; Carlier, R.Y.; Sabouraud, P.; Péréon, Y.; Chapon, F.; Thauvin-Robinet, C.; Laforêt, P.; Eymard, B.; et al. Phenotypic spectrum and incidence of TRPV4 mutations in patients with inherited axonal neuropathy. Neurology 2014, 82, 1919–1926. [Google Scholar] [CrossRef]
- Oonk, A.M.; Ekker, M.S.; Huygen, P.L.; Kunst, H.P.; Kremer, H.; Schelhaas, J.J.; Pennings, R.J. Intrafamilial variable hearing loss in TRPV4 induced spinal muscular atrophy. Ann. Otol. Rhinol. Laryngol. 2014, 123, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, T.; Bansagi, B.; Pyle, A.; Griffin, H.; Douroudis, K.; Polvikoski, T.; Antoniadi, T.; Bushby, K.; Straub, V.; Chinnery, P.F.; et al. Phenotypic variability of TRPV4 related neuropathies. Neuromuscul. Disord. 2015, 25, 516–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutsis, G.; Lynch, D.; Manole, A.; Karadima, G.; Reilly, M.M.; Houlden, H.; Panas, M. Charcot-Marie-Tooth disease type 2C and scapuloperoneal muscular atrophy overlap syndrome in a patient with the R232C TRPV4 mutation. J. Neurol. 2015, 262, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Ürel-Demir, G.; Şimşek-Kiper, P.Ö.; Öncel, İ.; Utine, G.E.; Haliloğlu, G.; Boduroğlu, K. Natural history of TRPV4-Related disorders: From skeletal dysplasia to neuromuscular phenotype. Eur. J. Paediatr. Neurol. 2021, 32, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.M.; Zimanyi, C.M.; Aisenberg, W.; Bears, B.; Chen, D.H.; Day, J.W.; Bird, T.D.; Siskind, C.E.; Gaudet, R.; Sumner, C.J. Novel mutations highlight the key role of the ankyrin repeat domain in TRPV4-mediated neuropathy. Neurol. Genet. 2015, 1, e29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolums, B.M.; McCray, B.A.; Sung, H.; Tabuchi, M.; Sullivan, J.M.; Ruppell, K.T.; Yang, Y.; Mamah, C.; Aisenberg, W.H.; Saavedra-Rivera, P.C.; et al. TRPV4 disrupts mitochondrial transport and causes axonal degeneration via a CaMKII-dependent elevation of intracellular Ca2+. Nat. Commun. 2020, 11, 2679. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.; Novak, K.; Denman, K.; Myers, J.H.; Sullivan, J.M.; Walker, P.V., 2nd. TRPV4 Antagonism Prevents Mechanically Induced Myotonia. Ann. Neurol. 2020, 88, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Goyal, N.; Skrdla, P.; Schroyer, R.; Kumar, S.; Fernando, D.; Oughton, A.; Norton, N.; Sprecher, D.L.; Cheriyan, J. Clinical Pharmacokinetics, Safety, and Tolerability of a Novel, First-in-Class TRPV4 Ion Channel Inhibitor, GSK2798745, in Healthy and Heart Failure Subjects. Am. J. Cardiovasc. Drugs 2019, 19, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, M.T.; McMillan, H.J.; Tomin, A.; Weiss, N. Compound heterozygous CACNA1H mutations associated with severe congenital amyotrophy. Channels 2019, 13, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Tawil, R.; Ptacek, L.J.; Pavlakis, S.G.; DeVivo, D.C.; Penn, A.S.; Ozdemir, C.; Griggs, R.C. Andersen’s syndrome: Potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann. Neurol. 1994, 35, 326–330. [Google Scholar] [CrossRef]
- Sansone, V.; Tawil, R. Management and treatment of Andersen-Tawil syndrome (ATS). Neurotherapeutics 2007, 4, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Riera, A.R.; Barbosa-Barros, R.; Samesina, N.; Pastore, C.A.; Scanavacca, M.; Daminello-Raimundo, R.; de Abreu, L.C.; Nikus, K.; Brugada, P. Andersen-Tawil Syndrome: A Comprehensive Review. Cardiol. Rev. 2021, 29, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.L.; Pieper, G.H.; Wilders, R. Andersen-Tawil syndrome: Clinical and molecular aspects. Int. J. Cardiol. 2013, 170, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Child, N.D.; Cleland, J.C.; Roxburgh, R. Andersen-Tawil syndrome presenting as a fixed myopathy. Muscle Nerve 2013, 48, 623. [Google Scholar] [CrossRef] [PubMed]
- Lefter, S.; Hardiman, O.; Costigan, D.; Lynch, B.; McConville, J.; Hand, C.K.; Ryan, A.M. Andersen-Tawil syndrome with early fixed myopathy. J. Clin. Neuromuscul. Dis. 2014, 16, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Oz Tuncer, G.; Teber, S.; Kutluk, M.G.; Albayrak, P.; Deda, G. Andersen-Tawil Syndrome with Early Onset Myopathy: 2 Cases. J. Neuromuscul. Dis. 2017, 4, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Fang, Y.; Zhang, B.R. Andersen-Tawil syndrome associated with myopathy. World J. Emerg. Med. 2020, 11, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Hiraide, T.; Fukumura, S.; Yamamoto, A.; Nakashima, M.; Saitsu, H. Familial periodic paralysis associated with a rare KCNJ5 variant that supposed to have incomplete penetrance. Brain. Dev. 2021, 43, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Plaster, N.M.; Tawil, R.; Tristani-Firouzi, M.; Canún, S.; Bendahhou, S.; Tsunoda, A.; Donaldson, M.R.; Iannaccone, S.T.; Brunt, E.; Barohn, R.; et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell 2001, 105, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.M.; Zhang, H.; Rohacs, T.; Jin, T.; Yang, J.; Logothetis, D.E. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 2002, 34, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Ballester, L.Y.; Benson, D.W.; Wong, B.; Law, I.H.; Mathews, K.D.; Vanoye, C.G.; George, A.L., Jr. Trafficking-competent and trafficking-defective KCNJ2 mutations in Andersen syndrome. Hum. Mutat. 2006, 27, 388. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Tang, X.D.; Rogers, T.B.; Welling, P.A. An andersen-Tawil syndrome mutation in Kir2.1 (V302M) alters the G-loop cytoplasmic K+ conduction pathway. J. Biol. Chem. 2007, 282, 5781–5789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handklo-Jamal, R.; Meisel, E.; Yakubovich, D.; Vysochek, L.; Beinart, R.; Glikson, M.; McMullen, J.R.; Dascal, N.; Nof, E.; Oz, S. Andersen-Tawil Syndrome Is Associated With Impaired PIP2 Regulation of the Potassium Channel Kir2.1. Front. Pharmacol. 2020, 11, 672. [Google Scholar] [CrossRef]
