
Extracellular Vesicles under Oxidative Stress Conditions: Biological Properties and Physiological Roles



 ,
,  ,
,  and
and 
Abstract
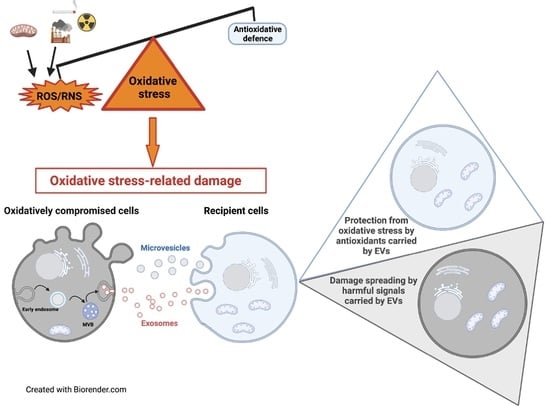
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Extracellular Vesicles (General Concepts)
2. Oxidative Stress (General Concepts)
3. Biochemical and Signaling Features of EVs Released under Oxidative Stress Conditions
3.1. Modulation of EVs Release by Oxidative Stress Conditions
3.2. Modulation of EV Cargo by Oxidative Stress Conditions
4. EV Role in Oxidative Stress-Related Diseases
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Urbanelli, L.; Magini, A.; Buratta, S.; Brozzi, A.; Sagini, K.; Polchi, A.; Tancini, B.; Emiliani, C. Signaling pathways in exosomes biogenesis, secretion and fate. Genes 2013, 4, 152–170. [Google Scholar] [CrossRef] [Green Version]
- Urbanelli, L.; Buratta, S.; Sagini, K.; Ferrara, G.; Lanni, M.; Emiliani, C. Exosome-based strategies for diagnosis and therapy. Recent Pat. CNS Drug Discov. Discontin. 2015, 10, 10–27. [Google Scholar] [CrossRef]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol. 2019, 17, e3000363. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Im, H.; Castro, C.M.; Breakefield, X.; Weissleder, R.; Lee, H. New technologies for analysis of extracellular vesicles. Chem. Rev. 2018, 118, 1917–1950. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.D.; Raposo, G. Extracellular vesicles: Exosomes and microvesicles, integrators of homeostasis. Physiology 2019, 34, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, R.; Hur, J.; Kim, Y.J.; Kim, J.; Chwae, Y.J. Apoptotic cell-derived exosomes: Messages from dying cells. Exp. Mol. Med. 2020, 52, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabeo, D.; Cvjetkovic, A.; Lässer, C.; Schorb, M.; Lötvall, J.; Höög, J.L. Exosomes purified from a single cell type have diverse morphology. J. Extracell. Vesicles 2017, 6, 1329476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crescitelli, R.; Lässer, C.; Jang, S.C.; Cvjetkovic, A.; Malmhäll, C.; Karimi, N.; Höög, J.L.; Johansson, I.; Fuchs, J.; Thorsell, A.; et al. Subpopulations of extracellular vesicles from human metastatic melanoma tissue identified by quantitative proteomics after optimized isolation. J. Extracell. Vesicles 2020, 9, 1722433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lyden, D. Asymmetric-flow field-flow fractionation technology for exomere and small extracellular vesicle separation and characterization. Nat. Protoc. 2019, 14, 1027–1053. [Google Scholar] [CrossRef] [PubMed]
- D’Acunzo, P.; Pérez-González, R.; Kim, Y.; Hargash, T.; Miller, C.; Alldred, M.J.; Erdjument-Bromage, H.; Penikalapati, S.C.; Pawlik, M.; Saito, M.; et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci. Adv. 2021, 7, eabe5085. [Google Scholar] [CrossRef] [PubMed]
- Minciacchi, V.R.; You, S.; Spinelli, C.; Morley, S.; Zandian, M.; Aspuria, P.J.; Cavallini, L.; Ciardello, C.; Sobreiro, M.R.; Morello, M.; et al. Large oncosomes contain distinct protein cargo and represent a separate functional class of tumorderived extracellular vesicles. Oncotarget 2015, 6, 1132741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royo, F.; Théry, C.; Falcón-Pérez, J.M.; Nieuwland, R.; Witwer, K.W. Methods for separation and characterization of extracellular vesicles: Results of a worldwide survey performed by the ISEV rigor and standardization subcommittee. Cells 2020, 9, 1955. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhang, W.; Zhang, H.; Zhang, F.; Chen, L.; Ma, L.; Larcher, L.M.; Chen, S.; Liu, N.; Zhao, Q.; et al. Progress, opportunity, and perspective on exosome isolation—Efforts for efficient exosome-based theranostics. Theranostics 2020, 10, 3684–3707. [Google Scholar] [CrossRef] [PubMed]
- Laulagnier, K.; Motta, C.; Hamdi, S.; Roy, S.; Fauvelle, F.; Pageaux, J.F.; Kobayashi, T.; Salles, J.P.; Perret, B.; Bonnerot, C.; et al. Mast cell- and dendritic cell-derived exosomes display a specific lipid composition and an unusual membrane organization. Biochem. J. 2004, 380, 161–171. [Google Scholar] [CrossRef]
- Llorente, A.; Skotland, T.; Sylvänne, T.; Kauhanen, D.; Róg, T.; Orłowski, A.; Vattulainen, I.; Ekroos, K.; Sandvig, K. Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2013, 1831, 1302–1309. [Google Scholar] [CrossRef]
- Lydic, T.A.; Townsend, S.; Adda, C.G.; Collins, C.; Mathivanan, S.; Reid, G.E. Rapid and comprehensive ‘shotgun’ lipidome profiling of colorectal cancer cell derived exosomes. Methods 2015, 87, 83–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagini, K.; Costanzi, E.; Emiliani, C.; Buratta, S.; Urbanelli, L. Extracellular vesicles as conveyors of membrane-derived bioactive lipids in immune system. Int. J. Mol. Sci. 2018, 19, 1227. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Juan, T.; Fürthauer, M. Biogenesis and function of ESCRT-dependent extracellular vesicles. Semin. Cell Dev. Biol. 2018, 74, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Larios, J.; Mercier, V.; Roux, A.; Gruenberg, J. ALIX- and ESCRT-III-dependent sorting of tetraspanins to exosomes. J. Cell. Biol. 2020, 219, e201904113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghossoub, R.; Lembo, F.; Rubio, A.; Gaillard, C.B.; Bouchet, J.; Vitale, N.; Slavík, J.; Machala, M. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat. Commun. 2014, 5, 3477. [Google Scholar] [CrossRef] [Green Version]
