
MicroRNAs in Prion Diseases—From Molecular Mechanisms to Insights in Translational Medicine

 and
and
Abstract
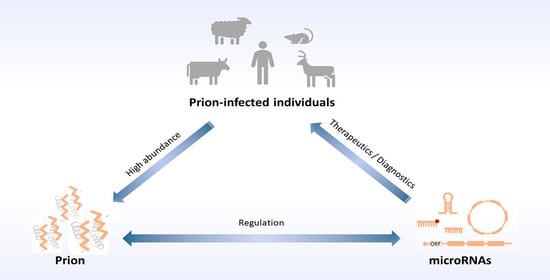
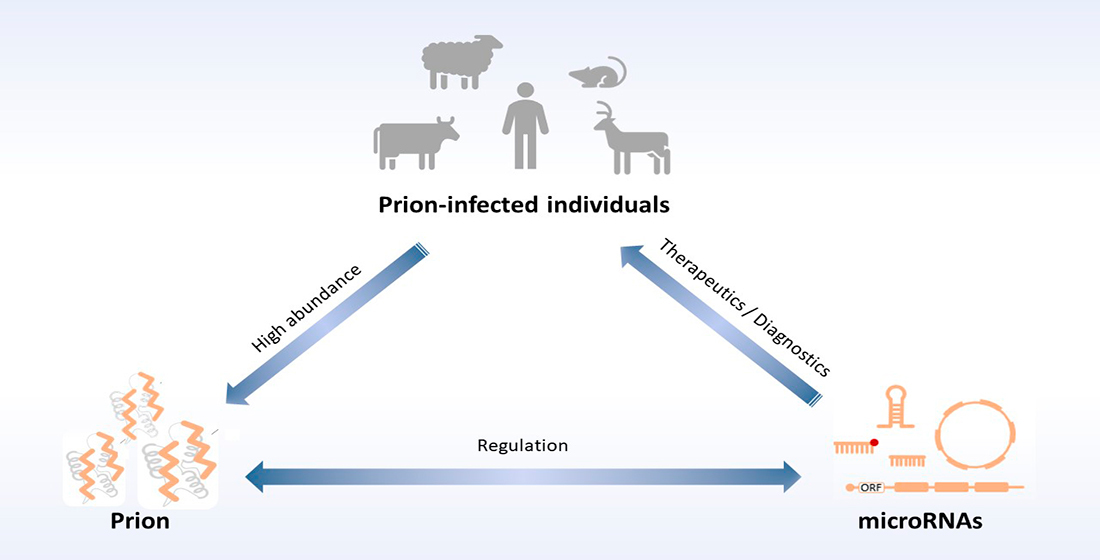
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
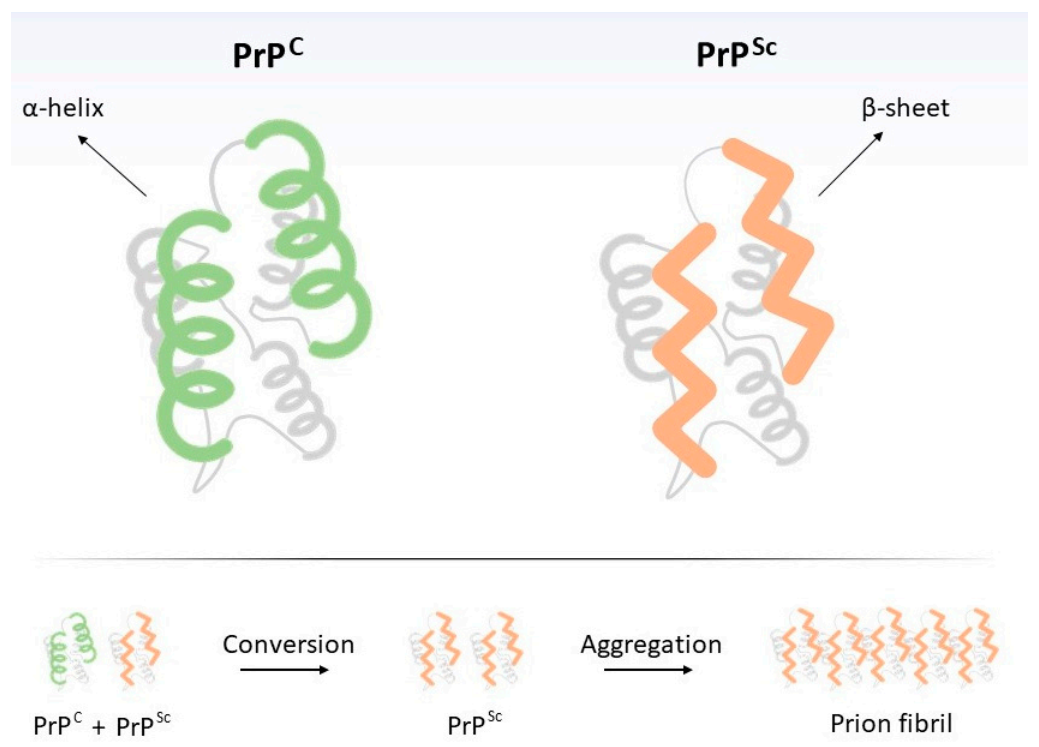
1.1. Prion: An Unconventional Infectious Agent
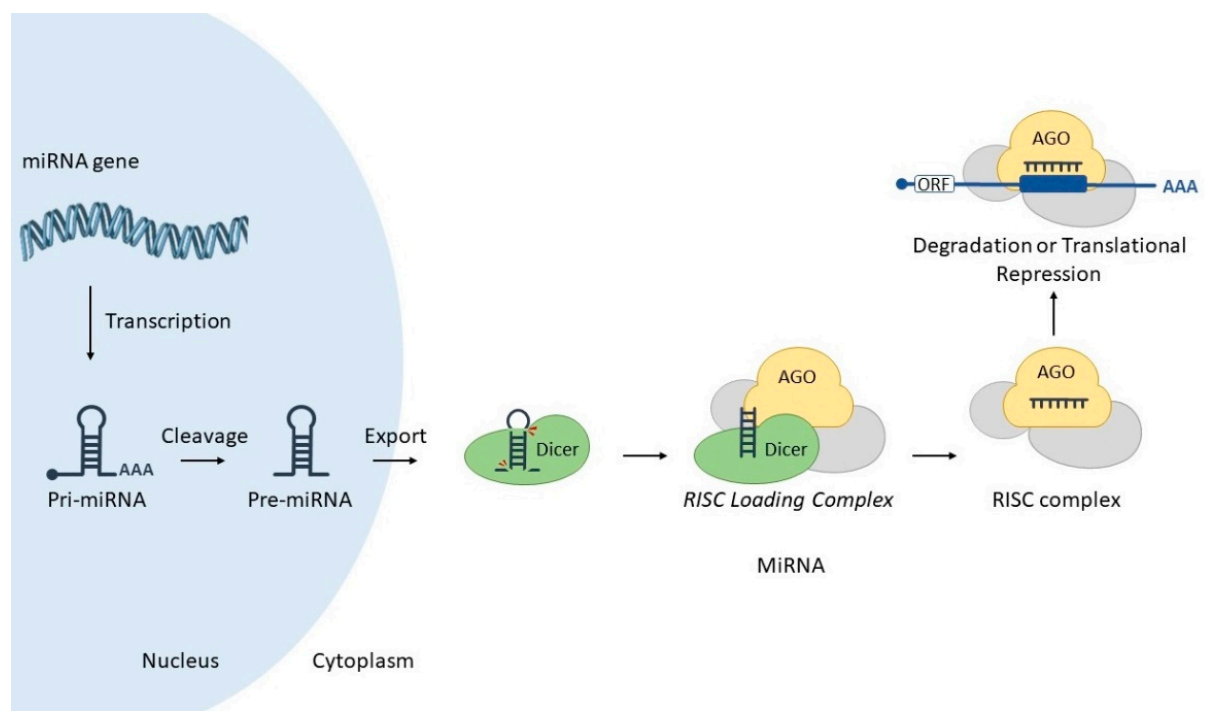
1.2. MicroRNAs: An Additional Layer of Gene Regulation in the Cell
2. Interplay between miRNAs and Cellular Prions in Health
2.1. miRNAs That Regulate Prion Expression
2.2. Prion Protein Affecting miRNAs’ Expression
3. Interplay between miRNAs and Prions in Disease
3.1. Dysregulated miRNAs in Prion Diseases
3.2. Potential Contributions of miRNAs Dysregulation to Pathology
4. Potential miRNA-Based Diagnosis
5. miRNA-Based Therapeutics
5.1. miRNA Replacement Approaches
5.2. miRNA Inhibitors and Others
5.3. Targeting PrP mRNA
5.3.1. Prnp-Targeting Artificial miRNA
5.3.2. RNA Interference
5.4. General Concerns on miRNA Therapies
6. Circular RNA, Sponges, and Competing Endogenous RNAs
7. MiRNAs in Other Prion-Like Diseases
8. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguzzi, A.; Polymenidou, M. Mammalian prion biology: One century of evolving concepts. Cell 2004, 116, 313–327. [Google Scholar] [CrossRef] [Green Version]
- Soto, C.; Castilla, J. The controversial protein-only hypothesis of prion propagation. Nat. Med. 2004, 10, S63–S67. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 2016, 539, 217–226. [Google Scholar] [CrossRef]
- Zhang, C.C.; Steele, A.D.; Lindquist, S.; Lodish, H.F. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc. Natl. Acad. Sci. USA 2006, 103, 2184–2189. [Google Scholar] [CrossRef] [Green Version]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Acevedo-Morantes, C.Y.; Wille, H. The structure of human prions: From biology to structural models—Considerations and pitfalls. Viruses 2014, 6, 3875–3892. [Google Scholar] [CrossRef]
- Collinge, J.; Sidle, K.C.L.; Meads, J.; Ironside, J.; Hill, A.F. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 1996, 383, 685–690. [Google Scholar] [CrossRef]
- Hill, A.F.; Desbruslais, M.; Joiner, S.; Sidle, K.C.L.; Gowland, I.; Collinge, J.; Doey, L.J.; Lantos, P. The same prion strain causes vCJD and BSE. Nature 1997, 389, 448–450. [Google Scholar] [CrossRef]
- Cohen, F.E.; Pan, K.M.; Huang, Z.; Baldwin, M.; Fletterick, R.J.; Prusiner, S.B. Structural clues to prion replication. Science 1994, 264, 530–531. [Google Scholar] [CrossRef]
- Wickner, R.B. Yeast and fungal prions. Cold Spring Harb. Perspect. Biol. 2016, 8, a023531. [Google Scholar] [CrossRef] [Green Version]
- Soto, C. Transmissible proteins: Expanding the prion heresy. Cell 2012, 149, 968–977. [Google Scholar] [CrossRef] [Green Version]
- Fabian, M.R.; Sonenberg, N. The mechanics of miRNA-mediated gene silencing: A look under the hood of miRISC. Nat. Struct. Mol. Biol. 2012, 19, 586–593. [Google Scholar] [CrossRef]
- Salmanidis, M.; Pillman, K.; Goodall, G.; Bracken, C. Direct transcriptional regulation by nuclear microRNAs. Int. J. Biochem. Cell Biol. 2014, 54, 304–311. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Pease, D.; Scheckel, C.; Schaper, E.; Eckhardt, V.; Emmenegger, M.; Xenarios, I.; Aguzzi, A. Genome-wide identification of microRNAs regulating the human prion protein. Brain Pathol. 2019, 29, 232–244. [Google Scholar] [CrossRef] [Green Version]
- Kalani, A.; Chaturvedi, P.; Maldonado, C.; Bauer, P.; Joshua, I.G.; Tyagi, S.C.; Tyagi, N. Dementia-like pathology in type-2 diabetes: A novel microRNA mechanism. Mol. Cell. Neurosci. 2017, 80, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wang, S.-Q.; Qing, L.-L.; Liu, L.-L.; Zhang, Y.-P. Expression of BSE-associated proteins in the CNS and lymphoreticular tissues of cattle and buffalo. Sci. Bull. 2016, 61, 1377–1383. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wang, S.; Guo, L.; Du, Y.; Liu, L.; Ma, T.; Otecko, N.O.; Li, C.; Zhang, Y. Fixed differences in the 3′UTR of buffalo PRNP gene provide binding sites for miRNAs post-transcriptional regulation. Oncotarget 2017, 8, 46006–46019. [Google Scholar] [CrossRef]