- Bendahhou, S.; Donaldson, M.R.; Plaster, N.M.; Tristani-Firouzi, M.; Fu, Y.H.; Ptácek, L.J. Defective potassium channel Kir2.1 trafficking underlies Andersen-Tawil syndrome. J. Biol. Chem. 2003, 278, 51779–51785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendahhou, S.; Fournier, E.; Sternberg, D.; Bassez, G.; Furby, A.; Sereni, C.; Donaldson, M.R.; Larroque, M.M.; Fontaine, B.; Barhanin, J. In vivo and in vitro functional characterization of Andersen’s syndrome mutations. J. Physiol. 2005, 565, 731–741. [Google Scholar] [CrossRef]
- Sacco, S.; Giuliano, S.; Sacconi, S.; Desnuelle, C.; Barhanin, J.; Amri, E.Z.; Bendahhou, S. The inward rectifier potassium channel Kir2.1 is required for osteoblastogenesis. Hum. Mol. Genet. 2015, 24, 471–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belus, M.T.; Rogers, M.A.; Elzubeir, A.; Josey, M.; Rose, S.; Andreeva, V.; Yelick, P.C.; Bates, E.A. Kir2.1 is important for efficient BMP signaling in mammalian face development. Dev. Biol. 2018, 444, S297–S307. [Google Scholar] [CrossRef] [PubMed]
- Pini, J.; Giuliano, S.; Matonti, J.; Gannoun, L.; Simkin, D.; Rouleau, M.; Bendahhou, S. Osteogenic and Chondrogenic Master Genes Expression Is Dependent on the Kir2.1 Potassium Channel Through the Bone Morphogenetic Protein Pathway. J. Bone Miner. Res. 2018, 33, 1826–1841. [Google Scholar] [CrossRef]
- Preisig-Müller, R.; Schlichthörl, G.; Goerge, T.; Heinen, S.; Brüggemann, A.; Rajan, S.; Derst, C.; Veh, R.W.; Daut, J. Heteromerization of Kir2.x potassium channels contributes to the phenotype of Andersen’s syndrome. Proc. Natl. Acad. Sci. USA 2002, 99, 7774–7779. [Google Scholar] [CrossRef] [Green Version]
- Decher, N.; Renigunta, V.; Zuzarte, M.; Soom, M.; Heinemann, S.H.; Timothy, K.W.; Keating, M.T.; Daut, J.; Sanguinetti, M.C.; Splawski, I. Impaired interaction between the slide helix and the C-terminus of Kir2.1: A novel mechanism of Andersen syndrome. Cardiovasc. Res. 2007, 75, 748–757. [Google Scholar] [CrossRef]
- Jagodzińska, M.; Szperl, M.; Ponińska, J.; Kosiec, A.; Gajda, R.; Kukla, P.; Biernacka, E.K. Coexistence of Andersen-Tawil Syndrome with Polymorphisms in hERG1 Gene (K897T) and SCN5A Gene (H558R) in One Family. Ann. Noninvasive Electrocardiol. 2016, 21, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyak, M.E.; Shestak, A.; Podolyak, D.; Zaklyazminskaya, E. Compound heterozygous mutations in KCNJ2 and KCNH2 in a patient with severe Andersen-Tawil syndrome. BMJ Case Rep. 2020, 13, e235703. [Google Scholar] [CrossRef]
- Priori, S.G.; Pandit, S.V.; Rivolta, I.; Berenfeld, O.; Ronchetti, E.; Dhamoon, A.; Napolitano, C.; Anumonwo, J.; Di Barletta, M.R.; Gudapakkam, S.; et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res. 2005, 96, 800–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, M.; Jin, Q.; Bendahhou, S.; He, Y.; Larroque, M.M.; Chen, Y.; Zhou, Q.; Yang, Y.; Liu, Y.; Liu, B.; et al. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem. Biophys. Res. Commun. 2005, 332, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Y.; Liang, B.; Liu, J.; Li, J.; Grunnet, M.; Olesen, S.P.; Rasmussen, H.B.; Ellinor, P.T.; Gao, L.; et al. Identification of a Kir3.4 mutation in congenital long QT syndrome. Am. J. Hum. Genet. 2010, 86, 872–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokunai, Y.; Nakata, T.; Furuta, M.; Sakata, S.; Kimura, H.; Aiba, T.; Yoshinaga, M.; Osaki, Y.; Nakamori, M.; Itoh, H.; et al. A Kir3.4 mutation causes Andersen-Tawil syndrome by an inhibitory effect on Kir2.1. Neurology 2014, 82, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, S.; Sakoda, S.; Koide, R.; Kawata, A.; Yuan, J.; Takashima, H.; Nakano, I. A case of Andersen-Tawil syndrome presenting periodic paralysis exacerbated by ACZ. J. Neurol. Sci. 2014, 347, 385–386. [Google Scholar] [CrossRef]
- Abott, G.W.; Butler, M.H.; Bendahhou, S.; Dalakas, M.C.; Ptacek, L.J.; Goldstein, S.A. MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell 2001, 104, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Dias Da Silva, M.R.; Cerutti, J.M.; Arnaldi, L.A.; Maciel, R.M. A mutation in the KCNE3 potassium channel gene is associated with susceptibility to thyrotoxic hypokalemic periodic paralysis. J. Clin. Endocrinol. Metab. 2002, 87, 4881–4884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternberg, D.; Tabti, N.; Fournier, E.; Hainque, B.; Fontaine, B. Lack of association of the potassium channel-associated peptide MiRP2-R83H variant with periodic paralysis. Neurology 2003, 61, 857–859. [Google Scholar] [CrossRef]
- Jurkat-Rott, K.; Lehmann-Horn, F. Periodic paralysis mutation MiRP2-R83H in controls: Interpretations and general recommendation. Neurology 2004, 62, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.L.; Chow, C.C.; Ko, G.T.; Tai, M.H.; Kwok, R.; Yao, X.Q.; Cockram, C.S. No mutation in the KCNE3 potassium channel gene in Chinese thyrotoxic hypokalaemic periodic paralysis patients. Clin. Endocrinol. 2004, 61, 109–112. [Google Scholar] [CrossRef]
- Abbott, G.W.; Butler, M.H.; Goldstein, S.A. Phosphorylation and protonation of neighboring MiRP2 sites: Function and pathophysiology of MiRP2-Kv3.4 potassium channels in periodic paralysis. FASEB J. 2006, 20, 293–301. [Google Scholar] [CrossRef]
