
ROCK Inhibition as Potential Target for Treatment of Pulmonary Hypertension

 ,
,  ,
,  ,
,  ,
,
Abstract
:1. Pulmonary Hypertension: Clinical Features and Limitations of Approved Drugs
2. Overview on ROCK Structure and Functions
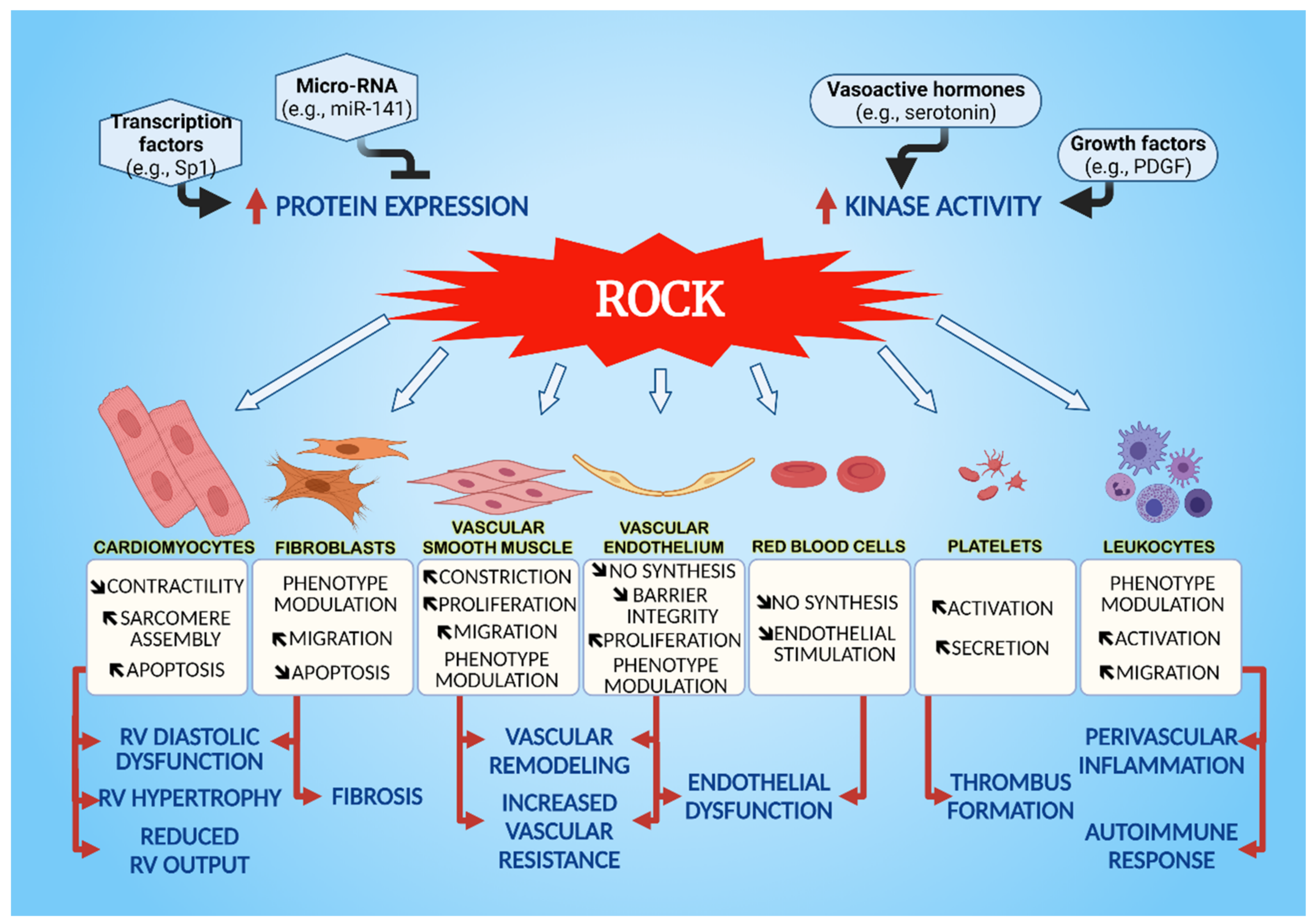
3. Cellular Effects of ROCK on the Cardiovascular System
3.1. Vascular Smooth Muscle Cells (VSMC)
3.2. Endothelial Cells
3.3. Cardiomyocytes
3.4. Fibroblasts
3.5. Leukocytes
3.5.1. Macrophages
3.5.2. Mast Cells
3.5.3. Neutrophils
3.5.4. Dendritic Cells
3.5.5. T and B Lymphocytes
3.6. Platelets and Red Blood Cells
3.6.1. Platelets
3.6.2. Red Blood Cells
4. ROCK in Preclinical Models of PH

{kind=link}
| Compound | Model | Effect | Reference |
|---|---|---|---|
| FDCA | Hypoxia in PAEC PASMC | ↓ TNF-α | [201] |
| PDGF-BB in PAEC/PASMC | ↓ TNF-α ↓ IL-6 | ||
| MCT-induced PH | ↓ mean PAP ↓ RVSP ↓ RV hypertrophy ↓Pulmonary vascular remodeling ↓ RV hypertrophy ↓ Collagen RV | ||
| SB772077-B | MCT-induced PH | ↓ PAP ↓ systemic arterial pressures | [199] |
| Azaindol-1 | PH induced by MCT or chronic hypoxia | ↓ RVSP ↓RV hypertrophy ↓ Pulmonary resistance ↓ muscularization ↓ pulmonary vasculature thickness ↓ expression of p-MYPT1, ↓ PCNA-positive vascular cells in the lungs | [198] |
| KD025 | PAH induced by MCT | ↓ RVSP | [211] |
| Y-27632 | PAH induced by MCT | ↓ RVSP ↓ RV remodeling ↑ Cardiac output | [196] |
| PAH induced by chronic hypoxia | ↓ RVSP ↓ RV hypertrophy ↓ PA wall thickness ↓ contraction in isolated PA | [35] | |
| ↓ RVSP | [162] | ||
| Hypoxia in PASMC | ↓ [Ca2+]i Suppression of SOCE and ROCE ↓ Expression of TRPC1, TRPC2, HIF-1α | [35] | |
| Compound 3 * | MCT-induced PH | Improves hemodynamics ↓ Vascular remodeling | [197] |
| Aloperine | MCT-induced PH | Improvement in hemodynamics ↓ Cardiac hypertrophy ↓ Pulmonary vascular remodeling ↓ Expression protein RhoA, ROCK1 and ROCK2 ↑ Expression protein p27kip1 and Bax ↓ Activation of MYPT1 | [195] |
| 18β-GA | MCT-induced PH | Improvement in hemodynamics ↓ Cardiac hypertrophy ↓ Pulmonary vascular remodeling | [49] |
| PDGF-BB in hPASMC | ↓ Expression protein RhoA, ROCK1 and ROCK2 ↑ Expression protein p27kip1 and Bax ↓ Activation of MYPT1 | [49] |
5. Clinical Trials of ROCK Inhibitors in PH
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Dunlap, B.; Weyer, G. Pulmonary hypertension: Diagnosis an-pd treatment. Am. Fam. Physician 2016, 94, 463–469. [Google Scholar]
- Taichman, D.B.; Mandel, J. Epidemiology of pulmonary arterial hypertension. Clin. Chest Med. 2013, 34, 619–637. [Google Scholar] [CrossRef]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, S13–S24. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.; Lau, E.M.T.; Montani, D.; Jaïs, X.; Sitbon, O.; Simonneau, G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation 2014, 130, 2189–2208. [Google Scholar] [CrossRef] [Green Version]
- Sahni, S.; Ojrzanowski, M.; Majewski, S.; Talwar, A. Pulmonary arterial hypertension: A current review of pharmacological management. Pneumonol. Alergol. Pol. 2016, 84, 47–61. [Google Scholar] [CrossRef] [Green Version]
- Schermuly, R.T.; Ghofrani, H.A.; Wilkins, M.R.; Grimminger, F. Mechanisms of disease: Pulmonary arterial hypertension. Nat. Rev. Cardiol. 2011, 8, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Guignabert, C.; Dorfmuller, P. Pathology and pathobiology of pulmonary hypertension. Semin. Respir. Crit. Care Med. 2013, 34, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Eddahibi, S.; Guignabert, C.; Barlier-Mur, A.-M.; Dewachter, L.; Fadel, E.; Dartevelle, P.; Humbert, M.; Simonneau, G.; Hanoun, N.; Saurini, F.; et al. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension. Circulation 2006, 113, 1857–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Tielemans, B.; Delcroix, M.; Belge, C.; Quarck, R. TGFβ and BMPRII signalling pathways in the pathogenesis of pulmonary arterial hypertension. Drug Discov. Today 2019, 24, 703–716. [Google Scholar] [CrossRef]
- Goldenberg, N.M.; Rabinovitch, M.; Steinberg, B.E. Inflammatory basis of pulmonary arterial hypertension. Anesthesiology 2019, 131, 898–907. [Google Scholar] [CrossRef]
- Parikh, V.; Bhardwaj, A.; Nair, A. Pharmacotherapy for pulmonary arterial hypertension. J. Thorac. Dis. 2019, 11, S1767–S1781. [Google Scholar] [CrossRef] [PubMed]
- Kylhammar, D.; Rådegran, G. The principal pathways involved in the in vivo modulation of hypoxic pulmonary vasoconstriction, pulmonary arterial remodelling and pulmonary hypertension. Acta Physiol. 2017, 219, 728–756. [Google Scholar] [CrossRef]
- Farber, H.W.; Miller, D.P.; Poms, A.D.; Badesch, D.B.; Frost, A.E.; Rouzic, E.M.-L.; Romero, A.J.; Benton, W.W.; Elliott, C.G.; McGoon, M.D.; et al. Five-year outcomes of patients enrolled in the REVEAL registry. Chest 2015, 148, 1043–1054. [Google Scholar] [CrossRef] [Green Version]
- Bourgeois, A.; Omura, J.; Habbout, K.; Bonnet, S.; Boucherat, O. Pulmonary arterial hypertension: New pathophysiological insights and emerging therapeutic targets. Int. J. Biochem. Cell Biol. 2018, 104, 9–13. [Google Scholar] [CrossRef]
- Lajoie, A.C.; Lauzière, G.; Lega, J.-C.; Lacasse, Y.; Martin, S.; Simard, S.; Bonnet, S.; Provencher, S. Combination therapy versus monotherapy for pulmonary arterial hypertension: A meta-analysis. Lancet Respir. Med. 2016, 4, 291–305. [Google Scholar] [CrossRef]
- Duong-Quy, S.; Bei, Y.; Liu, Z.; Dinh-Xuan, A.T. Role of Rho-kinase and its inhibitors in pulmonary hypertension. Pharmacol. Ther. 2013, 137, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, T.; Maekawa, M.; Fujisawa, K.; Okawa, K.; Iwamatsu, A.; Fujita, A.; Watanabe, N.; Saito, Y.; Kakizuka, A.; Morii, N.; et al. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 1996, 15, 1885–1893. [Google Scholar] [CrossRef]
- Leung, T.; Manser, E.; Tan, L.; Lim, L. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J. Biol. Chem. 1995, 270, 29051–29054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, F.; Ahmed, A.; Feroz, A.; Khaki, P.S.S.; Khan, M.S.; Tabrez, S.; Zaidi, S.K.; Abdulaal, W.H.; Shamsi, A.; Khan, W.; et al. An update on the association of protein kinases with cardiovascular diseases. Curr. Pharm. Des. 2019, 25, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Sladojevic, N.; Blair, J.E.; Liao, J.K. Targeting Rho-associated coiled-coil forming protein kinase (ROCK) in cardiovascular fibrosis and stiffening. Expert Opin. Ther. Targets 2020, 24, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Luo, W.; Chang, J. Rho kinase signaling and cardiac physiology. Curr. Opin. Physiol. 2018, 1, 14–20. [Google Scholar] [CrossRef]
- Hajdú, I.; Szilágyi, A.; Végh, B.M.; Wacha, A.; Györffy, D.; Gráczer, É.; Somogyi, M.; Gál, P.; Závodszky, P. Ligand-induced conformational rearrangements regulate the switch between membrane-proximal and distal functions of Rho kinase 2. Commun. Biol. 2020, 3, 721. [Google Scholar] [CrossRef] [PubMed]
- Sladojevic, N.; Yu, B.; Liao, J.K. ROCK as a therapeutic target for ischemic stroke. Expert Rev. Neurother. 2017, 17, 1167–1177. [Google Scholar] [CrossRef]
- Pernis, A.B.; Ricker, E.; Weng, C.-H.; Rozo, C.; Yi, W. Rho kinases in autoimmune diseases. Annu. Rev. Med. 2016, 67, 355–374. [Google Scholar] [CrossRef]