- Beaudoin, S.; Vanderperre, B.; Grenier, C.; Tremblay, I.; Leduc, F.; Roucou, X. A large ribonucleoprotein particle induced by cytoplasmic PrP shares striking similarities with the chromatoid body, an RNA granule predicted to function in posttranscriptional gene regulation. Biochim. Biophys. Acta BBA Mol. Cell Res. 2009, 1793, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Maundrell, K.; Soto, C. Is loss of function of the prion protein the cause of prion disorders? Trends Mol. Med. 2003, 9, 237–243. [Google Scholar] [CrossRef]
- Mouillet-Richard, S.; Ermonval, M.; Chebassier, C.; Laplanche, J.L.; Lehmann, S.; Launay, J.M.; Kellermann, O. Signal transduction through prion protein. Science 2000, 289, 1925–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, A.D.; Lindquist, S.; Aguzzi, A. The prion protein knockout mouse. Prion 2007, 1, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, F.; Yang, Y.; Wang, T.; Kouadir, M.; Zhao, D.; Hu, S. Cellular prion protein promotes neuronal differentiation of adipose-derived stem cells by upregulating miRNA-124. J. Mol. Neurosci. 2016, 59, 48–55. [Google Scholar] [CrossRef]
- Satoh, J.; Obayashi, S.; Misawa, T.; Sumiyoshi, K.; Oosumi, K.; Tabunoki, H. Protein microarray analysis identifies human cellular prion protein interactors. Neuropathol. Appl. Neurobiol. 2009, 35, 16–35. [Google Scholar] [CrossRef]
- Spielhaupter, C.; Schätzl, H.M. PrPC directly interacts with proteins involved in signaling pathways. J. Biol. Chem. 2001, 276, 44604–44612. [Google Scholar] [CrossRef] [Green Version]
- Gibbings, D.; Leblanc, P.; Jay, F.; Pontier, D.; Michel, F.; Schwab, Y.; Alais, S.; Lagrange, T.; Voinnet, O. Human prion protein binds Argonaute and promotes accumulation of microRNA effector complexes. Nat. Struct. Mol. Biol. 2012, 19, 517–524. [Google Scholar] [CrossRef]
- Vishnoi, A.; Rani, S. MiRNA biogenesis and regulation of diseases: An overview. In MicroRNA Profiling: Methods and Protocols; Methods in Molecular Biology; Rani, S., Ed.; Springer: New York, NY, USA, 2017; pp. 1–10. ISBN 978-1-4939-6524-3. [Google Scholar]
- Montag, J.; Hitt, R.; Opitz, L.; Schulz-Schaeffer, W.J.; Hunsmann, G.; Motzkus, D. Upregulation of miRNA hsa-miR-342-3p in experimental and idiopathic prion disease. Mol. Neurodegener. 2009, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Lukiw, W.J.; Dua, P.; Pogue, A.I.; Eicken, C.; Hill, J.M. Upregulation of Micro RNA-146a (miRNA-146a), a marker for inflammatory neurodegeneration, in sporadic Creutzfeldt–Jakob Disease (sCJD) and Gerstmann–Straussler–Scheinker (GSS) syndrome. J. Toxicol. Environ. Health A 2011, 74, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Llorens, F.; Thüne, K.; Martí, E.; Kanata, E.; Dafou, D.; Díaz-Lucena, D.; Vivancos, A.; Shomroni, O.; Zafar, S.; Schmitz, M.; et al. Regional and subtype-dependent miRNA signatures in sporadic Creutzfeldt-Jakob disease are accompanied by alterations in miRNA silencing machinery and biogenesis. PLoS Pathog. 2018, 14, e1006802. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Quek, C.; Li, X.; Bellingham, S.A.; Ellett, L.J.; Shambrook, M.; Zafar, S.; Zerr, I.; Lawson, V.A.; Hill, A.F. Distribution of microRNA profiles in pre-clinical and clinical forms of murine and human prion disease. Commun. Biol. 2021, 4, 1–12. [Google Scholar] [CrossRef]
- Saba, R.; Goodman, C.D.; Huzarewich, R.L.C.H.; Robertson, C.; Booth, S.A. A miRNA signature of prion induced neurodegeneration. PLoS ONE 2008, 3, e3652. [Google Scholar] [CrossRef] [Green Version]
- Majer, A.; Medina, S.J.; Niu, Y.; Abrenica, B.; Manguiat, K.J.; Frost, K.L.; Philipson, C.S.; Sorensen, D.L.; Booth, S.A. Early mechanisms of pathobiology are revealed by transcriptional temporal dynamics in hippocampal CA1 neurons of prion infected mice. PLoS Pathog. 2012, 8, e1003002. [Google Scholar] [CrossRef] [Green Version]
