
Cortactin Contributes to Activity-Dependent Modulation of Spine Actin Dynamics and Spatial Memory Formation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Cttn-Deficient Mice Show Deficits in Hippocampus-Dependent Spatial Memory Formation
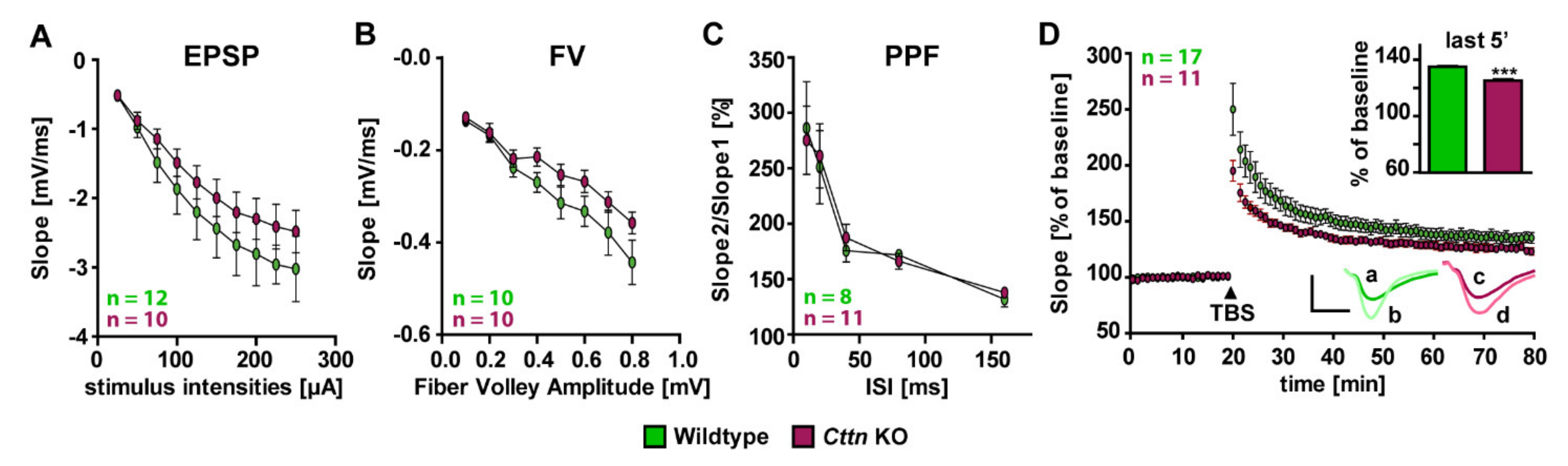
3.2. Cttn-Deficient Mice Show Impaired Synaptic Plasticity at The Hippocampal Schaffer Collateral Pathway
3.3. Basal Actin Dynamics and Dendritic Spine Motility Are Unaltered in Cttn-Deficient Hippocampal Neurons
3.4. Structural Spine Plasticity Is Impaired in the Absence of Cortactin
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Borovac, J.; Bosch, M.; Okamoto, K. Regulation of actin dynamics during structural plasticity of dendritic spines: Signaling messengers and actin-binding proteins. Mol. Cell. Neurosci. 2018, 91, 122–130. [Google Scholar] [CrossRef]
- Bosch, M.; Hayashi, Y. Structural plasticity of dendritic spines. Curr. Opin. Neurobiol. 2012, 22, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Caroni, P.; Donato, F.; Muller, D. Structural plasticity upon learning: Regulation and functions. Nat. Rev. Neurosci. 2012, 13, 478–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, M.; Kaech, S.; Knutti, D.; Matus, A. Rapid actin-based plasticity in dendritic spines. Neuron 1998, 20, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Fukazawa, Y.; Saitoh, Y.; Ozawa, F.; Ohta, Y.; Mizuno, K.; Inokuchi, K. Hippocampal ltp is accompanied by enhanced f-actin content within the dendritic spine that is essential for late ltp maintenance in vivo. Neuron 2003, 38, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Alicea, D.; Perez, M.; Maldonado, C.; Dominicci-Cotto, C.; Marie, B. Cortactin is a regulator of activity-dependent synaptic plasticity controlled by wingless. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 2203–2215. [Google Scholar] [CrossRef] [Green Version]
- Hering, H.; Sheng, M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 11759–11769. [Google Scholar] [CrossRef]
- Iki, J.; Inoue, A.; Bito, H.; Okabe, S. Bi-directional regulation of postsynaptic cortactin distribution by bdnf and nmda receptor activity. Eur. J. Neurosci. 2005, 22, 2985–2994. [Google Scholar] [CrossRef] [PubMed]
- MacGillavry, H.D.; Kerr, J.M.; Kassner, J.; Frost, N.A.; Blanpied, T.A. Shank-cortactin interactions control actin dynamics to maintain flexibility of neuronal spines and synapses. Eur. J. Neurosci. 2016, 43, 179–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racz, B.; Weinberg, R.J. The subcellular organization of cortactin in hippocampus. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 10310–10317. [Google Scholar] [CrossRef] [Green Version]
- Seese, R.R.; Babayan, A.H.; Katz, A.M.; Cox, C.D.; Lauterborn, J.C.; Lynch, G.; Gall, C.M. Ltp induction translocates cortactin at distant synapses in wild-type but not fmr1 knock-out mice. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 7403–7413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.K.; Hsueh, Y.P. Cortactin-binding protein 2 modulates the mobility of cortactin and regulates dendritic spine formation and maintenance. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnoor, M.; Stradal, T.E.; Rottner, K. Cortactin: Cell functions of a multifaceted actin-binding protein. Trends Cell Biol. 2018, 28, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Weed, S.A.; Karginov, A.V.; Schafer, D.A.; Weaver, A.M.; Kinley, A.W.; Cooper, J.A.; Parsons, J.T. Cortactin localization to sites of actin assembly in lamellipodia requires interactions with f-actin and the arp2/3 complex. J. Cell Biol. 2000, 151, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Bryce, N.S.; Clark, E.S.; Leysath, J.L.; Currie, J.D.; Webb, D.J.; Weaver, A.M. Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr. Biol. CB 2005, 15, 1276–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padrick, S.B.; Doolittle, L.K.; Brautigam, C.A.; King, D.S.; Rosen, M.K. Arp2/3 complex is bound and activated by two wasp proteins. Proc. Natl. Acad. Sci. USA 2011, 108, E472–E479. [Google Scholar] [CrossRef] [Green Version]
- Helgeson, L.A.; Nolen, B.J. Mechanism of synergistic activation of arp2/3 complex by cortactin and n-wasp. eLife 2013, 2, e00884. [Google Scholar] [CrossRef]
- Helgeson, L.A.; Prendergast, J.G.; Wagner, A.R.; Rodnick-Smith, M.; Nolen, B.J. Interactions with actin monomers, actin filaments, and arp2/3 complex define the roles of wasp family proteins and cortactin in coordinately regulating branched actin networks. J. Biol. Chem. 2014, 289, 28856–28869. [Google Scholar] [CrossRef] [Green Version]
- Lai, F.P.; Szczodrak, M.; Oelkers, J.M.; Ladwein, M.; Acconcia, F.; Benesch, S.; Auinger, S.; Faix, J.; Small, J.V.; Polo, S.; et al. Cortactin promotes migration and platelet-derived growth factor-induced actin reorganization by signaling to rho-gtpases. Mol. Biol. Cell 2009, 20, 3209–3223. [Google Scholar] [CrossRef] [Green Version]
- Biosse Duplan, M.; Zalli, D.; Stephens, S.; Zenger, S.; Neff, L.; Oelkers, J.M.; Lai, F.P.; Horne, W.; Rottner, K.; Baron, R. Microtubule dynamic instability controls podosome patterning in osteoclasts through eb1, cortactin, and src. Mol. Cell. Biol. 2014, 34, 16–29. [Google Scholar] [CrossRef] [Green Version]
- Kinnunen, T.; Kaksonen, M.; Saarinen, J.; Kalkkinen, N.; Peng, H.B.; Rauvala, H. Cortactin-src kinase signaling pathway is involved in n-syndecan-dependent neurite outgrowth. J. Biol. Chem. 1998, 273, 10702–10708. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Leung, S.; Mangoura, D. Transient suppression of cortactin ectopically induces large telencephalic neurons towards a gabaergic phenotype. J. Cell Sci. 2000, 113, 3161–3172. [Google Scholar] [CrossRef]
- Naisbitt, S.; Kim, E.; Tu, J.C.; Xiao, B.; Sala, C.; Valtschanoff, J.; Weinberg, R.J.; Worley, P.F.; Sheng, M. Shank, a novel family of postsynaptic density proteins that binds to the nmda receptor/psd-95/gkap complex and cortactin. Neuron 1999, 23, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Hattan, D.; Nesti, E.; Cachero, T.G.; Morielli, A.D. Tyrosine phosphorylation of kv1.2 modulates its interaction with the actin-binding protein cortactin. J. Biol. Chem. 2002, 277, 38596–38606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhaylova, M.; Bar, J.; van Bommel, B.; Schatzle, P.; YuanXiang, P.; Raman, R.; Hradsky, J.; Konietzny, A.; Loktionov, E.Y.; Reddy, P.P.; et al. Caldendrin directly couples postsynaptic calcium signals to actin remodeling in dendritic spines. Neuron 2018, 97, 1110–1125.e1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnoor, M.; Lai, F.P.; Zarbock, A.; Klaver, R.; Polaschegg, C.; Schulte, D.; Weich, H.A.; Oelkers, J.M.; Rottner, K.; Vestweber, D. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J. Exp. Med. 2011, 208, 1721–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelsen, K.; Murk, K.; Zagrebelsky, M.; Dreznjak, A.; Jockusch, B.M.; Rothkegel, M.; Korte, M. Fine-tuning of neuronal architecture requires two profilin isoforms. Proc. Natl. Acad. Sci. USA 2010, 107, 15780–15785. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.; Ballachey, E.L. A study of the rat’s behavior in a field. A contribution to method in comparative psychology. Univ. Calif. Publ. Psychol. 1932, 6, 1–12. [Google Scholar]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Takeuchi, T.; Duszkiewicz, A.J.; Morris, R.G. The synaptic plasticity and memory hypothesis: Encoding, storage and persistence. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2014, 369, 20130288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamprecht, R. The actin cytoskeleton in memory formation. Prog. Neurobiol. 2014, 117, 1–19. [Google Scholar] [CrossRef]
- Matsuzaki, M.; Honkura, N.; Ellis-Davies, G.C.; Kasai, H. Structural basis of long-term potentiation in single dendritic spines. Nature 2004, 429, 761–766. [Google Scholar] [CrossRef]
- Basu, S.; Lamprecht, R. The role of actin cytoskeleton in dendritic spines in the maintenance of long-term memory. Front. Mol. Neurosci. 2018, 11, 143. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H.; Lisman, J.E. A role of actin filament in synaptic transmission and long-term potentiation. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 4314–4324. [Google Scholar] [CrossRef] [Green Version]
- Szabo, E.C.; Manguinhas, R.; Fonseca, R. The interplay between neuronal activity and actin dynamics mimic the setting of an ltd synaptic tag. Sci. Rep. 2016, 6, 33685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Reynolds, A.B.; Kanner, S.B.; Vines, R.R.; Parsons, J.T. Identification and characterization of a novel cytoskeleton-associated pp60src substrate. Mol. Cell. Biol. 1991, 11, 5113–5124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, A.M.; Heuser, J.E.; Karginov, A.V.; Lee, W.L.; Parsons, J.T.; Cooper, J.A. Interaction of cortactin and n-wasp with arp2/3 complex. Curr. Biol. 2002, 12, 1270–1278. [Google Scholar] [CrossRef] [Green Version]
- Uruno, T.; Liu, J.; Zhang, P.; Fan, Y.; Egile, C.; Li, R.; Mueller, S.C.; Zhan, X. Activation of arp2/3 complex-mediated actin polymerization by cortactin. Nat. Cell Biol. 2001, 3, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.H.; Racz, B.; Wang, H.; Burianek, L.; Weinberg, R.; Yasuda, R.; Wetsel, W.C.; Soderling, S.H. Disruption of arp2/3 results in asymmetric structural plasticity of dendritic spines and progressive synaptic and behavioral abnormalities. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 6081–6092. [Google Scholar] [CrossRef] [Green Version]
- Wegner, A.M.; Nebhan, C.A.; Hu, L.; Majumdar, D.; Meier, K.M.; Weaver, A.M.; Webb, D.J. N-wasp and the arp2/3 complex are critical regulators of actin in the development of dendritic spines and synapses. J. Biol. Chem. 2008, 283, 15912–15920. [Google Scholar] [CrossRef] [Green Version]