- Lyle, M.A.; Davis, J.P.; Brozovich, F. V regulation of pulmonary vascular smooth muscle contractility in pulmonary arterial hypertension: Implications for therapy. Front. Physiol. 2017, 8, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Zhu, T.; Fang, Z. The role and regulation of pulmonary artery smooth muscle cells in pulmonary hypertension. Int. J. Hypertens. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Strassheim, D.; Gerasimovskaya, E.; Irwin, D.; Dempsey, E.C.; Stenmark, K.; Karoor, V. RhoGTPase in vascular disease. Cells 2019, 8, 0551. [Google Scholar] [CrossRef] [Green Version]
- Rowan, S.C.; Keane, M.P.; Gaine, S.; McLoughlin, P. Hypoxic pulmonary hypertension in chronic lung diseases: Novel vasoconstrictor pathways. Lancet. Respir. Med. 2016, 4, 225–236. [Google Scholar] [CrossRef]
- Sharma, P.; Roy, K. ROCK-2-selective targeting and its therapeutic outcomes. Drug Discov. Today 2020, 25, 446–455. [Google Scholar] [CrossRef]
- Shimokawa, H.; Satoh, K. 2015 ATVB plenary lecture: Translational research on rho-kinase in cardiovascular medicine. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1756–1769. [Google Scholar] [CrossRef]
- Gajecki, D.; Gawrys, J.; Szahidewicz-Krupska, E.; Doroszko, A. Novel molecular mechanisms of pulmonary hypertension: A search for biomarkers and novel drug targets—From bench to bed site. Oxid. Med. Cell. Longev. 2020, 2020, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-Y.; Mo, D.; Tian, W.; Liu, X.-X.; Zhou, Y.-G.; Sun, Y.; Feng, Y.-D.; Xiao, X.; Hao, X.-W.; Zhang, H.-N.; et al. Inhibition of RhoA/ROCK signaling pathway ameliorates hypoxic pulmonary hypertension via HIF-1alpha-dependent functional TRPC channels. Toxicol. Appl. Pharmacol. 2019, 369, 60–72. [Google Scholar] [CrossRef]
- McKenzie, C.; MacDonald, A.; Shaw, A.M. Mechanisms of U46619-induced contraction of rat pulmonary arteries in the presence and absence of the endothelium. Br. J. Pharmacol. 2009, 157, 581–596. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.-H.; Dai, Z.-K.; Wu, B.-N.; Yeh, J.-L.; Chai, C.-Y.; Chu, K.-S.; Liu, C.-P.; Chen, I.-J. KMUP-1 inhibits pulmonary artery proliferation by targeting serotonin receptors/transporter and NO synthase, inactivating RhoA and suppressing AKT/ERK phosphorylation. Vascul. Pharmacol. 2010, 53, 239–249. [Google Scholar] [CrossRef]
- Hartmann, S.; Ridley, A.J.; Lutz, S. The function of rho-associated kinases ROCK1 and ROCK2 in the pathogenesis of cardiovascular disease. Front. Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Shahbazi, R.; Baradaran, B.; Khordadmehr, M.; Safaei, S.; Baghbanzadeh, A.; Jigari, F.; Ezzati, H. Targeting ROCK signaling in health, malignant and non-malignant diseases. Immunol. Lett. 2020, 219, 15–26. [Google Scholar] [CrossRef]
- Jernigan, N.L.; Resta, T.C.; Gonzalez Bosc, L. V Altered redox balance in the development of chronic hypoxia-induced pulmonary hypertension. Adv. Exp. Med. Biol. 2017, 967, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Ghantous, C.M.; Kobeissy, F.H.; Soudani, N.; Rahman, F.A.; Al-Hariri, M.; Itani, H.A.; Sabra, R.; Zeidan, A. Mechanical stretch-induced vascular hypertrophy occurs through modulation of leptin synthesis-mediated ROS formation and GATA-4 nuclear translocation. Front. Pharmacol. 2015, 6, 240. [Google Scholar] [CrossRef]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Chi, L.; Kuebler, W.M.; Goldenberg, N.M. Perivascular inflammation in pulmonary arterial hypertension. Cells 2020, 9, 2338. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Tu, L.; Humbert, M.; Guignabert, C. Chronic inflammation within the vascular wall in pulmonary arterial hypertension: More than a spectator. Cardiovasc. Res. 2020, 116, 885–893. [Google Scholar] [CrossRef]
- Noblet, J.N.; Goodwill, A.G.; Sassoon, D.J.; Kiel, A.M.; Tune, J.D. Leptin augments coronary vasoconstriction and smooth muscle proliferation via a Rho-kinase-dependent pathway. Basic Res. Cardiol. 2016, 111, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Zhang, D.; Liu, L.; Xia, W.; Li, F. Rho signaling pathway enhances proliferation of PASMCs by suppressing nuclear translocation of Smad1 in PAH. Exp. Ther. Med. 2019, 17, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Dai, F.; Liu, Y.; Yu, X.; Huang, C.; Wang, Y.; Yao, W. RhoA/ROCK signaling regulates smooth muscle phenotypic modulation and vascular remodeling via the JNK pathway and vimentin cytoskeleton. Pharmacol. Res. 2018, 133, 201–212. [Google Scholar] [CrossRef]
- Cai, A.; Li, L.; Zhou, Y. Pathophysiological effects of RhoA and Rho-associated kinase on cardiovascular system. J. Hypertens. 2016, 34, 3–10. [Google Scholar] [CrossRef]
- Zhang, M.; Chang, Z.; Zhang, P.; Jing, Z.; Yan, L.; Feng, J.; Hu, Z.; Xu, Q.; Zhou, W.; Ma, P.; et al. Protective effects of 18beta-glycyrrhetinic acid on pulmonary arterial hypertension via regulation of Rho A/Rho kinsase pathway. Chem. Biol. Interact. 2019, 311, 108749. [Google Scholar] [CrossRef]
- Shi, W.; Wang, Q.; Wang, J.; Yan, X.; Feng, W.; Zhang, Q.; Zhai, C.; Chai, L.; Li, S.; Xie, X.; et al. Activation of yes-associated protein mediates sphingosine-1-phosphate-induced proliferation and migration of pulmonary artery smooth muscle cells and its potential mechanisms. J. Cell. Physiol. 2020, 236, 4694–4708. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K. Development of novel therapies for cardiovascular diseases by clinical application of basic research. Circ. J. 2017, 81, 1557–1563. [Google Scholar] [CrossRef] [Green Version]
- Prisco, S.Z.; Thenappan, T.; Prins, K.W. Treatment targets for right ventricular dysfunction in pulmonary arterial hypertension. JACC Basic Transl. Sci. 2020, 5, 1244–1260. [Google Scholar] [CrossRef]
- Sun, X.-Q.; Abbate, A.; Bogaard, H.-J. Role of cardiac inflammation in right ventricular failure. Cardiovasc. Res. 2017, 113, 1441–1452. [Google Scholar] [CrossRef] [Green Version]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic effects of statins on the cardiovascular system. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, J.; Chen, B.; Fan, L. A positive feedback loop of profilin-1 and RhoA/ROCK1 promotes endothelial dysfunction and oxidative stress. Oxid. Med. Cell. Longev. 2018, 2018, 4169575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xu, J.; Zhao, X.; Xie, W.; Wang, H.; Kong, H. Fasudil inhibits neutrophil-endothelial cell interactions by regulating the expressions of GRP78 and BMPR2. Exp. Cell Res. 2018, 365, 97–105. [Google Scholar] [CrossRef]
- Scott, H.A.; Quach, B.; Yang, X.; Ardekani, S.; Cabrera, A.P.; Wilson, R.; Messaoudi-Powers, I.; Ghosh, K. Matrix stiffness exerts biphasic control over monocyte-endothelial adhesion via Rho-mediated ICAM-1 clustering. Integr. Biol. 2016, 8, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Abedi, F.; Hayes, A.W.; Reiter, R.; Karimi, G. Acute lung injury: The therapeutic role of Rho kinase inhibitors. Pharmacol. Res. 2020, 155, 104736. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, H.; Shu, R.; Zhang, X.; Yu, Y.; Liu, X.; Xu, K. Hydrogen treatment prevents lipopolysaccharide-induced pulmonary endothelial cell dysfunction through RhoA inhibition. Biochem. Biophys. Res. Commun. 2020, 522, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Lv, L.; Zhai, P.; Long, T.; Zhou, Q.; Pan, H.; Botwe, G.; Wang, L.; Wang, Q.; Tan, L.; et al. Connexin 40 regulates lung endothelial permeability in acute lung injury via the ROCK1-MYPT1- MLC20 pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L35–L44. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, N.; Schimmel, L.; Oort, C.; van Rijssel, J.; Yin, T.; Ma, B.; van Unen, J.; Pitter, B.; Huveneers, S.; Goedhart, J.; et al. F-actin-rich contractile endothelial pores prevent vascular leakage during leukocyte diapedesis through local RhoA signalling. Nat. Commun. 2016, 7, 10493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia Ponce, A.; Citalan Madrid, A.F.; Vargas Robles, H.; Chanez Paredes, S.; Nava, P.; Betanzos, A.; Zarbock, A.; Rottner, K.; Vestweber, D.; Schnoor, M.; et al. Loss of cortactin causes endothelial barrier dysfunction via disturbed adrenomedullin secretion and actomyosin contractility. Sci. Rep. 2016, 6, 29003. [Google Scholar] [CrossRef]