- Boese, A.S.; Saba, R.; Campbell, K.; Majer, A.; Medina, S.; Burton, L.; Booth, T.F.; Chong, P.; Westmacott, G.; Dutta, S.M.; et al. MicroRNA abundance is altered in synaptoneurosomes during prion disease. Mol. Cell. Neurosci. 2016, 71, 13–24. [Google Scholar] [CrossRef]
- Gao, C.; Wei, J.; Zhang, B.-Y.; Shi, Q.; Chen, C.; Wang, J.; Shi, Q.; Dong, X.-P. MiRNA expression profiles in the brains of mice infected with scrapie agents 139A, ME7 and S15. Emerg. Microbes Infect. 2016, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Slota, J.A.; Medina, S.J.; Klassen, M.; Gorski, D.; Mesa, C.M.; Robertson, C.; Mitchell, G.; Coulthart, M.B.; Pritzkow, S.; Soto, C.; et al. Identification of circulating microRNA signatures as potential biomarkers in the serum of elk infected with chronic wasting disease. Sci. Rep. 2019, 9, 19705. [Google Scholar] [CrossRef] [Green Version]
- Toivonen, J.M.; Sanz-Rubio, D.; López-Pérez, Ó.; Marín-Moreno, A.; Bolea, R.; Osta, R.; Badiola, J.J.; Zaragoza, P.; Espinosa, J.-C.; Torres, J.-M.; et al. MicroRNA alterations in a Tg501 mouse model of prion disease. Biomolecules 2020, 10, 908. [Google Scholar] [CrossRef]
- Montag, J.; Brameier, M.; Schmädicke, A.-C.; Gilch, S.; Schätzl, H.M.; Motzkus, D. A genome-wide survey for prion-regulated miRNAs associated with cholesterol homeostasis. BMC Genomics 2012, 13, 486. [Google Scholar] [CrossRef] [Green Version]
- Bellingham, S.A.; Coleman, B.M.; Hill, A.F. Small RNA deep sequencing reveals a distinct miRNA signature released in exosomes from prion-infected neuronal cells. Nucleic Acids Res. 2012, 40, 10937–10949. [Google Scholar] [CrossRef] [Green Version]
- Bellingham, S.A.; Hill, A.F. Analysis of miRNA signatures in neurodegenerative prion disease. In Prions: Methods and Protocols; Methods in Molecular Biology; Lawson, V.A., Ed.; Springer: New York, NY, USA, 2017; pp. 67–80. ISBN 978-1-4939-7244-9. [Google Scholar]
- Rubio, D.S.; López-Pérez, Ó.; de Andrés Pablo, Á.; Bolea, R.; Osta, R.; Badiola, J.J.; Zaragoza, P.; Martín-Burriel, I.; Toivonen, J.M. 2017 increased circulating microRNAs miR-342-3p and miR-21-5p in natural sheep prion disease. J. Gen. Virol. 2017, 98, 305–310. [Google Scholar] [CrossRef]
- Burak, K.; Lamoureux, L.; Boese, A.; Majer, A.; Saba, R.; Niu, Y.; Frost, K.; Booth, S.A. MicroRNA-16 targets mRNA involved in neurite extension and branching in hippocampal neurons during presymptomatic prion disease. Neurobiol. Dis. 2018, 112, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.G.; Li, Y.Y.; Zhao, Y.; Bhattacharjee, S.; Lukiw, W.J. Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by microRNA-146a and NF-κB in stressed human astroglial cells and in Alzheimer disease. J. Biol. Chem. 2010, 285, 38951–38960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukiw, W.J.; Zhao, Y.; Cui, J.G. An NF-κB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J. Biol. Chem. 2008, 283, 31315–31322. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Chen, X.-P.; Li, Y.-J. MicroRNA-146a and human disease. Scand. J. Immunol. 2010, 71, 227–231. [Google Scholar] [CrossRef]
- Saba, R.; Gushue, S.; Huzarewich, R.L.C.H.; Manguiat, K.; Medina, S.; Robertson, C.; Booth, S.A. MicroRNA 146a (miR-146a) is over-expressed during prion disease and modulates the innate immune response and the microglial activation state. PLoS ONE 2012, 7, e30832. [Google Scholar] [CrossRef]
- Lugli, G.; Larson, J.; Demars, M.P.; Smalheiser, N.R. Primary microRNA precursor transcripts are localized at post-synaptic densities in adult mouse forebrain. J. Neurochem. 2012, 123, 459–466. [Google Scholar] [CrossRef]
- Nahalka, J. The role of the protein–RNA recognition code in neurodegeneration. Cell. Mol. Life Sci. 2019, 76, 2043–2058. [Google Scholar] [CrossRef]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef]
- Colby, D.W.; Zhang, Q.; Wang, S.; Groth, D.; Legname, G.; Riesner, D.; Prusiner, S.B. Prion detection by an amyloid seeding assay. Proc. Natl. Acad. Sci. USA 2007, 104, 20914–20919. [Google Scholar] [CrossRef] [Green Version]