- Andreu, Z.; Yáñez-Mó, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Egea-Jimenez, A.L.; Zimmermann, P. Phospholipase D and phosphatidic acid in the biogenesis and cargo loading of extracellular vesicles. J. Lipid Res. 2018, 59, 1554–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, F.; Perrotta, C.; Novellino, L.; Francolini, M.; Riganti, L.; Menna, E.; Saglietti, L.; Schuchman, E.H.; Furlan, R.; Clementi, E.; et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 2009, 28, 1043–1054. [Google Scholar] [CrossRef] [Green Version]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2017, 8, 220–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bister, N.; Pistono, C.; Huremagic, B.; Jolkkonen, J.; Giugno, R.; Malm, T. Hypoxia and extracellular vesicles: A review on methods, vesicular cargo and functions. J. Extracell. Vesicles 2020, 10, e12002. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M. Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell 2018, 17, e12734. [Google Scholar] [CrossRef]
- Urbanelli, L.; Buratta, S.; Sagini, K.; Tancini, B.; Emiliani, C. Extracellular vesicles as new players in cellular senescence. Int. J. Mol. Sci. 2016, 17, 1408. [Google Scholar] [CrossRef]
- Buratta, S.; Urbanelli, L.; Sagini, K.; Giovagnoli, S.; Caponi, S.; Fioretto, D.; Mitro, N.; Caruso, D.; Emiliani, C. Extracellular vesicles released by fibroblasts undergoing H-Ras induced senescence show changes in lipid profile. PLoS ONE 2017, 12, e0188840. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harris, S.L.; Levine, A.J. The regulation of exosome secretion: A novel function of the p53 protein. Cancer Res. 2006, 66, 4795–4801. [Google Scholar] [CrossRef] [Green Version]
- Yarana, C.; St Clair, D.K. Chemotherapy-induced tissue injury: An insight into the role of extracellular vesicles-mediated oxidative stress responses. Antioxidants 2017, 6, 75. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [Green Version]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Functional role of mitochondrial reactive oxygen species in physiology. Free Radic. Biol. Med. 2016, 100, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Gaffrey, M.J.; Li, X.; Qian, W.J. Characterization of cellular oxidative stress response by stoichiometric redox proteomics. Am. J. Physiol. Cell. Physiol. 2021, 320, C182–C194. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Mini-review: Oxidative stress, redox stress or redox success? Biochem. Biophys. Res. Commun. 2018, 502, 183–186. [Google Scholar]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Mitochondrial oxidative and nitrosative stress and Alzheimer disease. Antioxidants 2020, 9, 818. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E. Oxidative stress and cancer. Curr. Pharm. Des. 2019, 24, 4771–4778. [Google Scholar] [CrossRef]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; De Bittencourt, P.I.H. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Hecker, M.; Wagner, A.H. Role of protein carbonylation in diabetes. J. Inherit. Metab. Dis. 2018, 41, 29–38. [Google Scholar] [CrossRef]
- Selye, H. Stress and the general adaptation syndrome. Br. Med. J. 1950, 1, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.J. Protein oxidation and peroxidation. Biochem. J. 2016, 473, 805–825. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Spickett, C.M.; Pitt, A.R. Modification of proteins by reactive lipid oxidation products and biochemical effects of lipoxidation. Essays Biochem. 2021, 64, 19–31. [Google Scholar] [CrossRef]
- Castro, J.P.; Jung, T.; Grune, T.; Siems, W. 4-Hydroxynonenal (HNE) modified proteins in metabolic diseases. Free Radic. Biol. Med. 2017, 111, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.G.; Xu, X.; Xue, Y.M.; Zhao, S.L. Comparison of 4-Hydroxynonenal-induced P53-mediated apoptosis in prostate cancer cells LNCaP and DU145. Wspolczesna Onkol. 2014, 18, 22–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Sharma, R.; Chaudhary, P.; Vatsyayan, R.; Pearce, V.; Jeyabal, P.V.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. 4-Hydroxynonenal induces p53-mediated apoptosis in retinal pigment epithelial cells. Arch. Biochem. Biophys. 2008, 480, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Barrera, G.; Pizzimenti, S.; Laurora, S.; Briatore, F.; Toaldo, C.; Dianzani, M.U. 4-Hydroxynonenal and cell cycle. BioFactors 2005, 24, 151–157. [Google Scholar] [CrossRef]
- Dodson, M.; Wani, W.Y.; Redmann, M.; Benavides, G.A.; Johnson, M.S.; Ouyang, X.; Cofield, S.S.; Mitra, K.; Darley-Usmar, V.; Zhang, J. Regulation of autophagy, mitochondrial dynamics, and cellular bioenergetics by 4-hydroxynonenal in primary neurons. Autophagy 2017, 13, 1828–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wójcik, P.; Žarković, N.; Gęgotek, A.; Skrzydlewska, E. Involvement of metabolic lipid mediators in the regulation of apoptosis. Biomolecules 2020, 10, 402. [Google Scholar] [CrossRef] [Green Version]
- Milkovic, L.; Gasparovic, A.C.; Zarkovic, N. Overview on major lipid peroxidation bioactive factor 4-hydroxynonenal as pluripotent growth-regulating factor. Free Radic. Res. 2015, 49, 850–860. [Google Scholar] [CrossRef]
- Sharma, A.; Flora, S.J.S. Positive and negative regulation of ferroptosis and its role in maintaining metabolic and redox homeostasis. Oxid. Med. Cell. Longev. 2021, 2021, 9074206. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, C.L.; Davies, M.J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef] [Green Version]