- Portaro, S.; Altamura, C.; Licata, N.; Camerino, G.M.; Imbrici, P.; Musumeci, O.; Rodolico, C.; Camerino, D.C.; Toscano, A.; Desaphy, J.F. Clinical, Molecular, and Functional Characterization of CLCN1 Mutations in Three Families with Recessive Myotonia Congenita. Neuromol. Med. 2015, 17, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbrici, P.; Accogli, A.; Blunck, R.; Altamura, C.; Iacomino, M.; D’adamo, M.C.; Allegri, A.; Pedemonte, M.; Brolatti, N.; Vari, S.; et al. Musculoskeletal Features without Ataxia Associated with a Novel de novo Mutation in KCNA1 Impairing the Voltage Sensitivity of Kv1.1 Channel. Biomedicines 2021, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Salih, M.; van Kinschot, C.M.; Peeters, R.P.; de Herder, W.W.; Duschek, E.J.; Van Der Linden, J.; van Noord, C. Thyrotoxic periodic paralysis: An unusual presentation of hyperthyroidism. Neth. J. Med. 2017, 75, 315–320. [Google Scholar]
- Chu, P.Y.; Cheng, C.J.; Tseng, M.H.; Yang, S.S.; Chen, H.C.; Lin, S.H. Genetic variant rs623011 (17q24.3) associates with non-familial thyrotoxic and sporadic hypokalemic paralysis. Clin. Chim. Acta 2012, 414, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Song, I.W.; Sung, C.C.; Chen, C.H.; Cheng, C.J.; Yang, S.S.; Chou, Y.C.; Yang, J.H.; Chen, Y.T.; Wu, J.Y.; Lin, S.H. Novel susceptibility gene for nonfamilial hypokalemic periodic paralysis. Neurology 2016, 86, 1190–1198. [Google Scholar] [CrossRef]
- Nakaza, M.; Kitamura, Y.; Furuta, M.; Kubota, T.; Sasaki, R.; Takahashi, M.P. Analysis of the genetic background associated with sporadic periodic paralysis in Japanese patients. J. Neurol. Sci. 2020, 412, 116795. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.C. Further evidence for shared genetic susceptibility in both sporadic and thyrotoxic periodic paralysis. J. Neurol. Sci. 2020, 412, 116794. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Da Silva, M.R.; Soong, T.W.; Fontaine, B.; Donaldson, M.R.; Kung, A.W.; Jongjaroenprasert, W.; Liang, M.C.; Khoo, D.H.; Cheah, J.S.; et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell 2010, 140, 88–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.J.; Lin, S.H.; Lo, Y.F.; Yang, S.S.; Hsu, Y.J.; Cannon, S.C.; Huang, C.L. Identification and functional characterization of Kir2.6 mutations associated with non-familial hypokalemic periodic paralysis. J. Biol. Chem. 2011, 286, 27425–27435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Liang, Z.; Hou, Y.; Liu, F.; Hu, Y.; Lin, P.; Yan, C. A novel Kir2.6 mutation associated with hypokalemic periodic paralysis. Clin. Neurophysiol. 2016, 127, 2503–2508. [Google Scholar] [CrossRef] [PubMed]
- Paninka, R.M.; Carlos-Lima, E.; Lindsey, S.C.; Kunii, I.S.; Dias-da-Silva, M.R.; Arcisio-Miranda, M. Down-regulation of Kir2.6 channel by c-termini mutation D252N and its association with the susceptibility to Thyrotoxic Periodic Paralysis. Neuroscience 2017, 346, 197–202. [Google Scholar] [CrossRef]
- Cheung, C.L.; Lau, K.S.; Ho, A.Y.; Lee, K.K.; Tiu, S.C.; Lau, E.Y.; Leung, J.; Tsang, M.W.; Chan, K.W.; Yeung, C.Y.; et al. Genome-wide association study identifies a susceptibility locus for thyrotoxic periodic paralysis at 17q24.3. Nat. Genet. 2012, 44, 1026–1029. [Google Scholar] [CrossRef] [PubMed]
- Jongjaroenprasert, W.; Phusantisampan, T.; Mahasirimongkol, S.; Mushiroda, T.; Hirankarn, N.; Snabboon, T.; Chanprasertyotin, S.; Tantiwong, P.; Soonthornpun, S.; Rattanapichart, P.; et al. A genome-wide association study identifies novel susceptibility genetic variation for thyrotoxic hypokalemic periodic paralysis. J. Hum. Genet. 2012, 57, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yao, S.; Xiang, Y.; Zhang, X.; Wu, X.; Luo, L.; Huang, H.; Zhu, M.; Wan, H.; Hong, D. The clinical and genetic features in a cohort of mainland Chinese patients with thyrotoxic periodic paralysis. BMC Neurol. 2015, 15, 38. [Google Scholar] [CrossRef] [Green Version]
- Rolim, A.L.; Lindsey, S.C.; Kunii, I.S.; Crispim, F.; Moisés, R.C.; Maciel, R.M.; Dias-da-Silva, M.R. The insulin-sensitivity sulphonylurea receptor variant is associated with thyrotoxic paralysis. J. Mol. Endocrinol. 2014, 53, 295–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M.; Jurkat-Rott, K.; Lehmann-Horn, F. Rare KCNJ18 variants do not explain hypokalaemic periodic paralysis in 263 unrelated patients. J. Neurol. Neurosurg. Psychiatry 2016, 87, 49–52. [Google Scholar] [PubMed] [Green Version]
- Paninka, R.M.; Mazzotti, D.R.; Kizys, M.M.; Vidi, A.C.; Rodrigues, H.; Silva, S.P.; Kunii, I.S.; Furuzawa, G.K.; Arcisio-Miranda, M.; Dias-da-Silva, M.R. Whole genome and exome sequencing realignment supports the assignment of KCNJ12, KCNJ17, and KCNJ18 paralogous genes in thyrotoxic periodic paralysis locus: Functional characterization of two polymorphic Kir2.6 isoforms. Mol. Genet. Genom. 2016, 291, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Smeland, M.F.; McClenaghan, C.; Roessler, H.I.; Savelberg, S.; Hansen, G.Å.; Hjellnes, H.; Arntzen, K.A.; Müller, K.I.; Dybesland, A.R.; Harter, T.; et al. ABCC9-related Intellectual disability Myopathy Syndrome is a KATP channelopathy with loss-of-function mutations in ABCC9. Nat. Commun. 2019, 10, 4457. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, D.; Mele, A.; Lundquist, A.L.; Desai, R.R.; George, A.L., Jr.; Conte Camerino, D. Hybrid assemblies of ATP-sensitive K+ channels determine their muscle-type-dependent biophysical and pharmacological properties. Proc. Natl. Acad. Sci. USA 2006, 103, 1118–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flagg, T.P.; Enkvetchakul, D.; Koster, J.C.; Nichols, C.G. Muscle KATP channels: Recent insights to energy sensing and myoprotection. Physiol. Rev. 2010, 90, 799–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, D.L.; Gancher, S.T.; Nutt, J.G.; Brunt, E.R.; Smith, E.A.; Kramer, P.; Litt, M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat. Genet. 1994, 8, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Adelman, J.P.; Bond, C.T.; Pessia, M.; Maylie, J. Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron 1995, 15, 1449–1454. [Google Scholar] [CrossRef] [Green Version]