- Atarashi, R.; Satoh, K.; Sano, K.; Fuse, T.; Yamaguchi, N.; Ishibashi, D.; Matsubara, T.; Nakagaki, T.; Yamanaka, H.; Shirabe, S.; et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat. Med. 2011, 17, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Norsworthy, P.J.; Thompson, A.G.B.; Mok, T.H.; Guntoro, F.; Dabin, L.C.; Nihat, A.; Paterson, R.W.; Schott, J.M.; Collinge, J.; Mead, S.; et al. A blood miRNA signature associates with sporadic Creutzfeldt-Jakob disease diagnosis. Nat. Commun. 2020, 11, 3960. [Google Scholar] [CrossRef]
- Mead, S.; Uphill, J.; Beck, J.; Poulter, M.; Campbell, T.; Lowe, J.; Adamson, G.; Hummerich, H.; Klopp, N.; Rückert, I.-M.; et al. Genome-wide association study in multiple human prion diseases suggests genetic risk factors additional to PRNP. Hum. Mol. Genet. 2012, 21, 1897–1906. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Shi, Q.; Wei, J.; Zhou, W.; Xiao, K.; Wang, J.; Shi, Q.; Dong, X.-P. The associations of two SNPs in miRNA-146a and one SNP in ZBTB38-RASA2 with the disease susceptibility and the clinical features of the Chinese patients of sCJD and FFI. Prion 2018, 12, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Pegtel, D.M.; Peferoen, L.; Amor, S. Extracellular vesicles as modulators of cell-to-cell communication in the healthy and diseased brain. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130516. [Google Scholar] [CrossRef] [Green Version]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Cheng, L.; Doecke, J.D.; Sharples, R.A.; Villemagne, V.L.; Fowler, C.J.; Rembach, A.; Martins, R.N.; Rowe, C.C.; Macaulay, S.L.; Masters, C.L.; et al. Prognostic serum miRNA biomarkers associated with Alzheimer’s disease shows concordance with neuropsychological and neuroimaging assessment. Mol. Psychiatry 2015, 20, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef] [Green Version]
- Goold, R.; McKinnon, C.; Tabrizi, S.J. Prion degradation pathways: Potential for therapeutic intervention. Mol. Cell. Neurosci. 2015, 66, 12–20. [Google Scholar] [CrossRef]
- Heiseke, A.; Aguib, Y.; Riemer, C.; Baier, M.; Schätzl, H.M. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J. Neurochem. 2009, 109, 25–34. [Google Scholar] [CrossRef]
- Cortes, C.J.; Qin, K.; Cook, J.; Solanki, A.; Mastrianni, J.A. Rapamycin delays disease onset and prevents PrP plaque deposition in a mouse model of gerstmann–sträussler–scheinker disease. J. Neurosci. 2012, 32, 12396–12405. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Zhao, D.; Khan, S.H.; Yang, L. Role of autophagy in prion protein-induced neurodegenerative diseases. Acta Biochim. Biophys. Sin. 2013, 45, 494–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.Z.A.; Zhao, D.; Hussain, T.; Sabir, N.; Yang, L. Regulation of microRNAs-mediated autophagic flux: A new regulatory avenue for neurodegenerative diseases with focus on prion diseases. Front. Aging Neurosci. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, C.; Zhu, J.; Shu, P.; Yin, B.; Gong, Y.; Qiang, B.; Yuan, J.; Peng, X. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol. Aging 2012, 33, 522–534. [Google Scholar] [CrossRef]
- Müller, M.; Kuiperij, H.B.; Claassen, J.A.; Küsters, B.; Verbeek, M.M. MicroRNAs in Alzheimer’s disease: Differential expression in hippocampus and cell-free cerebrospinal fluid. Neurobiol. Aging 2014, 35, 152–158. [Google Scholar] [CrossRef]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; Strooper, B.D. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/β-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef] [Green Version]
- Hébert, S.S.; Papadopoulou, A.S.; Smith, P.; Galas, M.-C.; Planel, E.; Silahtaroglu, A.N.; Sergeant, N.; Buée, L.; De Strooper, B. Genetic ablation of Dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum. Mol. Genet. 2010, 19, 3959–3969. [Google Scholar] [CrossRef] [Green Version]
- Nunez-Iglesias, J.; Liu, C.-C.; Morgan, T.E.; Finch, C.E.; Zhou, X.J. Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PLoS ONE 2010, 5, e8898. [Google Scholar] [CrossRef] [Green Version]