- Grune, T.; Davies, K.J.A. The proteasomal system and HNE-modified proteins. Mol. Asp. Med. 2003, 24, 195–204. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Ichijo, H. Stress-responsive protein kinases in redox-regulated apoptosis signaling. Antioxid. Redox Signal. 2005, 7, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Woolley, J.F.; Corcoran, A.; Groeger, G.; Landry, W.D.; Cotter, T.G. Redox-regulated growth factor survival signaling. Antioxid. Redox Signal. 2013, 19, 1815–1827. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 2006, 10, 389–406. [Google Scholar] [CrossRef]
- Fedorova, M.; Bollineni, R.C.; Hoffmann, R. Protein carbonylation as a major hallmark of oxidative damage: Update of analytical strategies. Mass Spectrom. Rev. 2014, 33, 79–97. [Google Scholar] [CrossRef]
- Nyström, T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005, 24, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Gonos, E.S.; Kapetanou, M.; Sereikaite, J.; Bartosz, G.; Naparlo, K.; Grzesik, M.; Sadowska-Bartosz, I. Origin carbonylation, nitration and chlorination in age-related brain diseases and aging. Aging 2018, 10, 868–901. [Google Scholar] [CrossRef]
- Shen, L.; Chen, C.; Yang, A.; Chen, Y.; Liu, Q.; Ni, J. Redox proteomics identification of specifically carbonylated proteins in the hippocampi of triple transgenic Alzheimer’s disease mice at its earliest pathological stage. J. Proteom. 2015, 123, 101–113. [Google Scholar] [CrossRef]
- Rodríguez-García, A.; García-Vicente, R.; Morales, M.L.; Ortiz-Ruiz, A.; Martínez-López, J.; Linares, M. Protein carbonylation and lipid peroxidation in hematological malignancies. Antioxidants 2020, 9, 1212. [Google Scholar] [CrossRef] [PubMed]
- Colombo, G.; Reggiani, F.; Angelini, C.; Finazzi, S.; Astori, E.; Garavaglia, M.L.; Landoni, L.; Portinaro, N.M.; Giustarini, D.; Rossi, R.; et al. Plasma protein carbonyls as biomarkers of oxidative stress in chronic kidney disease, dialysis, and transplantation. Oxid. Med. Cell. Longev. 2020, 2020, 2975256. [Google Scholar] [CrossRef]
- Chiaradia, E.; Renzone, G.; Scaloni, A.; Caputo, M.; Costanzi, E.; Gambelunghe, A.; Muzi, G.; Avellini, L.; Emiliani, C.; Buratta, S. Protein carbonylation in dopaminergic cells exposed to rotenone. Toxicol. Lett. 2019, 309, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Tepe, J.J. Proteasome activation to combat proteotoxicity. Molecules 2019, 24, 2841. [Google Scholar] [CrossRef] [Green Version]
- Laskowska, E.; Kuczyńska-Wiśnik, D.; Lipińska, B. Proteomic analysis of protein homeostasis and aggregation. J. Proteom. 2019, 198, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.L.; Stanley, J.A.; Robinson, M.C.; Sher, J.W.; Li, Z.; Chan, Y.A.; Omdahl, A.R.; Wattiez, R.; Godzik, A.; Matallana-Surget, S. Protein structure, amino acid composition and sequence determine proteome vulnerability to oxidation-induced damage. EMBO J. 2020, 39, e104523. [Google Scholar] [CrossRef] [PubMed]
- Mannaa, A.; Hanisch, F.G. Redox proteomes in human physiology and disease mechanisms. J. Proteome Res. 2019, 19, 1–17. [Google Scholar] [CrossRef]
- Abello, N.; Kerstjens, H.A.M.; Postma, D.S.; Bischoff, R. Protein tyrosine nitration: Selectivity, physicochemical and biological consequences, denitration, and proteomics methods for the identification of tyrosine-nitrated proteins. J. Proteome Res. 2009, 8, 3222–3238. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Souza, J.M.; Peluffo, G.; Radi, R. Protein tyrosine nitration-Functional alteration or just a biomarker? Free Radic. Biol. Med. 2008, 45, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2018, 14, 618–625. [Google Scholar] [CrossRef]
- Franco, M.C.; Ye, Y.; Refakis, C.A.; Feldman, J.L.; Stokes, A.L.; Basso, M.; De Mera, R.M.M.F.; Sparrow, N.A.; Calingasan, N.Y.; Kiaei, M.; et al. Nitration of Hsp90 induces cell death. Proc. Natl. Acad. Sci. USA 2013, 110, E1102–E1111. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Moskovitz, J. The functions of the mammalian methionine sulfoxide reductase system and related diseases. Antioxidants 2018, 7, 122. [Google Scholar] [CrossRef] [Green Version]
- Pal, R.; Oien, D.B.; Ersen, F.Y.; Moskovitz, J. Elevated levels of brain-pathologies associated with neurodegenerative diseases in the methionine sulfoxide reductase a knockout mouse. Exp. Brain Res. 2007, 180, 765–774. [Google Scholar] [CrossRef]
- De Luca, A.; Sanna, F.; Sallese, M.; Ruggiero, C.; Grossi, M.; Sacchetta, P.; Rossi, C.; De Laurenzi, V.; Di Ilio, C.; Favaloro, B. Methionine sulfoxide reductase A down-regulation in human breast cancer cells results in a more aggressive phenotype. Proc. Natl. Acad. Sci. USA 2010, 107, 18628–18633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskovitz, J.; Oien, D.B. Protein carbonyl and the methionine sulfoxide reductase system. Antioxid. Redox Signal. 2010, 12, 405–415. [Google Scholar] [CrossRef]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-nitrosylation: An emerging paradigm of redox signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Oh, C.-K.; Zhang, X.; Tannenbaum, S.R.; Lipton, S.A. Protein transnitrosylation signaling networks contribute to inflammaging and neurodegenerative disorders. Antioxid. Redox Signal. 2021, 0081. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Fernando, V.; Letson, J.; Walia, Y.; Zheng, X.; Fackelman, D.; Furuta, S. S-nitrosylation in tumor microenvironment. Int. J. Mol. Sci. 2021, 22, 4600. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Zhang, Y.; Liu, B.; Zhou, Y.; Liao, D.; Qiao, X.; Gao, D.; Xie, T.; Yao, Q.; Zhang, Y.; et al. Protein S-nitrosylation regulates proteostasis and viability of hematopoietic stem cell during regeneration. Cell Rep. 2021, 34, 108922. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.T.; Stamler, J.S. Regulation by S-nitrosylation of protein post-translational modification. J. Biol. Chem. 2012, 287, 4411–4418. [Google Scholar] [CrossRef] [Green Version]