- Li, J.-R.; Zhao, Y.-S.; Chang, Y.; Yang, S.-C.; Guo, Y.-J.; Ji, E.-S. Fasudil improves endothelial dysfunction in rats exposed to chronic intermittent hypoxia through RhoA/ROCK/NFATc3 pathway. PLoS ONE 2018, 13, e0195604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryczka, J.; Przygodzka, P.; Bogusz, H.; Boncela, J. HMEC-1 adopt the mixed amoeboid-mesenchymal migration type during EndMT. Eur. J. Cell Biol. 2017, 96, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Harvey, L.D.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.-J.; de Jesus Perez, V. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 204589321775291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Cai, A.; Zheng, D.; Qiu, R.; Li, L.; Zhou, Y.; Feng, Y.; Mai, W. Amlodipine suppresses Ang-II-induced endothelium dysfunction by diminishing ROCK1 expression. Clin. Exp. Hypertens. 2016, 38, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Fan, S.; Wang, D.; Huyan, T.; Chen, J.; Chen, J.; Su, J.; Li, X.; Wang, Z.; Xie, S.; et al. FOXO1 inhibition potentiates endothelial angiogenic functions in diabetes via suppression of ROCK1/Drp1-mediated mitochondrial fission. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 2481–2494. [Google Scholar] [CrossRef] [PubMed]
- Qiao, F.; Zou, Z.; Liu, C.; Zhu, X.; Wang, X.; Yang, C.; Jiang, T.; Chen, Y. ROCK2 mediates the proliferation of pulmonary arterial endothelial cells induced by hypoxia in the development of pulmonary arterial hypertension. Exp. Ther. Med. 2016, 11, 2567–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, M.-J.; Zhang, C.; LeMaster, E.; Adamos, C.; Berdyshev, E.; Bogachkov, Y.; Kohler, E.E.; Baruah, J.; Fang, Y.; Schraufnagel, D.E.; et al. Oxidized LDL signals through Rho-GTPase to induce endothelial cell stiffening and promote capillary formation. J. Lipid Res. 2016, 57, 791–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boratkó, A.; Csortos, C.; Boratko, A.; Csortos, C. NHERF2 is crucial in ERM phosphorylation in pulmonary endothelial cells. Cell Commun. Signal. 2013, 11, 99. [Google Scholar] [CrossRef] [Green Version]
- Bowers, S.L.K.; Kemp, S.S.; Aguera, K.N.; Koller, G.M.; Forgy, J.C.; Davis, G.E. Defining an upstream VEGF (Vascular Endothelial Growth Factor) priming signature for downstream factor-induced endothelial cell-pericyte tube network coassembly. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2891–2909. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Tamosiuniene, R.; Nicolls, M.R. Challenges and opportunities in treating inflammation associated with pulmonary hypertension. Expert Rev. Cardiovasc. Ther. 2016, 14, 939–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.; Xiong, Q.; Zhang, X.; Zhang, L.; Shi, Y. Glucagon-like peptide 1 reverses myocardial hypertrophy through cAMP/PKA/RhoA/ROCK2 signaling. Acta Biochim. Biophys. Sin. 2020, 52, 612–619. [Google Scholar] [CrossRef]
- Anaruma, C.P.; Pereira, R.M.; da Cruz Rodrigues, K.C.; Ramos da Silva, A.S.; Cintra, D.E.; Ropelle, E.R.; Pauli, J.R.; Pereira de Moura, L. ROCK protein as cardiac hypertrophy modulator in obesity and physical exercise. Life Sci. 2020, 254, 116955. [Google Scholar] [CrossRef]
- Tsai, S.-H.; Lu, G.; Xu, X.; Ren, Y.; Hein, T.W.; Kuo, L. Enhanced endothelin-1/Rho-kinase signalling and coronary microvascular dysfunction in hypertensive myocardial hypertrophy. Cardiovasc. Res. 2017, 113, 1329–1337. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Liao, J.K. Rho kinases and cardiac remodeling. Circ. J. 2016, 80, 1491–1498. [Google Scholar] [CrossRef] [Green Version]
- Sunamura, S.; Satoh, K.; Kurosawa, R.; Ohtsuki, T.; Kikuchi, N.; Elias-Al-Mamun, M.; Shimizu, T.; Ikeda, S.; Suzuki, K.; Satoh, T.; et al. Different roles of myocardial ROCK1 and ROCK2 in cardiac dysfunction and postcapillary pulmonary hypertension in mice. Proc. Natl. Acad. Sci. USA 2018, 115, E7129–E7138. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Wei, S.-S.; Wang, H.; Wang, Q.; Li, W.; Li, G.; Hou, J.-W.; Chen, X.-M.; Chen, J.; Xu, W.-P.; et al. Crucial role of ROCK2-mediated phosphorylation and upregulation of FHOD3 in the pathogenesis of angiotensin II-induced cardiac hypertrophy. Hypertension 2017, 69, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Mera, C.; Godoy, I.; Ramírez, R.; Moya, J.; Ocaranza, M.P.; Jalil, J.E. Mechanisms of favorable effects of Rho kinase inhibition on myocardial remodeling and systolic function after experimental myocardial infarction in the rat. Ther. Adv. Cardiovasc. Dis. 2016, 10, 4–20. [Google Scholar] [CrossRef] [Green Version]
- Stam, K.; Cai, Z.; van der Velde, N.; van Duin, R.; Lam, E.; van der Velden, J.; Hirsch, A.; Duncker, D.J.; Merkus, D. Cardiac remodelling in a swine model of chronic thromboembolic pulmonary hypertension: Comparison of right vs. left ventricle. J. Physiol. 2019, 597, 4465–4480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yura, Y.; Amano, M.; Takefuji, M.; Bando, T.; Suzuki, K.; Kato, K.; Hamaguchi, T.; Hasanuzzaman Shohag, M.; Takano, T.; Funahashi, Y.; et al. Focused proteomics revealed a novel Rho-kinase signaling pathway in the heart. Cell Struct. Funct. 2016, 41, 105–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimura, T.; Muchir, A.; Kuwahara, M.; Morimoto, S.; Ishikawa, T.; Du, C.K.; Zhan, D.Y.; Nakao, S.; Machida, N.; Tanaka, R.; et al. Overexpression of heart-specific small subunit of myosin light chain phosphatase results in heart failure and conduction disturbance. Am. J. Physiol. Hear. Circ. Physiol. 2018, 314, H1192–H1202. [Google Scholar] [CrossRef]
- Lu, Y.-Y.; Lin, Y.-K.; Wen, Z.-H.; Chen, Y.-C.Y.-J.; Chen, S.-A.; Chen, Y.-C.Y.-J. Latrunculin B modulates electrophysiological characteristics and arrhythmogenesis in pulmonary vein cardiomyocytes. Clin. Sci. 2016, 130, 721–732. [Google Scholar] [CrossRef]
- Jovanovic, A.; Obradovic, M.; Milovanovic, E.S.; Stewart, A.J.; Pitt, S.J.; Alavantic, D.; Aleksic, E.; Isenovic, E.R. Changes in cardiac Na(+)/K(+)-ATPase expression and activity in female rats fed a high-fat diet. Mol. Cell. Biochem. 2017, 436, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soliman, H.; Nyamandi, V.; Garcia-Patino, M.; Zhang, P.C.; Lin, E.; Jia, Z.P.; Tibbits, G.F.; Hove-Madsen, L.; MacLeod, K.M. ROCK2 promotes ryanodine receptor phosphorylation and arrhythmic calcium release in diabetic cardiomyocytes. Int. J. Cardiol. 2019, 281, 90–98. [Google Scholar] [CrossRef]
- Lai, D.; Gao, J.; Bi, X.; He, H.; Shi, X.; Weng, S.; Chen, Y.; Yang, Y.; Ye, Y.; Fu, G. The Rho kinase inhibitor, fasudil, ameliorates diabetes-induced cardiac dysfunction by improving calcium clearance and actin remodeling. J. Mol. Med. 2017, 95, 155–165. [Google Scholar] [CrossRef]