- Shioya, M.; Obayashi, S.; Tabunoki, H.; Arima, K.; Saito, Y.; Ishida, T.; Satoh, J. Aberrant microRNA expression in the brains of neurodegenerative diseases: MiR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol. Appl. Neurobiol. 2010, 36, 320–330. [Google Scholar] [CrossRef]
- Wang, W.-X.; Huang, Q.; Hu, Y.; Stromberg, A.J.; Nelson, P.T. Patterns of microRNA expression in normal and early Alzheimer’s disease human temporal cortex: White matter versus gray matter. Acta Neuropathol. Berl. 2011, 121, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimers Dis. 2008, 14, 27–41. [Google Scholar] [CrossRef]
- Ai, J.; Sun, L.-H.; Che, H.; Zhang, R.; Zhang, T.-Z.; Wu, W.-C.; Su, X.-L.; Chen, X.; Yang, G.; Li, K.; et al. MicroRNA-195 protects against dementia induced by chronic brain hypoperfusion via its anti-amyloidogenic effect in rats. J. Neurosci. 2013, 33, 3989–4001. [Google Scholar] [CrossRef]
- Wang, W.-X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of β-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef]
- Nelson, P.T.; Wang, W.-X. MiR-107 is reduced in Alzheimer’s disease brain neocortex: Validation study. J. Alzheimers Dis. 2010, 21, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.; Hashimi, A.A.; Girard, J.; Delay, C.; Hébert, S.S. In vivo regulation of amyloid precursor protein neuronal splicing by microRNAs. J. Neurochem. 2011, 116, 240–247. [Google Scholar] [CrossRef]
- Zhu, H.-C.; Wang, L.-M.; Wang, M.; Song, B.; Tan, S.; Teng, J.-F.; Duan, D.-X. MicroRNA-195 downregulates Alzheimer’s disease amyloid-β production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef]
- Parsi, S.; Smith, P.Y.; Goupil, C.; Dorval, V.; Hébert, S.S. Preclinical evaluation of miR-15/107 family members as multifactorial drug targets for Alzheimer’s disease. Mol. Ther. Nucleic Acids 2015, 4, e256. [Google Scholar] [CrossRef]
- Zhou, R.; Li, X.; Hu, G.; Gong, A.-Y.; Drescher, K.M.; Chen, X.-M. MiR-16 targets transcriptional corepressor SMRT and modulates NF-kappaB-Regulated transactivation of interleukin-8 gene. PLoS ONE 2012, 7, e30772. [Google Scholar] [CrossRef] [Green Version]
- Frasca, D.; Diaz, A.; Romero, M.; Ferracci, F.; Blomberg, B.B. MicroRNAs miR-155 and miR-16 Decrease AID and E47 in B cells from elderly individuals. J. Immunol. 2015, 195, 2134–2140. [Google Scholar] [CrossRef] [Green Version]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T. Antisense Drug Technology: Principles, Strategies, and Applications, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007; ISBN 978-0-8493-8795-1. [Google Scholar]
- Hogan, D.J.; Vincent, T.M.; Fish, S.; Marcusson, E.G.; Bhat, B.; Chau, B.N.; Zisoulis, D.G. Anti-miRs competitively inhibit microRNAs in argonaute complexes. PLoS ONE 2014, 9, e100951. [Google Scholar] [CrossRef] [PubMed]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Ørum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef] [Green Version]
- Chau, B.N.; Xin, C.; Hartner, J.; Ren, S.; Castano, A.P.; Linn, G.; Li, J.; Tran, P.T.; Kaimal, V.; Huang, X.; et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 2012, 4, 121ra18. [Google Scholar] [CrossRef] [Green Version]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-L.; Hong, C.-G.; Yue, T.; Li, H.-M.; Duan, R.; Hu, W.-B.; Cao, J.; Wang, Z.-X.; Chen, C.-Y.; Hu, X.-K.; et al. Inhibition of miR-331-3p and miR-9-5p ameliorates Alzheimer’s disease by enhancing autophagy. Theranostics 2021, 11, 2395–2409. [Google Scholar] [CrossRef]
- Park, W.; Zhai, J.; Lee, J.-Y. Highly efficient gene silencing using perfect complementary artificial miRNA targeting AP1 or heteromeric artificial miRNA targeting AP1 and CAL genes. Plant Cell Rep. 2009, 28, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Parizotto, E.A.; Dunoyer, P.; Rahm, N.; Himber, C.; Voinnet, O. In vivo investigation of the transcription, processing, endonucleolytic activity, and functional relevance of the spatial distribution of a plant miRNA. Genes Dev. 2004, 18, 2237–2242. [Google Scholar] [CrossRef] [Green Version]