- Mannick, J.B.; Schonhoff, C.; Papeta, N.; Ghafourifar, P.; Szibor, M.; Fang, K.; Gaston, B. S-nitrosylation of mitochondrial caspases. J. Cell Biol. 2001, 154, 1111–1116. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Hao, J.R.; Li, X.Y.; Yin, X.H.; Zong, Y.Y.; Zhang, G.Y.; Gao, C. GluR6-FasL-Trx2 mediates denitrosylation and activation of procaspase-3 in cerebral ischemia/reperfusion in rats. Cell Death Dis. 2013, 4, e771. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; Zhang, D.F.; Zhang, B.F.; Li, H.Z.; Zhang, Q.; Li, L.T.; Pei, D.S.; Zheng, J.N. Melanoma differentiation associated gene-7/interleukin-24 induces caspase-3 denitrosylation to facilitate the activation of cancer cell apoptosis. J. Interf. Cytokine Res. 2015, 35, 157–167. [Google Scholar] [CrossRef]
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein S-nitrosylation: Determinants of specificity and enzymatic regulation of S-nitrosothiol-based signaling. Antioxid. Redox Signal. 2019, 30, 1331–1351. [Google Scholar] [CrossRef] [PubMed]
- Popov, D. Protein S-glutathionylation: From current basics to targeted modifications. Arch. Physiol. Biochem. 2014, 120, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Gill, R.; Young, A. Protein S-glutathionylation and the regulation of cellular functions. In Oxidative Stress: Eustress and Distress; Elsevier: Amsterdam, The Netherlands, 2019; pp. 217–247. [Google Scholar]
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-Glutathionylation: From molecular mechanisms to health outcomes. Antioxid. Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef] [Green Version]
- Aracena, P.; Tang, W.; Hamilton, S.L.; Hidalgo, C. Effects of S-glutathionylation and S-nitrosylation on calmodulin binding to triads and FKBP12 binding to type 1 calcium release channels. Antioxid. Redox Signal. 2005, 7, 870–881. [Google Scholar] [CrossRef]
- Muronetz, V.I.; Barinova, K.V.; Stroylova, Y.Y.; Semenyuk, P.I.; Schmalhausen, E.V. Glyceraldehyde-3-phosphate dehydrogenase: Aggregation mechanisms and impact on amyloid neurodegenerative diseases. Int. J. Biol. Macromol. 2017, 100, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Bago, Á.; Íñiguez, M.A.; Serrador, J.M. Nitric oxide and electrophilic cyclopentenone prostaglandins in redox signaling, regulation of cytoskeleton dynamics and intercellular communication. Front. Cell Dev. Biol. 2021, 9, 673973. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, M.; Bauri, A.K.; Chattopadhyay, S.; Patro, B.S. Thiol antioxidants sensitize malabaricone C induced cancer cell death via reprogramming redox sensitive p53 and NF-κB proteins in vitro and in vivo. Free Radic. Biol. Med. 2020, 148, 182–199. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef]
- Oestreicher, J.; Morgan, B. Glutathione: Subcellular distribution and membrane transport. Biochem. Cell Biol. 2019, 97, 270–289. [Google Scholar] [CrossRef] [Green Version]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The nrf2, thioredoxin, and glutathione system in tumorigenesis and anticancer therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Gao, L.; Zucker, I.H. Regulation of Nrf2 signaling pathway in heart failure: Role of extracellular vesicles and non-coding RNAs. Free Radic. Biol. Med. 2021, 167, 218–231. [Google Scholar] [CrossRef]
- Chen, H.H.; Chang, H.H.; Chang, J.Y.; Tang, Y.C.; Cheng, Y.C.; Lin, L.M.; Cheng, S.Y.; Huang, C.H.; Sun, M.W.; Chen, C.T.; et al. Enhanced B-Raf-mediated NRF2 gene transcription and HATs-mediated NRF2 protein acetylation contributes to ABCC1-mediated chemoresistance and glutathione-mediated survival in acquired topoisomerase II poison-resistant cancer cells. Free Radic. Biol. Med. 2017, 113, 505–518. [Google Scholar] [CrossRef]
- Pi, J.; Bai, Y.; Reece, J.M.; Williams, J.; Liu, D.; Freeman, M.L.; Fahl, W.E.; Shugar, D.; Liu, J.; Qu, W.; et al. Molecular mechanism of human Nrf2 activation and degradation: Role of sequential phosphorylation by protein kinase CK2. Free Radic. Biol. Med. 2007, 42, 1797–1806. [Google Scholar] [CrossRef] [Green Version]
- Saeed-Zidane, M.; Linden, L.; Salilew-Wondim, D.; Held, E.; Neuhoff, C.; Tholen, E.; Hoelker, M.; Schellander, K.; Tesfaye, D. Cellular and exosome mediated molecular defense mechanism in bovine granulosa cells exposed to oxidative stress. PLoS ONE 2017, 2, e0187569. [Google Scholar] [CrossRef] [Green Version]
- Kahroba, H.; Davatgaran-Taghipour, Y. Exosomal Nrf2: From anti-oxidant and anti-inflammation response to wound healing and tissue regeneration in aged-related diseases. Biochimie 2020, 171–172, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Benedikter, B.J.; Weseler, A.R.; Wouters, E.F.M.; Savelkoul, P.H.M.; Rohde, G.G.U.; Stassen, F.R.M. Redox-dependent thiol modifications: Implications for the release of extracellular vesicles. Cell. Mol. Life Sci. 2018, 75, 2321–2337. [Google Scholar] [CrossRef] [Green Version]
- Borras, C.; Mas-Bargues, C.; Sanz-Ros, J.; Román-Domínguez, A.; Gimeno-Mallench, L.; Inglés, M.; Gambini, J.; Viña, J. Free extracellular vesicles and redox modulation in aging. Radic. Biol Med. 2020, 149, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.C.; Hillery, C.A.; Hogg, N. Circulating membrane-derived microvesicles in redox biology. Free Radic. Biol. Med. 2014, 73, 214–228. [Google Scholar] [CrossRef] [Green Version]