- D’Adamo, M.C.; Liantonio, A.; Rolland, J.F.; Pessia, M.; Imbrici, P. Kv1.1 Channelopathies: Pathophysiological Mechanisms and Therapeutic Approaches. Int. J. Mol. Sci. 2020, 21, 2935. [Google Scholar] [CrossRef] [PubMed]
- Paulhus, K.; Ammerman, L.; Glasscock, E. Clinical Spectrum of KCNA1 Mutations: New Insights into Episodic Ataxia and Epilepsy Comorbidity. Int. J. Mol. Sci. 2020, 21, 2802. [Google Scholar] [CrossRef]
- Herson, P.S.; Virk, M.; Rustay, N.R.; Bond, C.T.; Crabbe, J.C.; Adelman, J.P.; Maylie, J. A mouse model of episodic ataxia type-1. Nat. Neurosci. 2003, 6, 378–383. [Google Scholar] [CrossRef]
- Begum, R.; Bakiri, Y.; Volynski, K.E.; Kullmann, D.M. Action potential broadening in a presynaptic channelopathy. Nat. Commun. 2016, 7, 12102. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, O.; Imbrici, P.; Botti, F.M.; Pettorossi, V.E.; D’Adamo, M.C.; Valentino, M.; Zammit, C.; Mora, M.; Gibertini, S.; Di Giovanni, G.; et al. Kv1.1 knock-in ataxic mice exhibit spontaneous myokymic activity exacerbated by fatigue, ischemia and low temperature. Neurobiol. Dis. 2012, 47, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Eunson, L.H.; Rea, R.; Zuberi, S.M.; Youroukos, S.; Panayiotopoulos, C.P.; Liguori, R.; Avoni, P.; McWilliam, R.C.; Stephenson, J.B.; Hanna, M.G.; et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann. Neurol. 2000, 48, 647–656. [Google Scholar] [CrossRef]
- Kinali, M.; Jungbluth, H.; Eunson, L.H.; Sewry, C.A.; Manzur, A.Y.; Mercuri, E.; Hanna, M.G.; Muntoni, F. Expanding the phenotype of potassium channelopathy: Severe neuromyotonia and skeletal deformities without prominent Episodic Ataxia. Neuromuscul. Disord. 2004, 14, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; von Hehn, C.; Kaczmarek, L.K.; Ment, L.R.; Pober, B.R.; Hisama, F.M. Functional analysis of a novel potassium channel (KCNA1) mutation in hereditary myokymia. Neurogenetics 2007, 8, 131–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, A.; Golumbek, P.; Cellini, E.; Doccini, V.; Guerrini, R.; Wallgren-Pettersson, C.; Thuresson, A.C.; Gurnett, C.A. De novo KCNA1 variants in the PVP motif cause infantile epileptic encephalopathy and cognitive impairment similar to recurrent KCNA2 variants. Am. J. Med. Genet. A 2018, 176, 1748–1752. [Google Scholar] [CrossRef]
- Russo, A.; Gobbi, G.; Pini, A.; Møller, R.S.; Rubboli, G. Encephalopathy related to status epilepticus during sleep due to a de novo KCNA1 variant in the Kv-specific Pro-Val-Pro motif: Phenotypic description and remarkable electroclinical response to ACTH. Epileptic. Disord. 2020, 22, 802–806. [Google Scholar] [PubMed]
- Trosclair, K.; Dhaibar, H.A.; Gautier, N.M.; Mishra, V.; Glasscock, E. Neuron-specific Kv1.1 deficiency is sufficient to cause epilepsy, premature death, and cardiorespiratory dysregulation. Neurobiol. Dis. 2020, 137, 104759. [Google Scholar] [CrossRef]
- Brugnoni, R.; Maggi, L.; Canioni, E.; Verde, F.; Gallone, A.; Ariatti, A.; Filosto, M.; Petrelli, C.; Logullo, F.O.; Esposito, M.; et al. Next-generation sequencing application to investigate skeletal muscle channelopathies in a large cohort of Italian patients. Neuromuscul. Disord. 2021, 31, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Graves, T.D.; Cha, Y.H.; Hahn, A.F.; Barohn, R.; Salajegheh, M.K.; Griggs, R.C.; Bundy, B.N.; Jen, J.C.; Baloh, R.W.; Hanna, M.G. Episodic ataxia type 1: Clinical characterization, quality of life and genotype-phenotype correlation. Brain 2014, 137, 1009–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Adamo, M.C.; Hasan, S.; Guglielmi, L.; Servettini, I.; Cenciarini, M.; Catacuzzeno, L.; Franciolini, F. New insights into the pathogenesis and therapeutics of episodic ataxia type 1. Front. Cell. Neurosci. 2015, 9, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jentsch, T.J.; Pusch, M. CLC Chloride Channels and Transporters: Structure, Function, Physiology, and Disease. Physiol. Rev. 2018, 98, 1493–1590. [Google Scholar] [CrossRef]
- Altamura, C.; Desaphy, J.F.; Conte, D.; De Luca, A.; Imbrici, P. Skeletal muscle ClC-1 chloride channels in health and diseases. Pflug. Arch. 2020, 472, 961–975. [Google Scholar] [CrossRef]
- Steinmeyer, K.; Ortland, C.; Jentsch, T.J. Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature 1991, 354, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; MacKinnon, R. Structure of the CLC-1 chloride channel from Homo sapiens. eLife 2018, 7, e36629. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Preisler, S.S.; Zhang, L.; Cui, Y.; Missel, J.W.; Grønberg, C.; Gotfryd, K.; Lindahl, E.; Andersson, M.; Calloe, K.; et al. Structure of the human ClC-1 chloride channel. PLoS Biol. 2019, 17, e3000218. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.C.; Steinmeyer, K.; Lorenz, C.; Ricker, K.; Wolf, F.; Otto, M.; Zoll, B.; Lehmann-Horn, F.; Grzeschik, K.H.; Jentsch, T.J. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science 1992, 257, 797–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, A.L., Jr.; Crackower, M.A.; Abdalla, J.A.; Hudson, A.J.; Ebers, G.C. Molecular basis of Thomsen’s disease (autosomal dominant myotonia congenita). Nat. Genet. 1993, 3, 305–310. [Google Scholar] [CrossRef]