- Olgar, Y.; Celen, M.C.; Yamasan, B.E.; Ozturk, N.; Turan, B.; Ozdemir, S. Rho-kinase inhibition reverses impaired Ca2+ handling and associated left ventricular dysfunction in pressure overload-induced cardiac hypertrophy. Cell Calcium 2017, 67, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xiang, C.; Yang, Z.; Miao, D.; Zhang, D. Rho kinase inhibition by fasudil attenuates adriamycin-induced chronic heart injury. Cardiovasc. Toxicol. 2020, 20, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-S.; Tang, L.-J.; Tu, H.; Wang, S.-J.; Liu, B.; Zhang, X.-J.; Li, N.-S.; Luo, X.-J.; Peng, J. Fasudil ameliorates the ischemia/reperfusion oxidative injury in rat hearts through suppression of myosin regulatory light chain/NADPH oxidase 2 pathway. Eur. J. Pharmacol. 2018, 822, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Waddingham, M.T.; Sonobe, T.; Tsuchimochi, H.; Edgley, A.J.; Sukumaran, V.; Chen, Y.C.; Hansra, S.S.; Schwenke, D.O.; Umetani, K.; Aoyama, K.; et al. Diastolic dysfunction is initiated by cardiomyocyte impairment ahead of endothelial dysfunction due to increased oxidative stress and inflammation in an experimental prediabetes model. J. Mol. Cell. Cardiol. 2019, 137, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Prysyazhna, O.; Burgoyne, J.R.; Scotcher, J.; Grover, S.; Kass, D.; Eaton, P. Phosphodiesterase 5 inhibition limits doxorubicin-induced heart failure by attenuating protein kinase G ialpha oxidation. J. Biol. Chem. 2016, 291, 17427–17436. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Sun, Y.; Zhang, L.; Kang, W.; Li, N.; Li, Y. The RhoA/ROCK pathway mediates high glucose-induced cardiomyocyte apoptosis via oxidative stress, JNK, and p38MAPK pathways. Diabetes Metab. Res. Rev. 2018, e3022. [Google Scholar] [CrossRef]
- Zhou, F.-T.; Ma, K. Fasudil protects against isoproterenol-induced myocardial infarction in mice via inhibiting Rho/ROCK signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 5659–5667. [Google Scholar] [CrossRef]
- Min, F.; Jia, X.J.; Gao, Q.; Niu, F.; Hu, Z.Y.; Han, Y.L.; Shi, H.J.; Yu, Y. Remote ischemic post-conditioning protects against myocardial ischemia/reperfusion injury by inhibiting the Rho-kinase signaling pathway. Exp. Ther. Med. 2020, 19, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Wang, Y.; Jiang, M.; Deng, X.; Pei, Z.; Li, F.; Xia, K.; Zhu, L.; Yang, T.; Chen, M. Downregulation of profilin-1 expression attenuates cardiomyocytes hypertrophy and apoptosis induced by advanced glycation end products in H9c2 cells. BioMed Res. Int. 2017, 2017, 9716087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernau, K.; Torr, E.E.; Evans, M.D.; Aoki, J.K.; Ngam, C.R.; Sandbo, N. Tensin 1 is essential for myofibroblast differentiation and extracellular matrix formation. Am. J. Respir. Cell Mol. Biol. 2017, 56, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Knipe, R.S.; Probst, C.K.; Lagares, D.; Franklin, A.; Spinney, J.J.; Brazee, P.L.; Grasberger, P.; Zhang, L.; Black, K.E.; Sakai, N.; et al. The Rho kinase isoforms ROCK1 and ROCK2 each contribute to the development of experimental pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 471–481. [Google Scholar] [CrossRef]
- Shimizu, T.; Narang, N.; Chen, P.; Yu, B.; Knapp, M.; Janardanan, J.; Blair, J.; Liao, J.K. Fibroblast deletion of ROCK2 attenuates cardiac hypertrophy, fibrosis, and diastolic dysfunction. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Chen, H.; Qu, J.; Huang, X.; Kurundkar, A.; Zhu, L.; Yang, N.; Venado, A.; Ding, Q.; Liu, G.; Antony, V.B.; et al. Mechanosensing by the alpha6-integrin confers an invasive fibroblast phenotype and mediates lung fibrosis. Nat. Commun. 2016, 7, 12564. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Murata, N.; Yamawaki, H. Canstatin stimulates migration of rat cardiac fibroblasts via secretion of matrix metalloproteinase-2. Am. J. Physiol. Cell Physiol. 2017, 312, C199–C208. [Google Scholar] [CrossRef]
- Li, Z.; Bratlie, K.M. Fibroblasts treated with macrophage conditioned medium results in phenotypic shifts and changes in collagen organization. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 122, 111915. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Xu, H.; Zhang, X.; Sun, Y.; Wang, R.; Brann, D.; Yang, F. Protective effect of Ac-SDKP on alveolar epithelial cells through inhibition of EMT via TGF-beta1/ROCK1 pathway in silicosis in rat. Toxicol. Appl. Pharmacol. 2016, 294, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Barnett, J.V.; Watanabe, M.; Ramírez-Bergeron, D. Hypoxia supports epicardial cell differentiation in vascular smooth muscle cells through the activation of the TGFβ pathway. J. Cardiovasc. Dev. Dis. 2018, 5, 0019. [Google Scholar] [CrossRef] [Green Version]
- Knipe, R.S.; Tager, A.M.; Liao, J.K. The Rho kinases: Critical mediators of multiple profibrotic processes and rational targets for new therapies for pulmonary fibrosis. Pharmacol. Rev. 2015, 67, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Xiao, P.; Liu, X.; Chen, Y.; Xu, Y.; Fan, J.; Yin, Y. Notch3 modulates cardiac fibroblast proliferation, apoptosis, and fibroblast to myofibroblast transition via negative regulation of the RhoA/ROCK/Hif1α axis. Front. Physiol. 2020, 11, 669. [Google Scholar] [CrossRef]
- Ouyang, P.; Wang, S.; Zhang, H.; Huang, Z.; Wei, P.; Zhang, Y.; Wu, Z.; Li, T. Microarray analysis of differentially expressed genes in L929 mouse fibroblast cells exposed to leptin and hypoxia. Mol. Med. Rep. 2017, 16, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Oh, R.S.; Haak, A.J.; Smith, K.M.J.; Ligresti, G.; Choi, K.M.; Xie, T.; Wang, S.; Walters, P.R.; Thompson, M.A.; Freeman, M.R.; et al. RNAi screening identifies a mechanosensitive ROCK-JAK2-STAT3 network central to myofibroblast activation. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Savai, R.; Pullamsetti, S.S.; Kolbe, J.; Bieniek, E.; Voswinckel, R.; Fink, L.; Scheed, A.; Ritter, C.; Dahal, B.K.; Vater, A.; et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 897–908. [Google Scholar] [CrossRef]
- Marsh, L.M.; Jandl, K.; Grünig, G.; Foris, V.; Bashir, M.; Ghanim, B.; Klepetko, W.; Olschewski, H.; Olschewski, A.; Kwapiszewska, G. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1701214. [Google Scholar] [CrossRef] [Green Version]
- Dewachter, L.; Dewachter, C. Inflammation in right ventricular failure: Does it matter? Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Biro, M.; Munoz, M.A.; Weninger, W. Targeting Rho-GTPases in immune cell migration and inflammation. Br. J. Pharmacol. 2014, 171, 5491–5506. [Google Scholar] [CrossRef]
- Kitano, K.; Usui, S.; Ootsuji, H.; Takashima, S.I.; Kobayashi, D.; Murai, H.; Furusho, H.; Nomura, A.; Kaneko, S.; Takamura, M. Rho-kinase activation in leukocytes plays a pivotal role in myocardial ischemia/reperfusion injury. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Zanin-Zhorov, A.; Flynn, R.; Waksal, S.D.; Blazar, B.R. Isoform-specific targeting of ROCK proteins in immune cells. Small GTPases 2016, 7, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Mallat, Z.; Gojova, A.; Sauzeau, V.; Brun, V.; Silvestre, J.-S.; Esposito, B.; Merval, R.; Groux, H.; Loirand, G.; Tedgui, A. Rho-associated protein kinase contributes to early atherosclerotic lesion formation in mice. Circ. Res. 2003, 93, 884–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaoita, N.; Satoh, K.; Shimokawa, H. Novel therapeutic targets of pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, e97–e102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nihei, T.; Takahashi, J.; Hao, K.; Kikuchi, Y.; Odaka, Y.; Tsuburaya, R.; Nishimiya, K.; Matsumoto, Y.; Ito, K.; Miyata, S.; et al. Prognostic impacts of Rho-kinase activity in circulating leucocytes in patients with vasospastic angina. Eur. Heart J. 2018, 39, 952–959. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, F.; Liu, X.-B.; Bi, S.-J.; Lu, Q.-H. Increased Rho kinase activity in patients with heart ischemia/reperfusion. Perfusion 2019, 34, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Dai, W.; Lin, Y.; Zhang, Z.; Pan, Y.; Han, H.; Jia, H.; Peng, J.; Zhao, J.; Xu, L. Leukocyte Rho kinase activity and serum cystatin C affect cardiovascular events in acute coronary syndrome. Medicine 2020, 99, e20060. [Google Scholar] [CrossRef]