- Atienzar-Aroca, S.; Flores-Bellver, M.; Serrano-Heras, G.; Martinez-Gil, N.; Barcia, J.M.; Aparicio, S.; Perez-Cremades, D.; Garcia-Verdugo, J.M.; Diaz-Llopis, M.; Romero, F.J.; et al. Oxidative stress in retinal pigment epithelium cells increases exosome secretion and promotes angiogenesis in endothelial cells. J. Cell. Mol. Med. 2016, 20, 1457–1466. [Google Scholar] [CrossRef]
- Takasugi, M.; Okada, R.; Takahashi, A.; Virya Chen, D.; Watanabe, S.; Hara, E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 2017, 8, 15729. [Google Scholar] [CrossRef] [PubMed]
- Manček-Keber, M.; Frank-Bertoncelj, M.; Hafner-Bratkovič, I.; Smole, A.; Zorko, M.; Pirher, N.; Hayer, S.; Kralj-Iglič, V.; Rozman, B.; Ilc, N.; et al. Toll-like receptor 4 senses oxidative stress mediated by the oxidation of phospholipids in extracellular vesicles. Sci. Signal. 2015, 8, ra60. [Google Scholar] [CrossRef]
- Van Meteren, N.; Lagadic-Gossmann, D.; Chevanne, M.; Gallais, I.; Gobart, D.; Burel, A.; Bucher, S.; Grova, N.; Fromenty, B.; Appenzeller, B.M.R.; et al. Polycyclic aromatic hydrocarbons can trigger hepatocyte release of extracellular vesicles by various mechanisms of action depending on their affinity for the aryl hydrocarbon receptor. Toxicol. Sci. 2019, 17, 1443–1462. [Google Scholar] [CrossRef]
- Van Meteren, N.; Lagadic-Gossmann, D.; Podechard, N.; Gobart, D.; Gallais, I.; Chevanne, M.; Collin, A.; Burel, A.; Dupont, A.; Rault, L.; et al. Extracellular vesicles released by polycyclic aromatic hydrocarbons-treated hepatocytes trigger oxidative stress in recipient hepatocytes by delivering iron. Free Radic. Biol. Med. 2020, 160, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Vatsyayan, R.; Kothari, H.; Pendurthi, U.R.; Rao, L.V. 4-Hydroxy-2-nonenal enhances tissue factor activity in human monocytic cells via p38 mitogen-activated protein kinase activation-dependent phosphatidylserine exposure. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1601–1611. [Google Scholar] [CrossRef] [Green Version]
- Benedikter, B.J.; Volgers, C.; van Eijck, P.H.; Wouters, E.F.M.; Savelkoul, P.H.M.; Reynaert, N.L.; Haenen, G.R.M.M.; Rohde, G.G.U.; Weseler, A.R.; Stassen, F.R.M. Cigarette smoke extract induced exosome release is mediated by depletion of exofacial thiols and can be inhibited by thiol-antioxidants. Free Radic. Biol. Med. 2017, 108, 334–344. [Google Scholar] [CrossRef]
- Liu, J.; Mao, W.; Ding, B.; Liang, C.S. ERKs/p53 signal transduction pathway is involved in doxorubicin-induced apoptosis in H9c2 cells and cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1956–H1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lespagnol, A.; Duflaut, D.; Beekman, C.; Blanc, L.; Fiucci, G.; Marine, J.C.; Vidal, M.; Amson, R.; Telerman, A. Exosome secretion, including the DNA damage-induced p53-dependent secretory pathway, is severely compromised in TSAP6/Steap3-null mice. Cell. Death Differ. 2008, 15, 1723–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Record, M.; Silvente-Poirot, S.; Poirot, M.; Wakelam, M.J.O. Extracellular vesicles: Lipids as key components of their biogenesis and functions. J. Lipid Res. 2018, 59, 1316–1324. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, J.J.; Trivedi, P.C.; Pulinilkunnil, T. Autophagic dysregulation in doxorubicin cardiomyopathy. J. Mol. Cell. Cardiol. 2017, 104, 1–8. [Google Scholar] [CrossRef]
- Dirks-Naylor, A.J. The role of autophagy in doxorubicin-induced cardiotoxicity. Life Sci. 2013, 93, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; López-Otín, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef] [Green Version]
- Fader, C.M.; Sánchez, D.; Furlán, M.; Colombo, M.I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 2008, 9, 230–250. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Øverbye, A.; Brech, A.; Torgersen, M.L.; Jakobsen, I.S.; Sandvig, K.; Llorente, A. PIKfyve inhibition increases exosome release and induces secretory autophagy. Cell. Mol. Life Sci. 2016, 73, 4717–4737. [Google Scholar] [CrossRef]
- Tancini, B.; Buratta, S.; Sagini, K.; Costanzi, E.; Delo, F.; Urbanelli, L.; Emiliani, C. Insight into the role of extracellular vesicles in lysosomal storage disorders. Genes 2019, 10, 510. [Google Scholar] [CrossRef] [Green Version]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Ortega, F.G.; Roefs, M.T.; de Miguel Perez, D.; Kooijmans, S.A.; de Jong, O.G.; Sluijter, J.P.; Schiffelers, R.M.; Vader, P. Interfering with endolysosomal trafficking enhances release of bioactive exosomes. Nanomedicine 2019, 20, 102014. [Google Scholar] [CrossRef]
- Boura, E.; Ivanov, V.; Carlson, L.A.; Mizuuchi, K.; Hurley, J.H. Endosomal sorting complex required for transport (ESCRT) complexes induce phase-separated microdomains in supported lipid bilayers. J. Biol. Chem. 2012, 287, 28144–28151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfrieger, F.W.; Vitale, N. Cholesterol and the journey of extracellular vesicles. J. Lipid Res. 2018, 59, 2255–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.T.; Kreutzberger, A.J.B.; Lee, J.; Kiessling, V.; Tamm, L.K. The role of cholesterol in membrane fusion. Chem. Phys. Lipids 2016, 199, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Szabó-Taylor, K.É.; Tóth, E.Á.; Balogh, A.M.; Sódar, B.W.; Kádár, L.; Pálóczi, K.; Fekete, N.; Németh, A.; Osteikoetxea, X.; Vukman, K.V.; et al. Monocyte activation drives preservation of membrane thiols by promoting release of oxidised membrane moieties via extracellular vesicles. Free Radic. Biol. Med. 2017, 108, 56–65. [Google Scholar] [CrossRef]