- Gallozzi, M.; Chapuis, J.; Le Provost, F.; Le Dur, A.; Morgenthaler, C.; Peyre, C.; Daniel-Carlier, N.; Pailhoux, E.; Vilotte, M.; Passet, B.; et al. Prnp knockdown in transgenic mice using RNA interference. Transgenic Res. 2008, 17, 783–791. [Google Scholar] [CrossRef]
- Kang, S.-G.; Kim, C.; Aiken, J.; Yoo, H.S.; McKenzie, D. Dual microRNA to cellular prion protein inhibits propagation of pathogenic prion protein in cultured cells. Mol. Neurobiol. 2018, 55, 2384–2396. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Hannon, G.J.; Rossi, J.J. Unlocking the potential of the human genome with RNA interference. Nature 2004, 431, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, Y.; Tamura, Y.; Yoshida, M.; Tokunaga, K.; Hohjoh, H. Enhancement of allele discrimination by introduction of nucleotide mismatches into siRNA in allele-specific gene silencing by RNAi. PLoS ONE 2008, 3, e2248. [Google Scholar] [CrossRef]
- Ridolfi, B.; Abdel-Haq, H. Neurodegenerative disorders treatment: The microRNA role. Curr. Gene Ther. 2017, 17, 327–363. [Google Scholar] [CrossRef]
- Satoh, J.; Yamamura, T. Gene expression profile following stable expression of the cellular prion protein. Cell. Mol. Neurobiol. 2004, 24, 793–814. [Google Scholar] [CrossRef]
- Qu, S.; Yang, X.; Li, X.; Wang, J.; Gao, Y.; Shang, R.; Sun, W.; Dou, K.; Li, H. Circular RNA: A new star of noncoding RNAs. Cancer Lett. 2015, 365, 141–148. [Google Scholar] [CrossRef]
- Akhter, R. Circular RNA and Alzheimer’s disease. In Circular RNAs: Biogenesis and Functions; Advances in Experimental Medicine and Biology; Xiao, J., Ed.; Springer: Singapore, 2018; pp. 239–243. ISBN 9789811314261. [Google Scholar]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- O’Nuallain, B.; Williams, A.D.; Westermark, P.; Wetzel, R. Seeding specificity in amyloid growth induced by heterologous fibrils. J. Biol. Chem. 2004, 279, 17490–17499. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.D.; Lansbury, P.T. Models of amyloid seeding in Alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar] [CrossRef]
- Soto, C.; Estrada, L.; Castilla, J. Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem. Sci. 2006, 31, 150–155. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Eisele, Y.S.; Obermüller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef] [Green Version]
- Langer, F.; Eisele, Y.S.; Fritschi, S.K.; Staufenbiel, M.; Walker, L.C.; Jucker, M. Soluble Aβ seeds are potent inducers of cerebral β-amyloid deposition. J. Neurosci. 2011, 31, 14488–14495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, R.; Duran-Aniotz, C.; Castilla, J.; Estrada, L.D.; Soto, C. De novo induction of amyloid-β deposition in vivo. Mol. Psychiatry 2012, 17, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Rosen, R.F.; Fritz, J.J.; Dooyema, J.; Cintron, A.F.; Hamaguchi, T.; Lah, J.J.; LeVine, H.; Jucker, M.; Walker, L.C. Exogenous seeding of cerebral β-amyloid deposition in βAPP-transgenic rats. J. Neurochem. 2012, 120, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Desplats, P.; Lee, H.-J.; Bae, E.-J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.-J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.Y. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Mougenot, A.-L.; Nicot, S.; Bencsik, A.; Morignat, E.; Verchère, J.; Lakhdar, L.; Legastelois, S.; Baron, T. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging 2012, 33, 2225–2228. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.; Selvakumar, G.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.; Zaheer, A. Neuroinflammation induces neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Slota, J.A.; Booth, S.A. MicroRNAs in neuroinflammation: Implications in disease pathogenesis, biomarker discovery and therapeutic applications. Non-Coding RNA 2019, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Junker, A.; Krumbholz, M.; Eisele, S.; Mohan, H.; Augstein, F.; Bittner, R.; Lassmann, H.; Wekerle, H.; Hohlfeld, R.; Meinl, E. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain 2009, 132, 3342–3352. [Google Scholar] [CrossRef] [Green Version]