- Ocaranza, M.P.; Moya, J.; Jalil, J.E.; Lavandero, S.; Kalergis, A.M.; Molina, C.; Gabrielli, L.; Godoy, I.; Córdova, S.; Castro, P.; et al. Rho-kinase pathway activation and apoptosis in circulating leucocytes in patients with heart failure with reduced ejection fraction. J. Cell. Mol. Med. 2020, 24, 1413–1427. [Google Scholar] [CrossRef]
- Fierro, C.; Novoa, U.; Gonzalez, V.; Ocaranza, M.P.; Jalil, J.E. Simultaneous Rho kinase inhibition in circulating leukocytes and in cardiovascular tissue in rats with high angiotensin converting enzyme levels. Int. J. Cardiol. 2016, 215, 309–317. [Google Scholar] [CrossRef]
- Bros, M.; Haas, K.; Moll, L.; Grabbe, S. RhoA as a key regulator of innate and adaptive immunity. Cells 2019, 8, 0733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niermann, C.; Gorressen, S.; Klier, M.; Gowert, N.S.; Billuart, P.; Kelm, M.; Merx, M.W.; Elvers, M. Oligophrenin1 protects mice against myocardial ischemia and reperfusion injury by modulating inflammation and myocardial apoptosis. Cell. Signal. 2016, 28, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Bzymek, R.; Horsthemke, M.; Isfort, K.; Mohr, S.; Tjaden, K.; Muller-Tidow, C.; Thomann, M.; Schwerdtle, T.; Bahler, M.; Schwab, A.; et al. Real-time two- and three-dimensional imaging of monocyte motility and navigation on planar surfaces and in collagen matrices: Roles of Rho. Sci. Rep. 2016, 6, 25016. [Google Scholar] [CrossRef] [Green Version]
- Takeda, Y.; Matoba, K.; Kawanami, D.; Nagai, Y.; Akamine, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. ROCK2 regulates monocyte migration and cell to cell adhesion in vascular endothelial cells. Int. J. Mol. Sci. 2019, 20, 1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Azreq, M.-A.; Kadiri, M.; Boisvert, M.; Page, N.; Tessier, P.A.; Aoudjit, F. Discoidin domain receptor 1 promotes Th17 cell migration by activating the RhoA/ROCK/MAPK/ERK signaling pathway. Oncotarget 2016, 7, 44975–44990. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Liu, P.-P.; Tang, D.-D.; Song, R.; Zhang, Y.-Q.; Lei, S.; Wu, S.-J. Targeting the RhoA-ROCK pathway to regulate T-cell homeostasis in hypoxia-induced pulmonary arterial hypertension. Pulm. Pharmacol. Ther. 2018, 50, 111–122. [Google Scholar] [CrossRef]
- West, J.; Chen, X.; Yan, L.; Gladson, S.; Loyd, J.; Rizwan, H.; Talati, M. Adverse effects of BMPR2 suppression in macrophages in animal models of pulmonary hypertension. Pulm. Circ. 2020, 10, 204589401985648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Hao, Y.; Gao, D.; Gao, L.; Li, G.; Zhang, Z. Phenotype and function of macrophage polarization in monocrotaline-induced pulmonary arterial hypertension rat model. Physiol. Res. 2021, 70, 213–226. [Google Scholar] [CrossRef]
- Yang, L.; Dai, F.; Tang, L.; Le, Y.; Yao, W. Macrophage differentiation induced by PMA is mediated by activation of RhoA/ROCK signaling. J. Toxicol. Sci. 2017, 42, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Miyagawa, Y.; Long, C.; Zhang, M.; Cooper, D.K.C.; Hara, H. Effect of Rho-kinase Inhibitor, Y27632, on porcine corneal endothelial cell culture, inflammation and immune regulation. Ocul. Immunol. Inflamm. 2016, 24, 579–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Mickael, C.; Kassa, B.; Sanders, L.; Hernandez-Saavedra, D.; Koyanagi, D.E.; Kumar, S.; Pugliese, S.C.; Thomas, S.; McClendon, J.; et al. Interstitial macrophage-derived thrombospondin-1 contributes to hypoxia-induced pulmonary hypertension. Cardiovasc. Res. 2020, 116, 2021–2030. [Google Scholar] [CrossRef]
- Florentin, J.; Coppin, E.; Vasamsetti, S.B.; Zhao, J.; Tai, Y.-Y.; Tang, Y.; Zhang, Y.; Watson, A.; Sembrat, J.; Rojas, M.; et al. Inflammatory macrophage expansion in pulmonary hypertension depends upon mobilization of blood-borne monocytes. J. Immunol. 2018, 200, 3612–3625. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, W.; Minze, L.J.; Kubiak, J.Z.; Li, X.C.; Ghobrial, R.M.; Kloc, M. Dissonant response of M0/M2 and M1 bone-marrow-derived macrophages to RhoA pathway interference. Cell Tissue Res. 2016, 366, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Gruber, E.; Heyward, C.; Cameron, J.; Leifer, C. Toll-like receptor signaling in macrophages is regulated by extracellular substrate stiffness and Rho-associated coiled-coil kinase (ROCK1/2). Int. Immunol. 2018, 30, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Abbadi, D.; Laroumanie, F.; Bizou, M.; Pozzo, J.; Daviaud, D.; Delage, C.; Calise, D.; Gaits-Iacovoni, F.; Dutaur, M.; Tortosa, F.; et al. Local production of tenascin-C acts as a trigger for monocyte/macrophage recruitment that provokes cardiac dysfunction. Cardiovasc. Res. 2018, 114, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Bewley, M.A.; Belchamber, K.B.R.; Chana, K.K.; Budd, R.C.; Donaldson, G.; Wedzicha, J.A.; Brightling, C.E.; Kilty, I.; Donnelly, L.E.; Barnes, P.J.; et al. Differential effects of p38, MAPK, PI3K or Rho kinase inhibitors on bacterial phagocytosis and efferocytosis by macrophages in COPD. PLoS ONE 2016, 11, e0163139. [Google Scholar] [CrossRef] [Green Version]
- Lescoat, A.; Ballerie, A.; Lelong, M.; Augagneur, Y.; Morzadec, C.; Jouneau, S.; Jégo, P.; Fardel, O.; Vernhet, L.; Lecureur, V. Crystalline silica impairs efferocytosis abilities of human and mouse macrophages: Implication for silica-associated systemic sclerosis. Front. Immunol. 2020, 11, 219. [Google Scholar] [CrossRef] [Green Version]
- Galvao, I.; Athayde, R.M.; Perez, D.A.; Reis, A.C.; Rezende, L.; de Oliveira, V.L.S.; Rezende, B.M.; Goncalves, W.A.; Sousa, L.P.L.P.; Teixeira, M.M.; et al. ROCK inhibition drives resolution of acute inflammation by enhancing neutrophil apoptosis. Cells 2019, 8, 0964. [Google Scholar] [CrossRef] [Green Version]
- Saclier, M.; Lapi, M.; Bonfanti, C.; Rossi, G.; Antonini, S.; Messina, G. The transcription factor Nfix requires RhoA-ROCK1 dependent phagocytosis to mediate macrophage skewing during skeletal muscle regeneration. Cells 2020, 9, 0708. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Wong, S.H.D.; Pan, Q.; Li, G.; Bian, L. Anisotropic ligand nanogeometry modulates the adhesion and polarization state of macrophages. Nano Lett. 2019, 19, 1963–1975. [Google Scholar] [CrossRef]
- Sridharan, R.; Cavanagh, B.; Cameron, A.R.; Kelly, D.J.; O’Brien, F.J. Material stiffness influences the polarization state, function and migration mode of macrophages. Acta Biomater. 2019, 89, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Zhou, H.; Wang, Q.; Zhou, S.; Li, C.; Liu, R.; Qiu, J.; Shi, C.; Lu, L. RIP3 deficiency alleviates liver fibrosis by inhibiting ROCK1-TLR4-NF-kappaB pathway in macrophages. FASEB J. 2019, fj201900752R. [Google Scholar] [CrossRef] [Green Version]