- Riddell, J.R.; Wang, X.Y.; Minderman, H.; Gollnick, S.O. Peroxiredoxin 1 stimulates secretion of proinflammatory cytokines by binding to TLR4. J. Immunol. 2010, 184, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Eldh, M.; Ekström, K.; Valadi, H.; Sjöstrand, M.; Olsson, B.; Jernås, M.; Lötvall, J. Exosomes communicate protective messages during oxidative stress; possible role of exosomal shuttle RNA. PLoS ONE 2010, 5, e15353. [Google Scholar] [CrossRef] [Green Version]
- Corsello, T.; Kudlicki, A.S.; Garofalo, R.P.; Casola, A. Cigarette smoke condensate exposure changes RNA content of extracellular vesicles released from small airway epithelial cells. Cells 2019, 8, 1652. [Google Scholar] [CrossRef] [Green Version]
- Benedikter, B.J.; Bouwman, F.G.; Heinzmann, A.C.A.; Vajen, T.; Mariman, E.C.; Wouters, E.F.M.; Savelkoul, P.H.M.; Koenen, R.R.; Rohde, G.G.U.; van Oerle, R.; et al. Proteomic analysis reveals procoagulant properties of cigarette smoke-induced extracellular vesicles. J. Extracell. Vesicles 2019, 8, 1585163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biasutto, L.; Chiechi, A.; Couch, R.; Liotta, L.A.; Espina, V. Retinal pigment epithelium (RPE) exosomes contain signaling phosphoproteins affected by oxidative stress. Exp. Cell Res. 2013, 319, 2113–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, K.D.; Zimmerman, G.A.; Prescott, S.M.; McIntyre, T.M. Novel leukocyte agonists are released by endothelial cells exposed to peroxide. J. Biol. Chem. 1992, 267, 15168–15175. [Google Scholar] [CrossRef]
- Huber, J.; Vales, A.; Mitulovic, G.; Blumer, M.; Schmid, R.; Witztum, J.L.; Binder, B.R.; Leitinger, N. Oxidized membrane vesicles and blebs from apoptotic cells contain biologically active oxidized phospholipids that induce monocyte-endothelial interactions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Bodega, G.; Alique, M.; Puebla, L.; Carracedo, J.; Ramírez, R.M. Microvesicles: ROS scavengers and ROS producers. J. Extracell. Vesicles 2019, 8, 1626654. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Xu, H.; Xu, W.; Wang, B.; Wu, H.; Tao, Y.; Zhang, B.; Wang, M.; Mao, F.; Yan, Y.; et al. Exosomes released by human umbilical cord mesenchymal stem cells protect against cisplatin-induced renal oxidative stress and apoptosis in vivo and in vitro. Stem Cell Res. Ther. 2013, 4, 34. [Google Scholar] [CrossRef] [Green Version]
- Alcayaga-Miranda, F.; González, P.L.; Lopez-Verrilli, A.; Varas-Godoy, M.; Aguila-Díaz, C.; Contreras, L.; Khoury, M. Prostate tumor-induced angiogenesis is blocked by exosomes derived from menstrual stem cells through the inhibition of reactive oxygen species. Oncotarget 2016, 7, 44462–44477. [Google Scholar] [CrossRef] [Green Version]
- Soleti, R.; Lauret, E.; Andriantsitohaina, R.; Carmen Martínez, M. Internalization and induction of antioxidant messages by microvesicles contribute to the antiapoptotic effects on human endothelial cells. Free Radic. Biol. Med. 2012, 53, 2159–2170. [Google Scholar] [CrossRef] [Green Version]
- Patel, G.K.; Khan, M.A.; Bhardwaj, A.; Srivastava, S.K.; Zubair, H.; Patton, M.C.; Singh, S.; Khushman, M.; Singh, A.P. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br. J. Cancer 2017, 116, 609–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, B.; Wang, Y.; Zhao, R.; Long, X.; Deng, W.; Wang, Z. Bone marrow mesenchymal stem cell-derived exosomal miR-21 protects C-kit+ cardiac stem cells from oxidative injury through the PTEN/PI3K/Akt axis. PLoS ONE 2018, 13, e0191616. [Google Scholar] [CrossRef] [Green Version]
- Mullen, L.; Hanschmann, E.M.; Lillig, C.H.; Herzenberg, L.A.; Ghezzi, P. Cysteine oxidation targets peroxiredoxins 1 and 2 for exosomal release through a novel mechanism of redox-dependent secretion. Mol. Med. 2015, 21, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Mahairaki, V.; Bai, H.; Ding, Z.; Li, J.; Witwer, K.W.; Cheng, L. Highly purified human extracellular vesicles produced by stem cells alleviate aging cellular phenotypes of senescent human cells. Stem Cells 2019, 37, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Burger, D.; Turner, M.; Munkonda, M.N.; Touyz, R.M. Endothelial microparticle-derived reactive oxygen species: Role in endothelial signaling and vascular function. Oxid. Med. Cell. Longev. 2016, 2016, 5047954. [Google Scholar] [CrossRef] [Green Version]
- Hervera, A.; De Virgiliis, F.; Palmisano, I.; Zhou, L.; Tantardini, E.; Kong, G.; Hutson, T.; Danzi, M.C.; Perry, R.B.; Santos, C.X.C.; et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat. Cell Biol. 2018, 20, 307–319. [Google Scholar] [CrossRef]
- Liguori, L.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 3, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Burger, D.; Kwart, D.G.; Montezano, A.C.; Read, N.C.; Kennedy, C.R.; Thompson, C.S.; Touyz, R.M. Microparticles induce cell cycle arrest through redox-sensitive processes in endothelial cells: Implications in vascular senescence. J. Am. Heart Assoc. 2012, 1, e001842. [Google Scholar] [CrossRef] [Green Version]
- Huber, V.; Fais, S.; Iero, M.; Lugini, L.; Canese, P.; Squarcina, P.; Zaccheddu, A.; Colone, M.; Arancia, G.; Gentile, M.; et al. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: Role in immune escape. Gastroenterology 2005, 128, 1796–1804. [Google Scholar] [CrossRef] [PubMed]