- Trivedi, J.R.; Bundy, B.; Statland, J.; Salajegheh, M.; Rayan, D.R.; Venance, S.L.; Wang, Y.; Fialho, D.; Matthews, E.; Cleland, J.; et al. Non-dystrophic myotonia: Prospective study of objective and patient reported outcomes. Brain 2013, 136, 2189–2200. [Google Scholar] [CrossRef] [Green Version]
- Lo Monaco, M.; D’Amico, A.; Luigetti, M.; Desaphy, J.F.; Modoni, A. Effect of mexiletine on transitory depression of compound motor action potential in recessive myotonia congenita. Clin. Neurophysiol. 2015, 126, 399–403. [Google Scholar] [CrossRef]
- Fialho, D.; Schorge, S.; Pucovska, U.; Davies, N.P.; Labrum, R.; Haworth, A.; Stanley, E.; Sud, R.; Wakeling, W.; Davis, M.B.; et al. Chloride channel myotonia: Exon 8 hot-spot for dominant-negative interactions. Brain 2007, 130, 3265–3274. [Google Scholar] [CrossRef] [Green Version]
- Brugnoni, R.; Kapetis, D.; Imbrici, P.; Pessia, M.; Canioni, E.; Colleoni, L.; De Rosbo, N.K.; Morandi, L.; Cudia, P.; Gashemi, N.; et al. A large cohort of myotonia congenita probands: Novel mutations and a high-frequency mutation region in exons 4 and 5 of the CLCN1 gene. J. Hum. Genet. 2013, 58, 581–587. [Google Scholar] [CrossRef]
- Altamura, C.; Ivanova, E.A.; Imbrici, P.; Conte, E.; Camerino, G.M.; Dadali, E.L.; Polyakov, A.V.; Kurbatov, S.A.; Girolamo, F.; Carratù, M.R.; et al. Pathomechanisms of a CLCN1 Mutation Found in a Russian Family Suffering From Becker’s Myotonia. Front. Neurol. 2020, 11, 1019. [Google Scholar] [CrossRef]
- Rayan, D.R.; Haworth, A.; Sud, R.; Matthews, E.; Fialho, D.; Burge, J.; Portaro, S.; Schorge, S.; Tuin, K.; Lunt, P.; et al. A new explanation for recessive myotonia congenita: Exon deletions and duplications in CLCN1. Neurology 2012, 78, 1953–1958. [Google Scholar] [CrossRef] [Green Version]
- Saviane, C.; Conti, F.; Pusch, M. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol. 1999, 113, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Imbrici, P.; Altamura, C.; Camerino, G.M.; Mangiatordi, G.F.; Conte, E.; Maggi, L.; Brugnoni, R.; Musaraj, K.; Caloiero, R.; Alberga, D.; et al. Multidisciplinary study of a new ClC-1 mutation causing myotonia congenita: A paradigm to understand and treat ion channelopathies. FASEB J. 2016, 30, 3285–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desaphy, J.F.; Gramegna, G.; Altamura, C.; Dinardo, M.M.; Imbrici, P.; George, A.L., Jr.; Modoni, A.; LoMonaco, M.; Camerino, D.C. Functional characterization of ClC-1 mutations from patients affected by recessive myotonia congenita presenting with different clinical phenotypes. Exp. Neurol. 2013, 248, 530–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altamura, C.; Lucchiari, S.; Sahbani, D.; Ulzi, G.; Comi, G.P.; D’Ambrosio, P.; Petillo, R.; Politano, L.; Vercelli, L.; Mongini, T.; et al. The analysis of myotonia congenita mutations discloses functional clusters of amino acids within the CBS2 domain and the C-terminal peptide of the ClC-1 channel. Hum. Mutat. 2018, 39, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Imbrici, P.; Maggi, L.; Mangiatordi, G.F.; Dinardo, M.M.; Altamura, C.; Brugnoni, R.; Alberga, D.; Pinter, G.L.; Ricci, G.; Siciliano, G.; et al. ClC-1 mutations in myotonia congenita patients: Insights into molecular gating mechanisms and genotype-phenotype correlation. J. Physiol. 2015, 593, 4181–4199. [Google Scholar] [CrossRef] [Green Version]
- Weinberger, S.; Wojciechowski, D.; Sternberg, D.; Lehmann-Horn, F.; Jurkat-Rott, K.; Becher, T.; Begemann, B.; Fahlke, C.; Fischer, M. Disease-causing mutations C277R and C277Y modify gating of human ClC-1 chloride channels in myotonia congenita. J. Physiol. 2012, 590, 3449–3464. [Google Scholar] [CrossRef] [Green Version]
- Jeng, C.J.; Fu, S.J.; You, C.Y.; Peng, Y.J.; Hsiao, C.T.; Chen, T.Y.; Tang, C.Y. Defective Gating and Proteostasis of Human ClC-1 Chloride Channel: Molecular Pathophysiology of Myotonia Congenita. Front. Neurol. 2020, 11, 76. [Google Scholar] [CrossRef] [Green Version]
- Myers, J.H.; Denman, K.; DuPont, C.; Hawash, A.A.; Novak, K.R.; Koesters, A.; Grabner, M.; Dayal, A.; Voss, A.A.; Rich, M.M. The mechanism underlying transient weakness in myotonia congenita. eLife 2021, 10, e65691. [Google Scholar] [CrossRef]
- Markhorst, J.M.; Stunnenberg, B.C.; Ginjaar, I.B.; Drost, G.; Erasmus, C.E.; Sie, L.T. Clinical experience with long-term ACZ treatment in children with nondystrophic myotonias: A three-case report. Pediatr. Neurol. 2014, 51, 537–541. [Google Scholar] [CrossRef]
- Moreira, S.D.; Barreto, R.; Roriz, J.M. Becker myotonia-a recently identified mutation in Iberian descendants with apparent ACZ-responsive phenotype. Muscle Nerve 2015, 51, 933–934. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, H.; Tsujino, A.; Kaibara, M.; Hayashi, H.; Shirabe, S.; Taniyama, K.; Eguchi, K. ACZ acts directly on the human skeletal muscle chloride channel. Muscle Nerve 2006, 34, 292–297. [Google Scholar] [CrossRef]
- Maggi, L.; Bernasconi, P.; D’Amico, A.; Brugnoni, R.; Fiorillo, C.; Garibaldi, M.; Astrea, G.; Bruno, C.; Santorelli, F.M.; Liguori, R.; et al. Italian recommendations for diagnosis and management of congenital myasthenic syndromes. Neurol. Sci. 2019, 40, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet. Neurol. 2015, 14, 420–434, Erratum in: Lancet Neurol. 2015, 14, 461. [Google Scholar] [CrossRef] [Green Version]