- Novotný, T.; Krejčí, J.; Malíková, J.; Švehlík, V.; Wasserbauer, R.; Uhlík, J.; Vajner, L. Mast cell stabilization with sodium cromoglycate modulates pulmonary vessel wall remodeling during four-day hypoxia in rats. Exp. Lung Res. 2015, 41, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Kosanovic, D.; Dahal, B.K.; Peters, D.M.; Seimetz, M.; Wygrecka, M.; Hoffmann, K.; Antel, J.; Reiss, I.; Ghofrani, H.A.; Weissmann, N.; et al. Histological characterization of mast cell chymase in patients with pulmonary hypertension and chronic obstructive pulmonary disease. Pulm. Circ. 2014, 4, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Farha, S.; Sharp, J.; Asosingh, K.; Park, M.; Comhair, S.A.A.; Tang, W.H.W.; Thomas, J.; Farver, C.; Hsieh, F.; Loyd, J.E.; et al. Mast cell number, phenotype, and function in human pulmonary arterial hypertension. Pulm. Circ. 2012, 2, 220–228. [Google Scholar] [CrossRef] [Green Version]
- Bartelds, B.; van Loon, R.L.E.; Mohaupt, S.; Wijnberg, H.; Dickinson, M.G.; Boersma, B.; Takens, J.; van Albada, M.; Berger, R.M.F. Mast cell inhibition improves pulmonary vascular remodeling in pulmonary hypertension. Chest 2012, 141, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Sendo, T.; Oishi, R. Physiology and pathophysiology of proteinase-activated receptors (PARs): Role of tryptase/PAR-2 in vascular endothelial barrier function. J. Pharmacol. Sci. 2005, 97, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Ashina, K.; Tsubosaka, Y.; Nakamura, T.; Omori, K.; Kobayashi, K.; Hori, M.; Ozaki, H.; Murata, T. Histamine induces vascular hyperpermeability by increasing blood flow and endothelial barrier disruption in vivo. PLoS ONE 2015, 10, e0132367. [Google Scholar] [CrossRef]
- Breitling, S.; Hui, Z.; Zabini, D.; Hu, Y.; Hoffmann, J.; Goldenberg, N.M.; Tabuchi, A.; Buelow, R.; Dos Santos, C.; Kuebler, W.M. The mast cell–B cell axis in lung vascular remodeling and pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2017, 312, L710–L721. [Google Scholar] [CrossRef] [PubMed]
- Kosanovic, D.; Luitel, H.; Dahal, B.K.; Cornitescu, T.; Janssen, W.; Danser, A.H.J.; Garrelds, I.M.; De Mey, J.G.R.; Fazzi, G.; Schiffers, P.; et al. Chymase: A multifunctional player in pulmonary hypertension associated with lung fibrosis. Eur. Respir. J. 2015, 46, 1084–1094. [Google Scholar] [CrossRef] [Green Version]
- Kwapiszewska, G.; Markart, P.; Dahal, B.K.; Kojonazarov, B.; Marsh, L.M.; Schermuly, R.T.; Taube, C.; Meinhardt, A.; Ghofrani, H.A.; Steinhoff, M.; et al. PAR-2 inhibition reverses experimental pulmonary hypertension. Circ. Res. 2012, 110, 1179–1191. [Google Scholar] [CrossRef]
- Kapur, R.; Shi, J.; Ghosh, J.; Munugalavadla, V.; Sims, E.; Martin, H.; Wei, L.; Mali, R.S. ROCK1 via LIM kinase regulates growth, maturation and actin based functions in mast cells. Oncotarget 2016, 7, 16936–16947. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Ohata, H.; Momose, K.; Honda, K. Lysophosphatidic acid induces histamine release from mast cells and skin fragments. Pharmacology 2005, 75, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Doe, Z.; Fukumoto, Y.; Takaki, A.; Tawara, S.; Ohashi, J.; Nakano, M.; Tada, T.; Saji, K.; Sugimura, K.; Fujita, H.; et al. Evidence for Rho-kinase activation in patients with pulmonary arterial hypertension. Circ. J. 2009, 73, 1731–1739. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.; Dirir, O.; Zamanian, R.T.; Rabinovitch, M.; Thompson, A.A.R. The role of neutrophils and neutrophil elastase in pulmonary arterial hypertension. Front. Med. 2018, 5. [Google Scholar] [CrossRef]
- Khandoga, A.G.; Khandoga, A.; Reichel, C.A.; Bihari, P.; Rehberg, M.; Krombach, F. In vivo imaging and quantitative analysis of leukocyte directional migration and polarization in inflamed tissue. PLoS ONE 2009, 4, e4693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jannat, R.A.; Dembo, M.; Hammer, D.A. Traction forces of neutrophils migrating on compliant substrates. Biophys. J. 2011, 101, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.; Harris, G.J.; Pinder, E.M.; Macfarlane, J.G.; Hellyer, T.P.; Rostron, A.J.; Conway Morris, A.; Thickett, D.R.; Perkins, G.D.; McAuley, D.F.; et al. Exchange protein directly activated by cyclic AMP (EPAC) activation reverses neutrophil dysfunction induced by beta2-agonists, corticosteroids, and critical illness. J. Allergy Clin. Immunol. 2016, 137, 535–544. [Google Scholar] [CrossRef]
- Filina, J.V.; Gabdoulkhakova, A.G.; Safronova, V.G. RhoA/ROCK downregulates FPR2-mediated NADPH oxidase activation in mouse bone marrow granulocytes. Cell. Signal. 2014, 26, 2138–2146. [Google Scholar] [CrossRef]
- Silveira, A.A.A.; Dominical, V.M.; Lazarini, M.; Costa, F.F.; Conran, N. Simvastatin abrogates inflamed neutrophil adhesive properties, in association with the inhibition of Mac-1 integrin expression and modulation of Rho kinase activity. Inflamm. Res. 2013, 62, 127–132. [Google Scholar] [CrossRef]
- Klinke, A.; Berghausen, E.; Friedrichs, K.; Molz, S.; Lau, D.; Remane, L.; Berlin, M.; Kaltwasser, C.; Adam, M.; Mehrkens, D.; et al. Myeloperoxidase aggravates pulmonary arterial hypertension by activation of vascular Rho-kinase. JCI Insight 2018, 3, 1–17. [Google Scholar] [CrossRef]
- Ikeda, K.T.; Hale, P.T.; Pauciulo, M.W.; Dasgupta, N.; Pastura, P.A.; Le Cras, T.D.; Pandey, M.K.; Nichols, W.C. Hypoxia-induced pulmonary hypertension in different mouse strains: Relation to transcriptome. Am. J. Respir. Cell Mol. Biol. 2019, 60, 106–116. [Google Scholar] [CrossRef]
- Nam, G.-H.; Lee, E.J.; Kim, Y.K.; Hong, Y.; Choi, Y.; Ryu, M.-J.; Woo, J.; Cho, Y.; Ahn, D.J.; Yang, Y.; et al. Combined Rho-kinase inhibition and immunogenic cell death triggers and propagates immunity against cancer. Nat. Commun. 2018, 9, 2165. [Google Scholar] [CrossRef]
- Qiu, H.; He, Y.; Ouyang, F.; Jiang, P.; Guo, S.; Guo, Y. The role of regulatory t cells in pulmonary arterial hypertension. J. Am. Heart Assoc. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Li, X.; Qiu, M.; Luo, R.; Lin, J.; Liu, B. LncRNA EGFR-AS1 upregulates ROCK1 by sponging miR-145 to promote esophageal squamous cell carcinoma cell invasion and migration. Cancer Biother. Radiopharm. 2020, 35, 66–71. [Google Scholar] [CrossRef]
- Ricker, E.; Chowdhury, L.; Yi, W.; Pernis, A.B. The RhoA-ROCK pathway in the regulation of T and B cell responses. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, W.; Nagpal, K.; Suárez-Fueyo, A.; Ferretti, A.; Yoshida, N.; Tsokos, M.G.; Tsokos, G.C. The regulatory subunit PPP2R2A of PP2A enhances Th1 and Th17 differentiation through activation of the GEF-H1/RhoA/ROCK signaling pathway. J. Immunol. 2021. [Google Scholar] [CrossRef]
- Takesono, A.; Heasman, S.J.; Wojciak-Stothard, B.; Garg, R.; Ridley, A.J. Microtubules regulate migratory polarity through Rho/ROCK signaling in T cells. PLoS ONE 2010, 5, e8774. [Google Scholar] [CrossRef]
- Chen, W.; Nyuydzefe, M.S.; Weiss, J.M.; Zhang, J.; Waksal, S.D.; Zanin-Zhorov, A. ROCK2, but not ROCK1 interacts with phosphorylated STAT3 and co-occupies TH17/TFH gene promoters in TH17-activated human T cells. Sci. Rep. 2018, 8, 16636. [Google Scholar] [CrossRef]
- Dai, K.; Wang, Y.; Tai, S.; Ni, H.; Lian, H.; Yu, Y.; Liao, W.; Zheng, C.; Chen, Q.; Kuver, A.; et al. Fasudil exerts a cardio-protective effect on mice with coxsackievirus B3-induced acute viral myocarditis. Cardiovasc. Ther. 2018, 36, e12477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badr, G.; Saad, H.; Waly, H.; Hassan, K.; Abdel-Tawab, H.; Alhazza, I.M.; Ahmed, E.A. Type I interferon (IFN-alpha/beta) rescues B-lymphocytes from apoptosis via PI3Kdelta/Akt, Rho-A, NFkappaB and Bcl-2/Bcl(XL). Cell. Immunol. 2010, 263, 31–40. [Google Scholar] [CrossRef]
- Ricker, E.; Verma, A.; Marullo, R.; Gupta, S.; Ye, C.; Pannellini, T.; Manni, M.; Tam, W.; Inghirami, G.; Elemento, O.; et al. Selective dysregulation of ROCK2 activity promotes aberrant transcriptional networks in ABC diffuse large B-cell lymphoma. Sci. Rep. 2020, 10, 13094. [Google Scholar] [CrossRef]