- Soderling, S.H.; Langeberg, L.K.; Soderling, J.A.; Davee, S.M.; Simerly, R.; Raber, J.; Scott, J.D. Loss of wave-1 causes sensorimotor retardation and reduced learning and memory in mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1723–1728. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Sung, J.Y.; Ceglia, I.; Lee, K.W.; Ahn, J.H.; Halford, J.M.; Kim, A.M.; Kwak, S.P.; Park, J.B.; Ho Ryu, S.; et al. Phosphorylation of wave1 regulates actin polymerization and dendritic spine morphology. Nature 2006, 442, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.D.; Mullins, R.D. Cellular control of actin nucleation. Annu. Rev. Cell Dev. Biol. 2002, 18, 247–288. [Google Scholar] [CrossRef]
- Abella, J.V.; Galloni, C.; Pernier, J.; Barry, D.J.; Kjaer, S.; Carlier, M.F.; Way, M. Isoform diversity in the arp2/3 complex determines actin filament dynamics. Nat. Cell Biol. 2016, 18, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Haditsch, U.; Leone, D.P.; Farinelli, M.; Chrostek-Grashoff, A.; Brakebusch, C.; Mansuy, I.M.; McConnell, S.K.; Palmer, T.D. A central role for the small gtpase rac1 in hippocampal plasticity and spatial learning and memory. Mol. Cell. Neurosci. 2009, 41, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Bongmba, O.Y.; Martinez, L.A.; Elhardt, M.E.; Butler, K.; Tejada-Simon, M.V. Modulation of dendritic spines and synaptic function by rac1: A possible link to fragile x syndrome pathology. Brain Res. 2011, 1399, 79–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, L.A.; Tejada-Simon, M.V. Pharmacological inactivation of the small gtpase rac1 impairs long-term plasticity in the mouse hippocampus. Neuropharmacology 2011, 61, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.H.; Wang, H.; Soderling, S.H.; Yasuda, R. Loss of cdc42 leads to defects in synaptic plasticity and remote memory recall. eLife 2014, 3, e02839. [Google Scholar] [CrossRef] [PubMed]
- Diana, G.; Valentini, G.; Travaglione, S.; Falzano, L.; Pieri, M.; Zona, C.; Meschini, S.; Fabbri, A.; Fiorentini, C. Enhancement of learning and memory after activation of cerebral rho gtpases. Proc. Natl. Acad. Sci. USA 2007, 104, 636–641. [Google Scholar] [CrossRef] [Green Version]
- Bhambhvani, H.P.; Simmons, M.; Haroutunian, V.; Meador-Woodruff, J.H. Decreased expression of cortactin in the schizophrenia brain. Neuroreport 2016, 27, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.J.; Hashimoto, T.; Lewis, D.A. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 2006, 11, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornelius, J.; Rottner, K.; Korte, M.; Michaelsen-Preusse, K. Cortactin Contributes to Activity-Dependent Modulation of Spine Actin Dynamics and Spatial Memory Formation. Cells 2021, 10, 1835. https://doi.org/10.3390/cells10071835
Cornelius J, Rottner K, Korte M, Michaelsen-Preusse K. Cortactin Contributes to Activity-Dependent Modulation of Spine Actin Dynamics and Spatial Memory Formation. Cells. 2021; 10(7):1835. https://doi.org/10.3390/cells10071835
Chicago/Turabian StyleCornelius, Jonas, Klemens Rottner, Martin Korte, and Kristin Michaelsen-Preusse. 2021. "Cortactin Contributes to Activity-Dependent Modulation of Spine Actin Dynamics and Spatial Memory Formation" Cells 10, no. 7: 1835. https://doi.org/10.3390/cells10071835
APA StyleCornelius, J., Rottner, K., Korte, M., & Michaelsen-Preusse, K. (2021). Cortactin Contributes to Activity-Dependent Modulation of Spine Actin Dynamics and Spatial Memory Formation. Cells, 10(7), 1835. https://doi.org/10.3390/cells10071835