- Finlayson, S.; Beeson, D.; Palace, J. Congenital myasthenic syndromes: An update. Pract. Neurol. 2013, 13, 80–91. [Google Scholar] [CrossRef]
- Abicht, A.; Dusl, M.; Gallenmüller, C.; Guergueltcheva, V.; Schara, U.; Della Marina, A.; Wibbeler, E.; Almaras, S.; Mihaylova, V.; von der Hagen, M.; et al. Congenital myasthenic syndromes: Achievements and limitations of phenotype-guided gene-after-gene sequencing in diagnostic practice: A study of 680 patients. Hum. Mutat. 2012, 33, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Müller, J.S.; Stricker, S.; Megarbane, A.; Rajab, A.; Lindner, T.H.; Cohen, M.; Chouery, E.; Adaimy, L.; Ghanem, I.; et al. Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor fetal gamma subunit. Am. J. Hum. Genet. 2006, 79, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, J.; Harrison, B.J.; Spearman, H.; Cossins, J.; Vermeer, S.; ten Cate, L.N. Mutation analysis of CHRNA1, CHRNB1, CHRND, and RAPSN genes in multiple pterygium syndrome/fetal akinesia patients. Am. J. Hum. Genet. 2008, 82, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalk, A.; Stricker, S.; Becker, J.; Rupps, R.; Pantzar, T.; Miertus, J.; Botta, G.; Naretto, V.G.; Janetzki, C.; Yaqoob, N.; et al. Acetylcholine receptor pathway mutations explain various fetal akinesia deformation sequence disorders. Am. J. Hum. Genet. 2008, 82, 464–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santovito, L.S.; Brugnoni, R.; Banfi, P.; Maggi, L. Salbutamol as effective treatment in slow-channel syndrome—first report. Neurol. Sci. 2021, 42, 1611–1612. [Google Scholar] [CrossRef]
- Nigro, V.; Savarese, M. Next-generation sequencing approaches for the diagnosis of skeletal muscle disorders. Curr. Opin. Neurol. 2016, 29, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Ferradini, V.; Cassone, M.; Nuovo, S.; Bagni, I.; D’Apice, M.R.; Botta, A.; Novelli, G.; Sangiuolo, F. Targeted Next Generation Sequencing in patients with Myotonia Congenita. Clin. Chim. Acta 2017, 470, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandam, V.; Männikkö, R.; Matthews, E.; Hanna, M.G. Improving genetic diagnostics of skeletal muscle channelopathies. Expert. Rev. Mol. Diagn. 2020, 20, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.M.; Liao, M.H.; Chu, C.L.; Lin, Y.H.; Sun, W.Z.; Lai, L.P.; Chen, P.L. Next-generation sequencing and bioinformatics to identify genetic causes of malignant hyperthermia. J. Formos. Med. Assoc. 2021, 120, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Morales, F.; Pusch, M. An Up-to-Date Overview of the Complexity of Genotype-Phenotype Relationships in Myotonic Channelopathies. Front. Neurol. 2020, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Ravaglia, S.; Farinato, A.; Brugnoni, R.; Altamura, C.; Imbrici, P.; Camerino, D.C.; Padovani, A.; Mantegazza, R.; Bernasconi, P.; et al. Coexistence of CLCN1 and SCN4A mutations in one family suffering from myotonia. Neurogenetics 2017, 18, 219–225. [Google Scholar] [CrossRef]
- Thor, M.G.; Vivekanandam, V.; Sampedro-Castañeda, M.; Tan, S.V.; Suetterlin, K.; Sud, R.; Durran, S.; Schorge, S.; Kullmann, D.M.; Hanna, M.G.; et al. Myotonia in a patient with a mutation in an S4 arginine residue associated with hypokalaemic periodic paralysis and a concomitant synonymous CLCN1 mutation. Sci. Rep. 2019, 9, 17560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenes, O.; Barbieri, R.; Vásquez, M.; Vindas-Smith, R.; Roig, J.; Romero, A.; Valle, G.D.; Bermúdez-Guzmán, L.; Bertelli, S.; Pusch, M.; et al. Functional and Structural Characterization of ClC-1 and Nav1.4 Channels Resulting from CLCN1 and SCN4A Mutations Identified Alone and Coexisting in Myotonic Patients. Cells 2021, 10, 374. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.A.; Mayer-Hamblett, N.; Rowe, S.M. Cystic Fibrosis: Emergence of Highly Effective Targeted Therapeutics and Potential Clinical Implications. Am. J. Respir. Crit. Care Med. 2020, 201, 1193–1208. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Gene | Inheritance | Clinical Phenotype | Phenotype MIM | Age at Onset | Muscle Biopsy | Specific Features | Treatment |
|---|---|---|---|---|---|---|---|---|
| SODIUM CHANNEL SUBUNITS | ||||||||
| Nav1.4 | SCN4A | AD | Sodium channel myotonia (SCM; includes potassium-aggravated myotonia, myotonia fluctuans, myotonia permanens, ACZ-responsive myotonia, and SNEL) | 608,390 | Highly variable (neonatal–early childhood–adulthood) | non-specific myopathic pattern | Predominance in cranial muscles, precipitated by cold, presence of warm-up, muscle weakness absent or mild with late-onset | Mexiletine, lamotrigine, carbamazepine, ACZ, flecainide, and propafenone |
| AD | Paramyotonia congenita (PMC) | 168,300 | First decade | non-specific myopathic pattern | Myotonia and episodic muscle weakness precipitated by cold, paradoxical myotonia, predominant in cranial muscles, possible fixed muscle weakness in late disease stages. | As for SCM | ||
| AD | Hyperkalemic periodic paralysis (hyperPP) | 170,500 | First decade | Vacuolar and tubular aggregate myopathy | Episodic flaccid muscle weakness lasting up to 2 h accompanied by hyperkalemia > 4.5 mEq/L. Associated with myotonia. | ACZ, diclorophenamide | ||
| AD | Hypokalemic periodic paralysis type 2 (hypoPP2) | 613,345 | Childhood–third decade | Vacuolar and tubular aggregate myopathy | Episodic flaccid muscle weakness lasting up to 24 h accompanied by hypokalemia < 3.5 mEq/L. | ACZ, K+ sparing diuretics (spironolactone, triamterene) | ||