- Cho, S.-Y.; Sung, C.O.; Chae, J.; Lee, J.; Na, D.; Kang, W.; Kang, J.; Min, S.; Lee, A.; Kwak, E.; et al. Alterations in the Rho pathway contribute to Epstein-Barr virus-induced lymphomagenesis in immunosuppressed environments. Blood 2018, 131, 1931–1941. [Google Scholar] [CrossRef] [Green Version]
- Ricker, E.; Chinenov, Y.; Pannellini, T.; Flores-Castro, D.; Ye, C.; Gupta, S.; Manni, M.; Liao, J.K.; Pernis, A.B. Serine-threonine kinase ROCK2 regulates germinal center B cell positioning and cholesterol biosynthesis. J. Clin. Investig. 2020, 130, 3654–3670. [Google Scholar] [CrossRef]
- Bouafia, A.; Lofek, S.; Bruneau, J.; Chentout, L.; Lamrini, H.; Trinquand, A.; Deau, M.-C.; Heurtier, L.; Meignin, V.; Picard, C.; et al. Loss of ARHGEF1 causes a human primary antibody deficiency. J. Clin. Investig. 2019, 129, 1047–1060. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, C.; Li, L.; Tu, X.; Lu, Z. Red blood cell distribution width predicts pulmonary hypertension secondary to chronic obstructive pulmonary disease. Can. Respir. J. 2019, 2019, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, K.C.; Gladwin, M.T.; Straub, A.C. Sickle cell disease: At the crossroads of pulmonary hypertension and diastolic heart failure. Heart 2020, 106, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Al-Qadi, M.; LeVarge, B.; Ford, H.J. Epidemiology, pathogenesis, and clinical approach in group 5 pulmonary hypertension. Front. Med. 2021, 7. [Google Scholar] [CrossRef]
- Olschewski, H.; Rich, S. Are anticoagulants still indicated in pulmonary arterial hypertension? Pulm. Circ. 2018, 8, 204589401880768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbar, H.; Duan, X.; Saleem, S.; Davis, A.K.; Zheng, Y. RhoA and Rac1 GTPases differentially regulate agonist-receptor mediated reactive oxygen species generation in platelets. PLoS ONE 2016, 11, e0163227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, Y.; Doi, T.; Tokuda, H.; Matsushima-Nishiwaki, R.; Tsujimoto, M.; Kuroyanagi, G.; Yamamoto, N.; Enomoto, Y.; Tanabe, K.; Otsuka, T.; et al. Rho-kinase regulates human platelet activation induced by thromboxane A2 independently of p38 MAP kinase. Prostaglandins. Leukot. Essent. Fat. Acids 2015, 94, 73–81. [Google Scholar] [CrossRef]
- Feghhi, S.; Tooley, W.W.; Sniadecki, N.J. Nonmuscle myosin IIA regulates platelet contractile forces through rho kinase and myosin light-chain kinase. J. Biomech. Eng. 2016, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sladojevic, N.; Oh, G.T.; Kim, H.-H.; Beaulieu, L.M.; Falet, H.; Kaminski, K.; Freedman, J.E.; Liao, J.K. Decreased thromboembolic stroke but not atherosclerosis or vascular remodelling in mice with ROCK2-deficient platelets. Cardiovasc. Res. 2017, 113, 1307–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, P.; Coupland, D.; Nombalais, M.; MWalsh, G.; Devine, D. V RhoA/ROCK signaling contributes to sex differences in the activation of human platelets. Thromb. Res. 2016, 139, 50–55. [Google Scholar] [CrossRef]
- Guilluy, C.; Eddahibi, S.; Agard, C.; Guignabert, C.; Izikki, M.; Tu, L.; Savale, L.; Humbert, M.; Fadel, E.; Adnot, S.; et al. RhoA and Rho kinase activation in human pulmonary hypertension: Role of 5-HT signaling. Am. J. Respir. Crit. Care Med. 2009, 179, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Hsu, L.-W.; Chen, P.-W.; Chang, W.-T.; Lee, W.-H.; Liu, P.-Y. The role of ROCK in platelet-monocyte collaborative induction of thromboinflammation during acute coronary syndrome. Thromb. Haemost. 2020, 120, 1417–1431. [Google Scholar] [CrossRef]
- Smukowska-Gorynia, A.; Tomaszewska, I.; Malaczynska-Rajpold, K.; Marcinkowska, J.; Komosa, A.; Janus, M.; Olasinska-Wisniewska, A.; Slawek, S.; Araszkiewicz, A.; Jankiewicz, S.; et al. Red blood cells distribution width as a potential prognostic biomarker in patients with pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Hear. Lung Circ. 2018, 27, 842–848. [Google Scholar] [CrossRef]
- Geenen, L.W.; Baggen, V.J.M.; Koudstaal, T.; Boomars, K.A.; Eindhoven, J.A.; Boersma, E.; Roos-Hesselink, J.W.; van den Bosch, A.E. The prognostic value of various biomarkers in adults with pulmonary hypertension; a multi-biomarker approach. Am. Heart J. 2019, 208, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Schreier, D.A.; Forouzan, O.; Hacker, T.A.; Sheehan, J.; Chesler, N. increased red blood cell stiffness increases pulmonary vascular resistance and pulmonary arterial pressure. J. Biomech. Eng. 2016, 138. [Google Scholar] [CrossRef] [Green Version]
- Racine, M.L.; Dinenno, F.A. Reduced deformability contributes to impaired deoxygenation-induced ATP release from red blood cells of older adult humans. J. Physiol. 2019, 597, 4503–4519. [Google Scholar] [CrossRef] [PubMed]
- Ulker, P.; Ozen, N.; Abduleyeva, G.; Koksoy, S.; Yaras, N.; Basrali, F. Rho-kinase is a negative regulator of red blood cell eNOS under basal conditions. Clin. Hemorheol. Microcirc. 2019. [Google Scholar] [CrossRef] [PubMed]
- Basarici, I.; Özen, N.; Kilavuz, E.; Kısak, F.; Basrali, F.; Yaras, N.; Koksoy, S.; Celik, M.L.; Ulker, P. Concealed role of red blood cells in pathogenesis of pulmonary arterial hypertension: Decreased red blood cell nitric oxide generation and effect of Rho-Kinase inhibitor fasudil. Clin. Hemorheol. Microcirc. 2020. [Google Scholar] [CrossRef]
- Chapados, R.; Abe, K.; Ihida-Stansbury, K.; McKean, D.; Gates, A.T.; Kern, M.; Merklinger, S.; Elliott, J.; Plant, A.; Shimokawa, H.; et al. ROCK controls matrix synthesis in vascular smooth muscle cells: Coupling vasoconstriction to vascular remodeling. Circ. Res. 2006, 99, 837–844. [Google Scholar] [CrossRef]
- Wu, F.; Yao, W.; Yang, J.; Zhang, M.; Xu, Y.; Hao, Y.; Yan, L.; Niu, Y.; Sun, T.; Yu, J.; et al. Protective effects of aloperin on monocroline-induced pulmonary hypertension via regulation of Rho A/Rho kinsase pathway in rats. Biomed. Pharmacother. 2017, 95, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Novelli, D.; Fumagalli, F.; Staszewsky, L.; Ristagno, G.; Olivari, D.; Masson, S.; De Giorgio, D.; Ceriani, S.; Affatato, R.; De Logu, F.; et al. Monocrotaline-induced pulmonary arterial hypertension: Time-course of injury and comparative evaluation of macitentan and Y-27632, a Rho kinase inhibitor. Eur. J. Pharmacol. 2019, 865, 172777. [Google Scholar] [CrossRef]
- Lei, S.; Peng, F.; Li, M.L.; Duan, W.B.; Peng, C.Q.; Wu, S.J. LncRNA-SMILR modulates RhoA/ROCK signaling by targeting miR-141 to regulate vascular remodeling in pulmonary arterial hypertension. Am. J. Physiol. Hear. Circ. Physiol. 2020, 319, H377–H391. [Google Scholar] [CrossRef]
- Cantoni, S.; Cavalli, S.; Pastore, F.; Accetta, A.; Pala, D.; Vaccaro, F.; Cesari, N.; De Logu, F.; Nassini, R.; Villetti, G.; et al. Pharmacological characterization of a highly selective Rho kinase (ROCK) inhibitor and its therapeutic effects in experimental pulmonary hypertension. Eur. J. Pharmacol. 2019, 850, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Dahal, B.K.; Kosanovic, D.; Pamarthi, P.K.; Sydykov, A.; Lai, Y.-J.; Kast, R.; Schirok, H.; Stasch, J.-P.; Ghofrani, H.A.; Weissmann, N.; et al. Therapeutic efficacy of azaindole-1 in experimental pulmonary hypertension. Eur. Respir. J. 2010, 36, 808–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhaliwal, J.S.; Badejo, A.M.J.; Casey, D.B.; Murthy, S.N.; Kadowitz, P.J. Analysis of pulmonary vasodilator responses to SB-772077-B [4-(7-((3-amino-1-pyrrolidinyl)carbonyl)-1-ethyl-1H-imidazo(4,5-c)pyridin-2-yl)-1,2,5-oxadiazol-3-amine], a novel aminofurazan-based Rho kinase inhibitor. J. Pharmacol. Exp. Ther. 2009, 330, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Shimokawa, H.; Morikawa, K.; Uwatoku, T.; Oi, K.; Matsumoto, Y.; Hattori, T.; Nakashima, Y.; Kaibuchi, K.; Sueishi, K.; et al. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ. Res. 2004, 94, 385–393. [Google Scholar] [CrossRef]