- Loyer, X.; Vion, A.C.; Tedgui, A.; Boulanger, C.M. Microvesicles as cell-cell messengers in cardiovascular diseases. Circ. Res. 2014, 114, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.Y.; Ke, Z.Y.; Xiao, M.; Quazi, S.H. Exosomes: A novel therapeutic target for Alzheimer’s disease? Neural Regen. Res. 2018, 13, 930–935. [Google Scholar] [CrossRef]
- Fafián-Labora, J.A.; O’Loghlen, A. Classical and nonclassical intercellular communication in senescence and ageing. Trends Cell Biol. 2020, 30, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Borghesan, M.; Fafián-Labora, J.; Eleftheriadou, O.; Carpintero-Fernández, P.; Paez-Ribes, M.; Vizcay-Barrena, G.; Swisa, A.; Kolodkin-Gal, D.; Ximénez-Embún, P.; Lowe, R.; et al. Small extracellular vesicles are key regulators of non-cell autonomous intercellular communication in senescence via the interferon protein IFITM3. Cell Rep. 2019, 27, 3956–3971.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fafián-Labora, J.A.; Rodríguez-Navarro, J.A.; O’Loghlen, A. Small extracellular vesicles have GST activity and ameliorate senescence-related tissue damage. Cell Metab. 2020, 32, 71–86.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Sun, Y.; Zhang, J.; Zhu, Q.; Yang, Y.; Niu, X.; Deng, Z.; Li, Q.; Wang, Y. Human embryonic stem cell-derived exosomes promote pressure ulcer healing in aged mice by rejuvenating senescent endothelial cells. Stem Cell Res. Ther. 2019, 10, 142. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhu, L.; Hu, Q.; Li, J.; Ma, J.; Yan, J. Loss of exosomal MALAT1 from ox-LDL treated vascular endothelial cells induces maturation of dendritic cells in atherosclerosis development. Cell Cycle 2019, 18, 2255–2267. [Google Scholar] [CrossRef]
- Ishii, E.; Warabi, G.E.; Mann, G.E. Circadian control of BDNF-mediated Nrf2 activation in astrocytes protects dopaminergic neurons from ferroptosis. Free Radic. Biol. Med. 2019, 133, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Leroyer, A.S.; Isobe, H.; Lesèche, G.; Castier, Y.; Wassef, M.; Mallat, Z.; Binder, B.R.; Tedgui, A.; Boulanger, C.M. Cellular origins and thrombogenic activity of microparticles isolated from human atherosclerotic plaques. J. Am. Coll. Cardiol. 2007, 49, 772–777. [Google Scholar] [CrossRef] [Green Version]
- Rautou, P.E.; Vion, A.C.; Amabile, N.; Chironi, G.; Simon, A.; Tedgui, A.; Boulanger, C.M. Microparticles, vascular function, and atherothrombosis. Circ. Res. 2011, 109, 593–606. [Google Scholar] [CrossRef]
- Burger, D.; Montezano, A.C.; Nishigaki, N.; He, Y.; Carter, A.; Touyz, R.M. Endothelial microparticle formation by angiotensin II is mediated via AngII receptor type I/NADPH oxidase/Rho kinase pathways targeted to lipid rafts. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1898–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janas, A.M.; Sapon, K.; Janas, T.; Stowell, M.H.; Janas, T. Exosomes and other extracellular vesicles in neural cells and neurodegenerative diseases. Biochim. Biophys. Acta BBA Biomembr. 2016, 1858, 1139–1151. [Google Scholar] [CrossRef]
- Vella, L.J.; Hill, A.; Cheng, L. Focus on extracellular vesicles: Exosomes and their role in protein tracking and biomarker potential in Alzheimer’s and Parkinson’s disease. Int. J. Mol. Sci. 2016, 17, 173. [Google Scholar] [CrossRef]
- Schneider, A.; Simons, M. Exosomes: Vesicular carriers for intercellular communication in neurodegenerative disorders. Cell Tissue Res. 2013, 352, 33–47. [Google Scholar] [CrossRef] [Green Version]
- Kanamaru, T.; Kamimura, N.; Yokota, T.; Iuchi, K.; Nishimaki, K.; Takami, S.; Akashiba, H.; Shitaka, Y.; Katsura, K.I.; Kimura, K. Oxidative stress accelerates amyloid deposition and memory impairment in a double-transgenic mouse model of Alzheimer’s disease. Neurosci. Lett. 2015, 587, 126–131. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kalani, A.; Rai, S.; Swarnkar, S.; Tota, S.; Nath, C.; Tyagi, N. Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of Alzheimer’s disease: Understanding the therapeutics strategies. Mol. Neurobiol. 2014, 53, 648–661. [Google Scholar] [CrossRef] [Green Version]
- Bonda, D.J.; Wang, X.; Lee, H.-G.; Smith, M.A.; Perry, G.; Zhu, X. Neuronal failure in Alzheimer’s disease: A view through the oxidative stress looking-glass. Neurosci. Bull. 2014, 30, 243–252. [Google Scholar] [CrossRef]
- Persson, T.; Sharples, R.A.; Vella, L.J.; Nisbet, R.M.; Naylor, R.; Perez, K.; Barnham, K.J.; Masters, C.L.; Hill, A. Inhibition of γ-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J. 2008, 22, 1469–1478. [Google Scholar]
- Saman, S.; Lee, N.C.; Inoyo, I.; Jin, J.; Li, Z.; Doyle, T.; McKee, A.C.; Hall, G.F. Proteins recruited to exosomes by tau overexpression implicate novel cellular mechanisms linking tau secretion with Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 40, S47–S70. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Mustapic, M.; Kapogiannis, D.; Eitan, E.; Lobach, I.V.; Goetzl, L.; Schwartz, J.B.; Miller, B.L. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. FASEB J. 2016, 30, 3853–3859. [Google Scholar] [CrossRef] [Green Version]
- Yuyama, K.; Igarashi, Y. Exosomes as carriers of Alzheimer’s amyloid-ß. Front. Neurosci. 2017, 11, 229. [Google Scholar] [CrossRef] [Green Version]
- Yuyama, K.; Sun, H.; Sakai, S.; Mitsutake, S.; Okada, M.; Tahara, H.; Furukawa, J.; Fujitani, N.; Shinohara, Y.; Igarashi, Y. Decreased amyloid-beta pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J. Biol. Chem. 2014, 289, 24488–24498. [Google Scholar] [CrossRef] [Green Version]