- Goldmann, T.; Prinz, M. Role of microglia in CNS autoimmunity. Clin. Dev. Immunol. 2013, 2013, e208093. [Google Scholar] [CrossRef] [Green Version]
- Guedes, J.R.; Custódia, C.M.; Silva, R.J.; de Almeida, L.P.; Pedroso de Lima, M.C.; Cardoso, A.L. Early miR-155 upregulation contributes to neuroinflammation in Alzheimer’s disease triple transgenic mouse model. Hum. Mol. Genet. 2014, 23, 6286–6301. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Ramirez, M.A.; Wu, D.; Pryce, G.; Simpson, J.E.; Reijerkerk, A.; King-Robson, J.; Kay, O.; de Vries, H.E.; Hirst, M.C.; Sharrack, B.; et al. MicroRNA-155 negatively affects blood–brain barrier function during neuroinflammation. FASEB J. 2014, 28, 2551–2565. [Google Scholar] [CrossRef]
- Park, R.; Lee, W.J.; Ji, J.D. Association between the three functional miR-146a single-nucleotide polymorphisms, rs2910164, rs57095329, and rs2431697, and autoimmune disease susceptibility: A meta-analysis. Autoimmunity 2016, 49, 451–458. [Google Scholar] [CrossRef]
- Thome, A.D.; Harms, A.S.; Volpicelli-Daley, L.A.; Standaert, D.G. microRNA-155 Regulates alpha-synuclein-induced inflammatory responses in models of parkinson disease. J. Neurosci. 2016, 36, 2383–2390. [Google Scholar] [CrossRef]
- Martin, N.A.; Molnar, V.; Szilagyi, G.T.; Elkjaer, M.L.; Nawrocki, A.; Okarmus, J.; Wlodarczyk, A.; Thygesen, E.K.; Palkovits, M.; Gallyas, F.J.; et al. Experimental demyelination and axonal loss are reduced in microRNA-146a deficient mice. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Chen, M.; Simpson, S.; Lucas, R.M.; Charlesworth, J.C.; Blackburn, N.; van der Mei, I.; Ponsonby, A.-L.; Lucas, R.M.; Dear, K.; et al. Common genetic variation within miR-146a predicts disease onset and relapse in multiple sclerosis. Neurol. Sci. 2018, 39, 297–304. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Z.G.; Lu, M.; Zhang, Y.; Shang, X.; Chopp, M. MiR-146a promotes oligodendrocyte progenitor cell differentiation and enhances remyelination in a model of experimental autoimmune encephalomyelitis. Neurobiol. Dis. 2019, 125, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Alexandrov, P.N. Regulation of Complement Factor H (CFH) by multiple miRNAs in Alzheimer’s disease (AD) brain. Mol. Neurobiol. 2012, 46, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.M.; Krüger, C.; Park, B.; Derkow, K.; Rosenberger, K.; Baumgart, J.; Trimbuch, T.; Eom, G.; Hinz, M.; Kaul, D.; et al. An unconventional role for miRNA: Let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat. Neurosci. 2012, 15, 827–835. [Google Scholar] [CrossRef]
- Juźwik, C.A.; Drake, S.S.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A.; Douglas, C.; Morquette, B.; Moore, C.S.; Fournier, A.E. MicroRNA dysregulation in neurodegenerative diseases: A systematic review. Prog. Neurobiol. 2019, 182, 101664. [Google Scholar] [CrossRef] [PubMed]
- Soreq, H.; Wolf, Y. NeurimmiRs: MicroRNAs in the neuroimmune interface. Trends Mol. Med. 2011, 17, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.; Keon, M.; Liu, B.; Su, Z.; Saksena, N.K. Panoramic visualization of circulating microRNAs across neurodegenerative diseases in humans. Mol. Neurobiol. 2019, 56, 7380–7407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deleault, N.R.; Lucassen, R.W.; Supattapone, S. RNA molecules stimulate prion protein conversion. Nature 2003, 425, 717–720. [Google Scholar] [CrossRef]
- Harvey, Z.H.; Chen, Y.; Jarosz, D.F. Protein-based inheritance: Epigenetics beyond the chromosome. Mol. Cell 2018, 69, 195–202. [Google Scholar] [CrossRef]
- Nizhnikov, A.A.; Ryzhova, T.A.; Volkov, K.V.; Zadorsky, S.P.; Sopova, J.V.; Inge-Vechtomov, S.G.; Galkin, A.P. Interaction of prions causes heritable traits in saccharomyces cerevisiae. PLoS Genet. 2016, 12, e1006504. [Google Scholar] [CrossRef] [Green Version]