- Qi, L.; Lv, T.; Cheng, Y.; Yu, M.; Han, H.; Kong, H.; Xie, W.; Wang, H.; Zhang, Y.; Huang, Z. Fasudil dichloroacetate (FDCA), an orally available agent with potent therapeutic efficiency on monocrotaline-induced pulmonary arterial hypertension rats. Bioorg. Med. Chem. Lett. 2019, 29, 1812–1818. [Google Scholar] [CrossRef]
- Liu, M.; Wang, Y.; Zheng, L.; Zheng, W.; Dong, K.; Chen, S.; Zhang, B.; Li, Z. Fasudil reversed MCT-induced and chronic hypoxia-induced pulmonary hypertension by attenuating oxidative stress and inhibiting the expression of Trx1 and HIF-1α. Respir. Physiol. Neurobiol. 2014, 201, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Jasińska-Stroschein, M.; Oszajca, K.; Świtlik, W.; Ruchwa, J.; Orszulak-Michalak, D. Treatment with platelet-derived growth factor (PDGF) and ROCK inhibitors is related to declined nerve growth factor (NGF) signaling in an experimental model of rat pulmonary hypertension. Pharmacol. Rep. 2017, 69, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Fukumoto, Y.; Tanaka, S.-I.; Satoh, K.; Ikeda, S.; Shimokawa, H. Crucial role of ROCK2 in vascular smooth muscle cells for hypoxia-induced pulmonary hypertension in mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2780–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, Y.S.; Lee, J.; Zhang, X.; Lindholm, P.F. Lysophosphatidic acid activates the RhoA and NF-κB through Akt/IκBα signaling and promotes prostate cancer invasion and progression by enhancing functional invadopodia formation. Tumor Biol. 2016, 37, 6775–6785. [Google Scholar] [CrossRef] [PubMed]
- Weise-Cross, L.; Sands, M.A.; Sheak, J.R.; Broughton, B.R.S.; Snow, J.B.; Gonzalez Bosc, L.V.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Actin polymerization contributes to enhanced pulmonary vasoconstrictor reactivity after chronic hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H1011–H1021. [Google Scholar] [CrossRef]
- Guan, R.; Xu, X.; Chen, M.; Hu, H.; Ge, H.; Wen, S.; Zhou, S.; Pi, R. Advances in the studies of roles of Rho/Rho-kinase in diseases and the development of its inhibitors. Eur. J. Med. Chem. 2013, 70, 613–622. [Google Scholar] [CrossRef]
- Freund-Michel, V.; Cardoso Dos Santos, M.; Guignabert, C.; Montani, D.; Phan, C.; Coste, F.; Tu, L.; Dubois, M.; Girerd, B.; Courtois, A.; et al. Role of nerve growth factor in development and persistence of experimental pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 342–355. [Google Scholar] [CrossRef]
- Shlyonsky, V.; Naeije, R.; Mies, F. Possible role of lysophosphatidic acid in rat model of hypoxic pulmonary vascular remodeling. Pulm. Circ. 2014, 4, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Doe, C.; Bentley, R.; Behm, D.J.; Lafferty, R.; Stavenger, R.; Jung, D.; Bamford, M.; Panchal, T.; Grygielko, E.; Wright, L.L.; et al. Novel Rho kinase inhibitors with anti-inflammatory and vasodilatory activities. J. Pharmacol. Exp. Ther. 2007, 320, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, A.; Nayeem, M.J.; Sato, M. The Rho kinase 2 (ROCK2)-specific inhibitor KD025 ameliorates the development of pulmonary arterial hypertension. Biochem. Biophys. Res. Commun. 2021, 534, 795–801. [Google Scholar] [CrossRef]
- Mouchaers, K.T.B.; Schalij, I.; de Boer, M.A.; Postmus, P.E.; van Hinsbergh, V.W.M.; van Nieuw Amerongen, G.P.; Vonk Noordegraaf, A.; van der Laarse, W.J. Fasudil reduces monocrotaline-induced pulmonary arterial hypertension: Comparison with bosentan and sildenafil. Eur. Respir. J. 2010, 36, 800–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wu, S. Effects of fasudil on pulmonary hypertension in clinical practice. Pulm. Pharmacol. Ther. 2017, 46, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, Y.-F.; Zhao, Q.-H.; Jiang, R.; Wu, Y.; Peng, F.-H.; Xu, X.-Q.; Wang, L.; He, J.; Jing, Z.-C. Acute hemodynamic response of infused fasudil in patients with pulmonary arterial hypertension: A randomized, controlled, crossover study. Int. J. Cardiol. 2014, 177, 61–65. [Google Scholar] [CrossRef]
- Ruan, H.; Zhang, Y.; Liu, R.; Yang, X. The acute effects of 30 mg vs 60 mg of intravenous Fasudil on patients with congenital heart defects and severe pulmonary arterial hypertension. Congenit. Heart Dis. 2019, 14, 645–650. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Wang, S.; Luo, J.; Zhao, Z.; Zheng, C.; Shen, J. Effects of Fasudil on patients with pulmonary hypertension associated with left ventricular heart failure with preserved ejection fraction: A prospective intervention study. Can. Respir. J. 2018, 2018, 3148259. [Google Scholar] [CrossRef]
- Defert, O.; Boland, S. Rho kinase inhibitors: A patent review (2014–2016). Expert Opin. Ther. Pat. 2017, 27, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, Y.; Yamada, N.; Matsubara, H.; Mizoguchi, M.; Uchino, K.; Yao, A.; Kihara, Y.; Kawano, M.; Watanabe, H.; Takeda, Y.; et al. Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ. J. 2013, 77, 2619–2625. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Wu, Y.; Li, Q.; Wang, X.; Liu, Y.; Di, X. Aldehyde oxidase-dependent species difference in hepatic metabolism of fasudil to hydroxyfasudil. Xenobiotica 2018, 48, 170–177. [Google Scholar] [CrossRef]
- Argikar, U.A.; Potter, P.M.; Hutzler, J.M.; Marathe, P.H. Challenges and Opportunities with Non-CYP Enzymes Aldehyde Oxidase, Carboxylesterase, and UDP-Glucuronosyltransferase: Focus on Reaction Phenotyping and Prediction of Human Clearance. AAPS J. 2016, 18, 1391–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beedham, C. Aldehyde oxidase; new approaches to old problems. Xenobiotica 2020, 50, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-W.; Sherman, B.; Moore, L.A.; Laethem, C.L.; Lu, D.-W.; Pattabiraman, P.P.; Rao, P.V.; deLong, M.A.; Kopczynski, C.C. Discovery and Preclinical Development of Netarsudil, a Novel Ocular Hypotensive Agent for the Treatment of Glaucoma. J. Ocul. Pharmacol. Ther. 2018, 34, 40–51. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montagnoli, T.L.; da Silva, J.S.; Sudo, S.Z.; Santos, A.D.; Gomide, G.F.; de Sá, M.P.L.; Zapata-Sudo, G. ROCK Inhibition as Potential Target for Treatment of Pulmonary Hypertension. Cells 2021, 10, 1648. https://doi.org/10.3390/cells10071648
Montagnoli TL, da Silva JS, Sudo SZ, Santos AD, Gomide GF, de Sá MPL, Zapata-Sudo G. ROCK Inhibition as Potential Target for Treatment of Pulmonary Hypertension. Cells. 2021; 10(7):1648. https://doi.org/10.3390/cells10071648
Chicago/Turabian StyleMontagnoli, Tadeu L., Jaqueline S. da Silva, Susumu Z. Sudo, Aimeé D. Santos, Gabriel F. Gomide, Mauro P. L. de Sá, and Gisele Zapata-Sudo. 2021. "ROCK Inhibition as Potential Target for Treatment of Pulmonary Hypertension" Cells 10, no. 7: 1648. https://doi.org/10.3390/cells10071648
APA StyleMontagnoli, T. L., da Silva, J. S., Sudo, S. Z., Santos, A. D., Gomide, G. F., de Sá, M. P. L., & Zapata-Sudo, G. (2021). ROCK Inhibition as Potential Target for Treatment of Pulmonary Hypertension. Cells, 10(7), 1648. https://doi.org/10.3390/cells10071648