- Lei, L.; Lei, Z.; Zhang, Z.; Zhang, Z.; Cheng, Y. Downregulated miR-29c correlates with increased BACE1 expression in sporadic Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2015, 8, 1565–1574. [Google Scholar] [PubMed]
- Jaouen, F.; Gascon, E. Understanding the role of miR-33 in brain lipid metabolism: Implications for Alzheimer’s disease. J. Neurosci. 2016, 36, 2558–2560. [Google Scholar] [CrossRef] [PubMed]
- Konovalova, J.; Gerasymchuk, D.; Parkkinen, I.; Chmielarz, P.; Domanskyi, A. Interplay between MicroRNAs and oxidative stress in neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 6055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.J.; Dolios, G.; Wang, R.; Liao, F.F. Soluble beta-amyloid peptides, but not insoluble fibrils, have specific effect on neuronal microRNA expression. PLoS ONE 2014, 9, e90770. [Google Scholar] [CrossRef] [PubMed]
- Varadarajan, S.; Yatin, S.; Aksenova, M.; Butterfield, D.A. Alzheimer’s amyloid β-peptide-associated free radical oxidative stress and neurotoxicity. J. Struct. Biol. 2000, 130, 184–208. [Google Scholar] [CrossRef]
- McKeever, P.M.; Schneider, R.; Taghdiri, F.; Weichert, A.; Multani, N.; Brown, R.A.; Boxer, A.L.; Karydas, A.; Miller, B.; Robertson, J.; et al. MicroRNA expression levels are altered in the cerebrospinal fluid of patients with young-onset Alzheimer’s disease. Mol. Neurobiol. 2018, 55, 8826–8841. [Google Scholar] [CrossRef] [Green Version]
- Banzhaf-Strathmann, J.; Benito, E.; May, S.; Arzberger, T.; Tahirovic, S.; Kretzschmar, H.; Fischer, A.; Edbauer, D. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 2014, 33, 1667–1680. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Tu, Q.; Liu, M. MicroRNA125b regulates Alzheimer’s disease through SphK1 regulation. Mol. Med. Rep. 2018, 18, 2373–2380. [Google Scholar] [PubMed]
- Li, P.; Xu, Y.; Wang, B.; Huang, J.; Li, Q. miR-34a-5p and miR-125b-5p attenuate Aβ-induced neurotoxicity through targeting BACE1. J. Neurol. Sci. 2020, 413, 116793. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial dysfunction, oxidative stress, and neuroinflammation: Intertwined roads to neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Wu, X.; Zheng, T.; Zhang, B. Exosomes in Parkinson’s disease. Neurosci. Bull. 2017, 33, 331–338. [Google Scholar] [CrossRef]
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X.Y. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043–37053. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Liu, J.; Chen, L.; Jin, Y.; Zhang, G.; Lin, Z.; Du, S.; Fu, Z.; Chen, T.; Qin, Y.; et al. Serum secreted miR-137-containing exosomes affects oxidative stress of neurons by regulating OXR1 in Parkinson’s disease. Brain Res. 2019, 1722, 146331. [Google Scholar] [CrossRef]
- Narayanan, D.; Ma, S.; Özcelik, D. Targeting the redox landscape in cancer therapy. Cancers 2020, 12, 1706. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer. Implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Chen, M.; Jiang, R.; Guo, Y.; Wu, M.; Zhang, X. Exosome-related tumor microenvironment. J. Cancer 2018, 9, 3084–3092. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhou, L.; Lv, D.; Zhu, X.; Tang, H. Exosome-mediated communication in the tumor microenvironment contributes to hepatocellular carcinoma development and progression. J. Hematol. Oncol. 2019, 12, 53. [Google Scholar] [CrossRef]
- Azmi, A.S.; Bao, B.; Sarkar, F.H. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013, 32, 623–642. [Google Scholar] [CrossRef] [Green Version]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef]
- Wang, J.S.; Wang, H.J.; Qian, H.L. Biological effects of radiation on cancer cells. Mil. Med. Res. 2018, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, N.; Nawaz, M.; Rezaie, J. Ionizing radiation increases the activity of exosomal secretory pathway in MCF-7 human breast cancer cells: A possible way to communicate resistance against radiotherapy. Int. J. Mol. Sci. 2019, 20, 3649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mayah, A.; Bright, S.; Chapman, K.; Irons, S.; Luo, P.; Carter, D.; Goodwin, E.; Kadhim, M. The non-targeted effects of radiation are perpetuated by exosomes. Mutat. Res. 2015, 772, 38–45. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiaradia, E.; Tancini, B.; Emiliani, C.; Delo, F.; Pellegrino, R.M.; Tognoloni, A.; Urbanelli, L.; Buratta, S. Extracellular Vesicles under Oxidative Stress Conditions: Biological Properties and Physiological Roles. Cells 2021, 10, 1763. https://doi.org/10.3390/cells10071763
Chiaradia E, Tancini B, Emiliani C, Delo F, Pellegrino RM, Tognoloni A, Urbanelli L, Buratta S. Extracellular Vesicles under Oxidative Stress Conditions: Biological Properties and Physiological Roles. Cells. 2021; 10(7):1763. https://doi.org/10.3390/cells10071763
Chicago/Turabian StyleChiaradia, Elisabetta, Brunella Tancini, Carla Emiliani, Federica Delo, Roberto Maria Pellegrino, Alessia Tognoloni, Lorena Urbanelli, and Sandra Buratta. 2021. "Extracellular Vesicles under Oxidative Stress Conditions: Biological Properties and Physiological Roles" Cells 10, no. 7: 1763. https://doi.org/10.3390/cells10071763
APA StyleChiaradia, E., Tancini, B., Emiliani, C., Delo, F., Pellegrino, R. M., Tognoloni, A., Urbanelli, L., & Buratta, S. (2021). Extracellular Vesicles under Oxidative Stress Conditions: Biological Properties and Physiological Roles. Cells, 10(7), 1763. https://doi.org/10.3390/cells10071763