| AR | Congenital myasthenic syndrome type 16 (CMS16) | 614,198 | Neonatal or early infancy | non-specific myopathic pattern | Decremental response of the CMAP on RNS. Predominant involvement of bulbar and respiratory muscles. | Pyridostigmine and ACZ may be beneficial | ||
| AR | Congenital myopathy | n.a. | Neonatal or early infancy | No evident nemaline rods or structural abnormalities | Predominant axial and pelvic muscle weakness, delayed motor milestones, improvement in strength over time | ACZ | ||
| CALCIUM CHANNEL SUBUNITS | ||||||||
| Cav1.1 | CACNA1S | AD | Hypokalemic periodic paralysis type 1 (hypoPP1) | 170,400 | Childhood–second decade | Vacuolar myopathy (non-rimmed) | Episodic flaccid muscle weakness lasting up to 24 h accompanied by hypokalemia < 3.5 mEq/L. Fixed myopathy often developing in late disease stages. | ACZ, diclorphenamide |
| AD | Malignant hyperthermia susceptibility type 5 (MHS5) | 601,887 | When exposed to volatile anesthetics or succinylcholine | No structural abnormalities | MHS | Dantrolene (antidote) | ||
| AR | Congenital myopathy | n.a. | Neonatal or early infancy | Centronuclear or core myopathy | Hypotonia, delayed motor milestones, facial involvement (ophthalmoplegia), progressive muscle weakness (mainly axial). | ACZ | ||
| Stac3 | STAC3 | AR | Bailey-Bloch congenital myopathy/Native American Myopathy (NAM) | 255,995 | Neonatal or early infancy | non-specific myopathic pattern | Dysmorphic facial features and facial weakness (ptosis) susceptibility to MHS; multiple joint contractures | n.a. |
| Orai 1 | ORAI1 | AD | Tubular aggregate myopathy (TAM2) | 615,883 | Childhood | tubular aggregates in type II fibers, predominance of type I fibers | Slowly progressive | n.a |
| AR | CRAC channelopathies | 612,782 | <1 year | (1 patient) myopathic pattern; no evident nemaline rods or structural abnormalities. | Congenital non-progressive myopathy; immunodeficiency as the main feature. | n.a. | ||
| Stim 1 | STIM1 | AD | Stormorken syndrome (STRMK), York platelet syndrome | 185,070 | Childhood–early adult | tubular aggregates in type II fibers, predominance of type I fibers | Thrombocytopenia, anemia, asplenia, congenital miosis, and ichthyosis, asymptomatic to slowly progressive proximal muscle weakness | n.a. |
| AD | Non-syndromic tubular aggregate myopathy (TAM1) | 160,565 | Childhood | tubular aggregates mainly in type II fibers; predominance type I fibers | Slowly progressive, elevated CPK | n.a. | ||
| AR | CRAC channelopathies | 612,783 | <1 year | n.a. | Congenital non-progressive myopathy; immunodeficiency as the main feature. | n.a. | ||
| Ryr1 | RYR1 | AD | Malignant hyperthermia susceptibility type 1 (MHS1) | 145,600 | When exposed to volatile anesthetics or depolarizing muscle relaxants | Central core, multiminicore myopathy | MHS | Dantrolene (antidote) |
| AD/AR | Central core disease | 117,000 | First decade, rarely in adulthood | Central cores in type 1 fibers, predominance of type 1 fibers | Floppy infant; non-progressive/slowly progressive myopathy; joint contractures | n.a | ||
| AR | Multiminicore myopathy with external ophthalmoplegia | 255,320 | Neonatal or early infancy | Dystrophic signs and minicores | Hypotonia, delayed motor milestones, dysmorphic facial features and facial weakness, and progressive muscle weakness | n.a | ||
| Ryr3 | RYR3 | AR | Nemaline myopathy (NEM3) | 161,800 | 1 case, infantile | Perinuclear and subsarcolemmal nemaline bodies, wide variation in fiber size with type 1 fiber predominance and atrophy, increased internal nuclei | Dysmorphic face; normal CPK; myopathic EMG. Slowly progressive proximal limb weakness | n.a. |
| TRPV4 | TRPV4 | AD (reduced penetrance) | scapuloperoneal spinal muscular atrophy (SPSMA) | 181,405 | Neonatal or early infancy | Grouped type 1 and 2 fiber atrophy | Non-progressive or slowly progressive scapular and peroneal muscle weakness and atrophy, and peripheral motor neuropathy | n.a. |
| AD (reduced penetrance) | congenital distal spinal muscular atrophy (CDSMA) | 600,175 | Neonatal or early infancy | neurogenic muscle damage | Variable from lower-limb muscle weakness to severe neurogenic weakness and arthrogryposis | n.a. | ||
| AD | Hereditary motor ans sensory neuropathy (HMSN2C) | 606,071 | Variable (infancy, childhood, adulthood) | Neurogenic muscle damage and atrophy | Axonal polyneuropathy, diaphragmatic and vocal cord paresis, and distal muscle weakness | n.a. | ||
| Cav3.2 | CACNA1H | AR | Congenital amyotrophy | 1 case, neonatal | n.a. | Severe amyotrophy at birth | n.a. | |
| POTASSIUM CHANNEL SUBUNITS | ||||||||
| Kir2.1 | KCNJ2 | AD | Andersen–Tawil syndrome type 1 (ATS1, LQT7) | 170,390 | Variable onset of periodic paralysis (early childhood-adulthood) | non-specific myopathic pattern (few cases) | Potassium-sensitive periodic paralysis, cardiac arrhythmia, and facial and skeletal malformations. Prolonged QT (plus other arrhythmias). Episodic flaccid paralysis lasting up to 24h and more, usually accompanied by hypokalemia. | ACZ, dichlorphenamide, ß-blockers, If hypokaliemic, K+ supplementation or K+ sparing diuretics. If hyperkaliemic, be careful with K+ wasting diuretics for cardiac risk. |
| Kir3.4 | KCNJ5 | AD | Andersen–Tawil syndrome type 2 (ATS2, LQT13) | 613,785 | Variable onset of periodic paralysis as for ATS1 | n.a. | Same triad of symptoms of ATS1 | As for ATS1 |