- Dogini, D.B.; Pascoal, V.D.B.; Avansini, S.H.; Vieira, A.S.; Pereira, T.C.; Lopes-Cendes, I. The new world of RNAs. Genet. Mol. Biol. 2014, 37, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Baronti, L.; Guzzetti, I.; Ebrahimi, P.; Friebe Sandoz, S.; Steiner, E.; Schlagnitweit, J.; Fromm, B.; Silva, L.; Fontana, C.; Chen, A.A.; et al. Base-pair conformational switch modulates miR-34a targeting of Sirt1 mRNA. Nature 2020, 583, 139–144. [Google Scholar] [CrossRef]
- Han, J.; LaVigne, C.A.; Jones, B.T.; Zhang, H.; Gillett, F.; Mendell, J.T. A ubiquitin ligase mediates target-directed microRNA decay independently of tailing and trimming. Science 2020, 370. [Google Scholar] [CrossRef]
- Seok, H.; Lee, H.; Lee, S.; Ahn, S.H.; Lee, H.-S.; Kim, G.-W.D.; Peak, J.; Park, J.; Cho, Y.K.; Jeong, Y.; et al. Position-specific oxidation of miR-1 encodes cardiac hypertrophy. Nature 2020, 584, 279–285. [Google Scholar] [CrossRef]
- Shi, C.Y.; Kingston, E.R.; Kleaveland, B.; Lin, D.H.; Stubna, M.W.; Bartel, D.P. The ZSWIM8 ubiquitin ligase mediates target-directed microRNA degradation. Science 2020, 370. [Google Scholar] [CrossRef]
- Aakula, A.; Kohonen, P.; Leivonen, S.-K.; Mäkelä, R.; Hintsanen, P.; Mpindi, J.P.; Martens-Uzunova, E.; Aittokallio, T.; Jenster, G.; Perälä, M.; et al. Systematic identification of microRNAs that impact on proliferation of prostate cancer cells and display changed expression in tumor tissue. Eur. Urol. 2016, 69, 1120–1128. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Jiang, L.; Sun, D.; Hou, J.; Ji, Z. CircRNA: A novel type of biomarker for cancer. Breast Cancer 2018, 25, 1–7. [Google Scholar] [CrossRef]
- Liu, X.; Chen, F.; Tan, F.; Li, F.; Yi, R.; Yang, D.; Zhao, X. Construction of a potential breast cancer-related miRNA-mRNA regulatory network. BioMed Res. Int. 2020, 2020, e6149174. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Li, G.-S.; Li, J.-D.; Pan, W.-Y.; Shi, Q.; Xiong, D.-D.; Mo, C.-H.; Zeng, J.-J.; Chen, G.; Feng, Z.-B.; et al. The role of upregulated miR-375 expression in breast cancer: An in vitro and in silico study. Pathol. Res. Pract. 2020, 216, 152754. [Google Scholar] [CrossRef] [PubMed]
- Caudron, F.; Barral, Y. A Super-assembly of Whi3 encodes memory of deceptive encounters by single cells during yeast courtship. Cell 2013, 155, 1244–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabortee, S.; Byers, J.S.; Jones, S.; Garcia, D.M.; Bhullar, B.; Chang, A.; She, R.; Lee, L.; Fremin, B.; Lindquist, S.; et al. Intrinsically disordered proteins drive emergence and inheritance of biological traits. Cell 2016, 167, 369–381.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevacqua, R.J.; Fernandez-Martín, R.; Savy, V.; Canel, N.G.; Gismondi, M.I.; Kues, W.A.; Carlson, D.F.; Fahrenkrug, S.C.; Niemann, H.; Taboga, O.A.; et al. Efficient edition of the bovine PRNP prion gene in somatic cells and IVF embryos using the CRISPR/Cas9 system. Theriogenology 2016, 86, 1886–1896.e1. [Google Scholar] [CrossRef] [PubMed]
- Mead, S.; Stumpf, M.P.H.; Whitfield, J.; Beck, J.A.; Poulter, M.; Campbell, T.; Uphill, J.B.; Goldstein, D.; Alpers, M.; Fisher, E.M.C.; et al. Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Science 2003, 300, 640–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contiliani, D.F.; Ribeiro, Y.d.A.; de Moraes, V.N.; Pereira, T.C. MicroRNAs in Prion Diseases—From Molecular Mechanisms to Insights in Translational Medicine. Cells 2021, 10, 1620. https://doi.org/10.3390/cells10071620
Contiliani DF, Ribeiro YdA, de Moraes VN, Pereira TC. MicroRNAs in Prion Diseases—From Molecular Mechanisms to Insights in Translational Medicine. Cells. 2021; 10(7):1620. https://doi.org/10.3390/cells10071620
Chicago/Turabian StyleContiliani, Danyel Fernandes, Yasmin de Araújo Ribeiro, Vitor Nolasco de Moraes, and Tiago Campos Pereira. 2021. "MicroRNAs in Prion Diseases—From Molecular Mechanisms to Insights in Translational Medicine" Cells 10, no. 7: 1620. https://doi.org/10.3390/cells10071620
APA StyleContiliani, D. F., Ribeiro, Y. d. A., de Moraes, V. N., & Pereira, T. C. (2021). MicroRNAs in Prion Diseases—From Molecular Mechanisms to Insights in Translational Medicine. Cells, 10(7), 1620. https://doi.org/10.3390/cells10071620