| Kir2.6 | KCNJ18 | AD | Susceptibility to thyrotoxic periodic paralysis type 2 (TTPP2) | 613,239 | Early adulthood | Variable, vacuolation, mitochondrial changes, glycogen granules accumulation. | Episodic flaccid paralysis during thyrotoxicosis and hypokalemia. More frequent in Asian males. | Treatment of hyperthyroidism |
| Sur2 | ABCC9 | AR | Intellectual disability myopathy syndrome (IDMS) | n.a. | Variable onset of muscle symptoms (childhood-adulthood) | (1 patient) non-specific changes (mitochondrial aggregation, fiber caliber variation) | Hypotonia, muscle weakness and fatigability, dysmorphic features, intellectual disability and developmental delay, and cardiac systolic dysfunction | n.a. |
| Kv1.1 | KCNA1 | AD | Episodic ataxia/myokymia syndrome (EA1) | 160,120 | Childhood | non-specific, denervation findings | Muscle symptoms (preponderant in a few patients): constant myokymia and acute episodes of muscle contractions (face and limbs), stiffness, cramps, weakness, and episodic cerebellar ataxia, seizures, and hypomagnesemia. | ACZ antiepileptic drugs |
| CHLORIDE CHANNEL SUBUNITS | ||||||||
| ClC-1 | CLCN1 | AD | Myotonia congenita, Thomsen’s disease | 160,800 | First decade | No structural abnormalities | Myotonia with the warm-up phenomenon, cold sensitivity, predominant in limb muscles. | Mexiletine lamotrigine, carbamazepine, ACZ |
| AR | Recessive generalized myotonia, Becker’s disease | 255,700 | First decade | No structural abnormalities | More severe than Thomsen’s disease; transient weakness and “Herculean” appearance | As for Thomsen’s disease | ||
| NICOTINIC RECEPTOR | ||||||||
| AChR | CHRNE | AR | Congenital myasthenic syndrome (CMS4C) associated with AChR deficiency | 608,931 | Variable, infancy–adulthood | non-specific | Variable, from arthrogryposis multiplex congenital and fetal akinesia, ocular, bulbar and respiratory symptoms, delayed motor milestones, to mild adult muscle weakness. Slowly progressive with possible exacerbations. Decremental CMAP in response to RNS-EMG. | Pyridostigmine, 3,4 diaminopyridine, salbutamol/albuterol, and ephedrine |
| AD/AR (rare) | Slow-channel CMS (CMS4A) | 605,809 | Infancy–young adult | non-specific | Neonatal hypotonia; ocular, bulbar, respiratory muscle involvement, with predominant weakness of cervical, wrist, finger, and finger extensor muscles. Double CMAP on single nerve stimuli ENG. | Quinidine, fluoxetine, Worsening with pyridostigmine | ||
| AR | Fast-channel CMS (CMS4B) | 616,324 | Infancy | non-specific | Neonatal hypotonia, recurrent respiratory crises. Ocular, neck, and limb progressive muscle weakness and fatigability. Decremental CMAP in response to RNS-EMG. | Pyridostigmine, 3,4 diaminopyridine | ||
| CHRNA1 | AD | Slow-channel CMS (CMS1A) | 601,462 | Infancy–young adult | non-specific | Ocular, bulbar, and respiratory muscle involvement, with predominant weakness of cervical, wrist, finger, and finger extensor muscles. Double CMAP on single nerve stimuli ENG. | Quinidine, fluoxetine, Worsening with pyridostigmine | |
| AD (rare)/AR | Fast-channel CMS (CMS1B) | 608,930 | Infancy | non-specific | Neonatal hypotonia; ocular, bulbar (dysarthria), neck, recurrent respiratory crises, and limb progressive muscle weakness and fatigability. Decremental CMAP in response to RNS-EMG. | Pyridostigmine, 3,4 diaminopyridine | ||
| CHRNB1 | AR | CMS2C associated with AChR deficiency | 616,314 | Birth | non-specific | Neonatal hypotonia; respiratory and limb progressive muscle weakness. Decremental CMAP in response to RNS-EMG | Pyridostigmine | |
| AD | Slow-channel CMS2A | 616,313 | Infancy–young adult | non-specific | Ocular, bulbar, respiratory muscle involvement, with predominant weakness of cervical, wrist, finger, and finger extensor muscles. Double CMAP on single nerve stimuli ENG. | Quinidine, fluoxetine, Worsening with pyridostigmine | ||
| CHRND | AD | Slow-channel CMS3A | 616,321 | Infancy—young adult | non-specific | Ocular, bulbar, respiratory muscle involvement, with predominant weakness of cervical, wrist, finger, and finger extensor muscles. Double CMAP on single nerve stimuli ENG | Quinidine, fluoxetine, Worsening with pyridostigmine | |
| AR | CMS3C associated with AChR deficiency | 616,323 | Birth | non-specific | Neonatal hypotonia; ocular, episodic respiratory insufficiency, bulbar (swallowing), and proximal limb muscle weakness. Decremental CMAP in response to RNS-EMG. | Pyridostigmine | ||
| AR | Fast-channel CMS3B | 616,322 | Infancy | non-specific | Neonatal hypotonia; ocular, neck, respiratory, muscle weakness and fatigability. Decremental CMAP in response to RNS-EMG. | Pyridostigmine, 3,4-diaminopyridine | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maggi, L.; Bonanno, S.; Altamura, C.; Desaphy, J.-F. Ion Channel Gene Mutations Causing Skeletal Muscle Disorders: Pathomechanisms and Opportunities for Therapy. Cells 2021, 10, 1521. https://doi.org/10.3390/cells10061521
Maggi L, Bonanno S, Altamura C, Desaphy J-F. Ion Channel Gene Mutations Causing Skeletal Muscle Disorders: Pathomechanisms and Opportunities for Therapy. Cells. 2021; 10(6):1521. https://doi.org/10.3390/cells10061521
Chicago/Turabian StyleMaggi, Lorenzo, Silvia Bonanno, Concetta Altamura, and Jean-François Desaphy. 2021. "Ion Channel Gene Mutations Causing Skeletal Muscle Disorders: Pathomechanisms and Opportunities for Therapy" Cells 10, no. 6: 1521. https://doi.org/10.3390/cells10061521
APA StyleMaggi, L., Bonanno, S., Altamura, C., & Desaphy, J. -F. (2021). Ion Channel Gene Mutations Causing Skeletal Muscle Disorders: Pathomechanisms and Opportunities for Therapy. Cells, 10(6), 1521. https://doi.org/10.3390/cells10061521