
BNIP3-Dependent Mitophagy via PGC1α Promotes Cartilage Degradation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Osteoarthritis (OA)
2.2. Human Cartilage and Chondrocyte Culture
2.3. Lentivirus Packaging and Delivery
2.4. Histological Analysis
2.5. Immature Mice Articular Chondrocytes (iMACs) Culture
2.6. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.7. Western Blotting
2.8. Monitoring of Autophagy and Mitophagy
2.9. Annexin V & Dead Cell and MitoPotential Assay
2.10. Small Interfering RNA (siRNA) and microRNA (miRNA, miR) Transfection
2.11. Luciferase Reporter Assay
3. Results
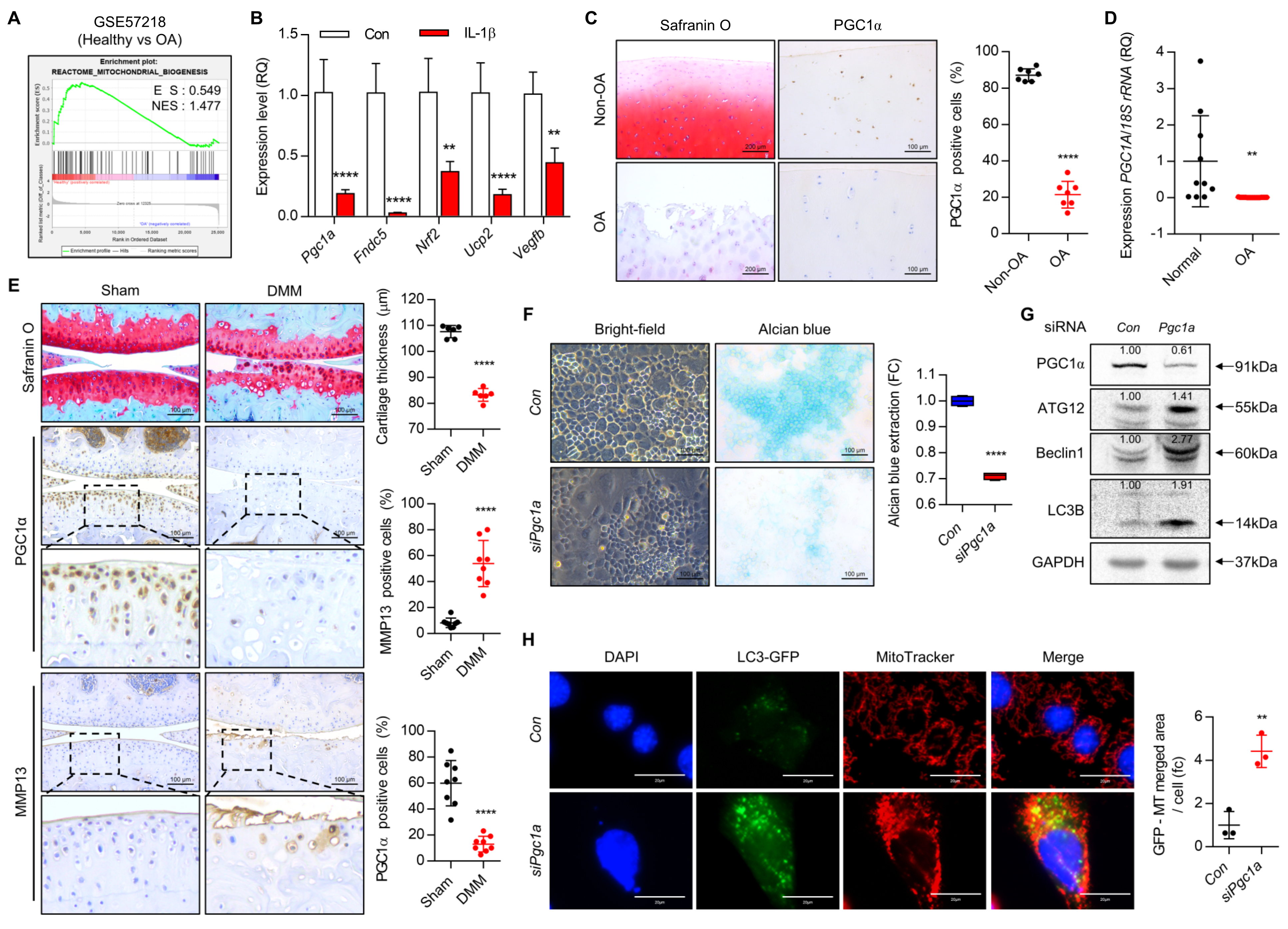
3.1. Upregulated PCG1a Is Responsible for OA Pathogenesis
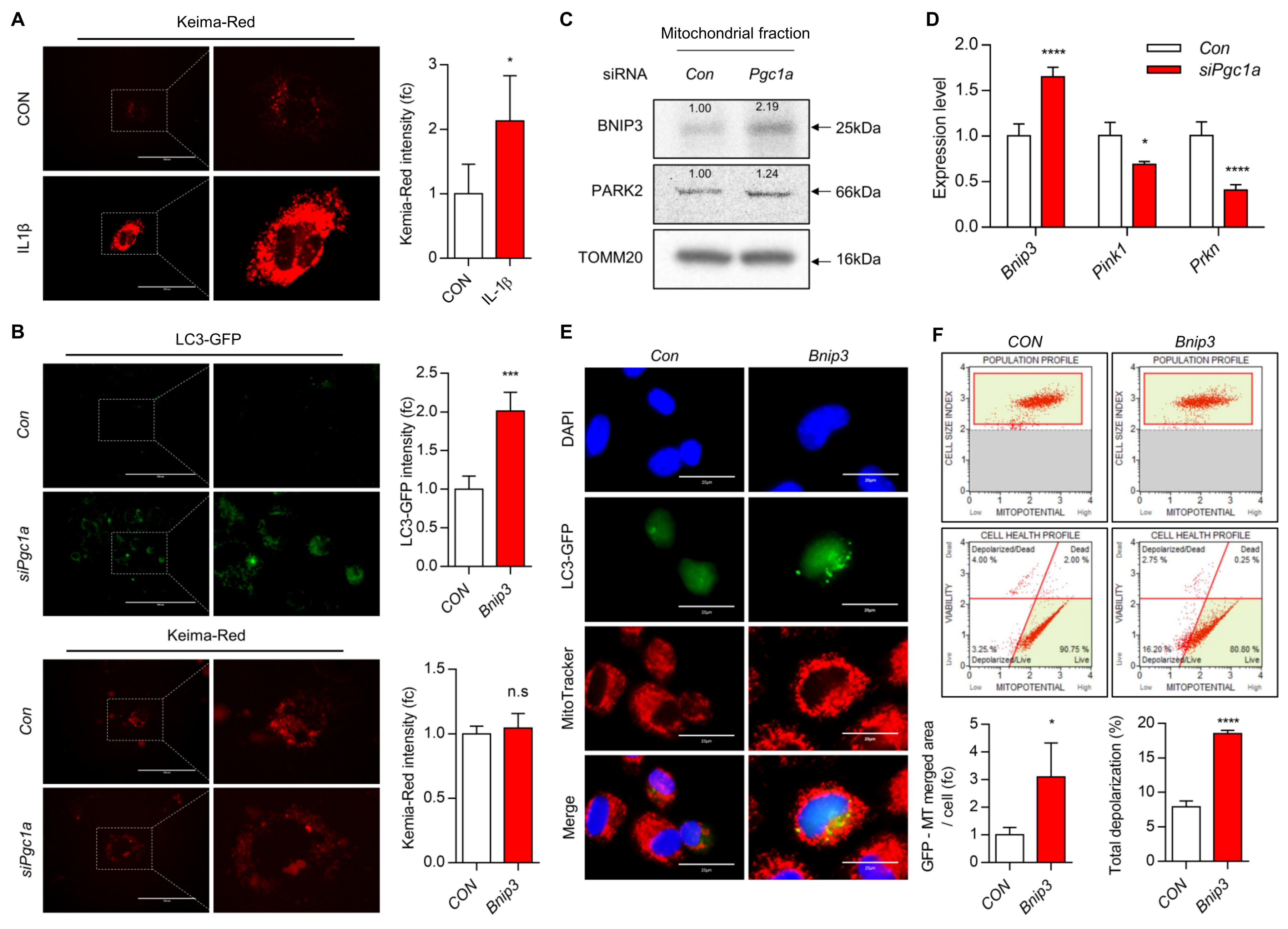
3.2. Suppression of PGC1a Activates PRKN-Independent Mitophagy through Upregulation of BNIP3
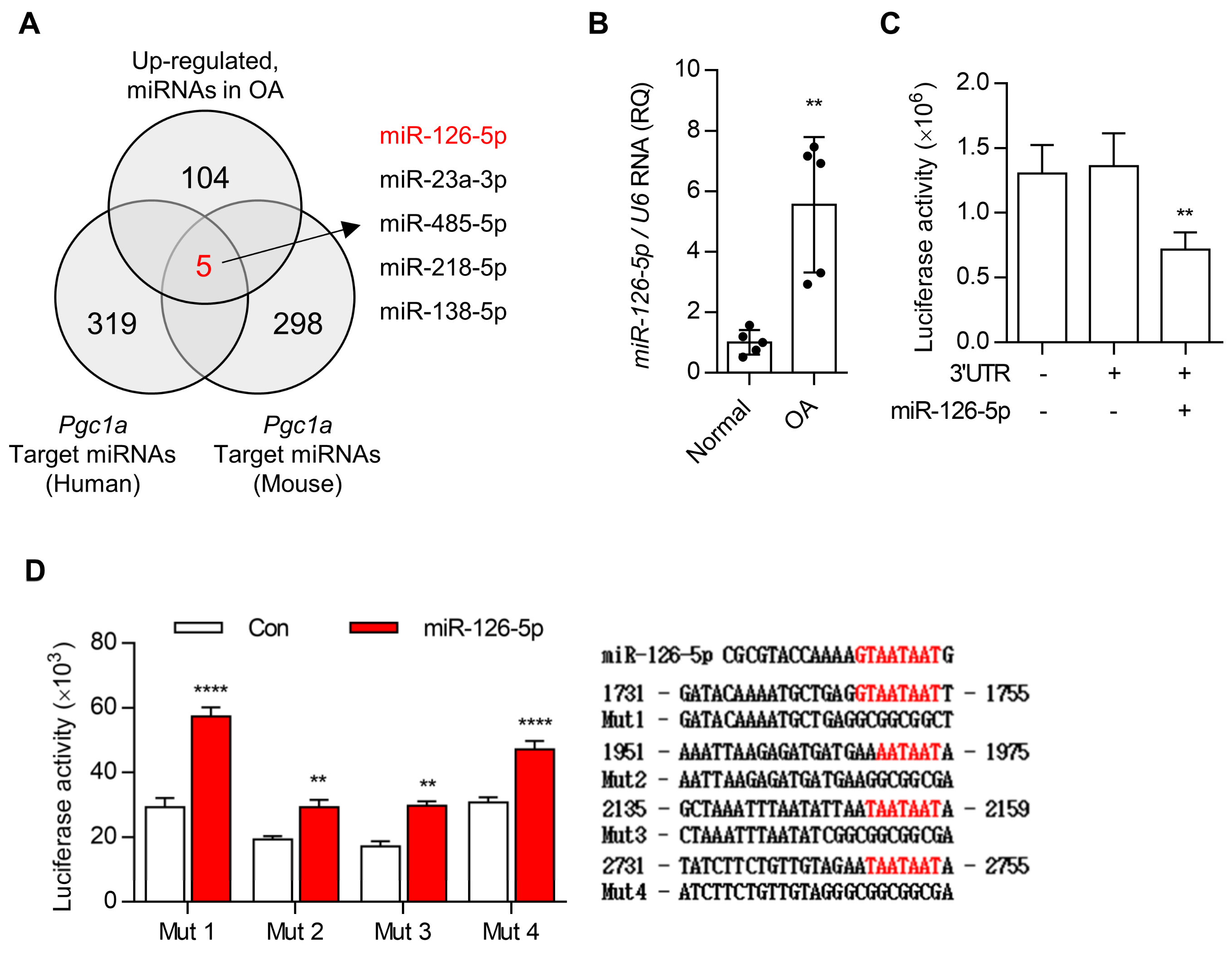
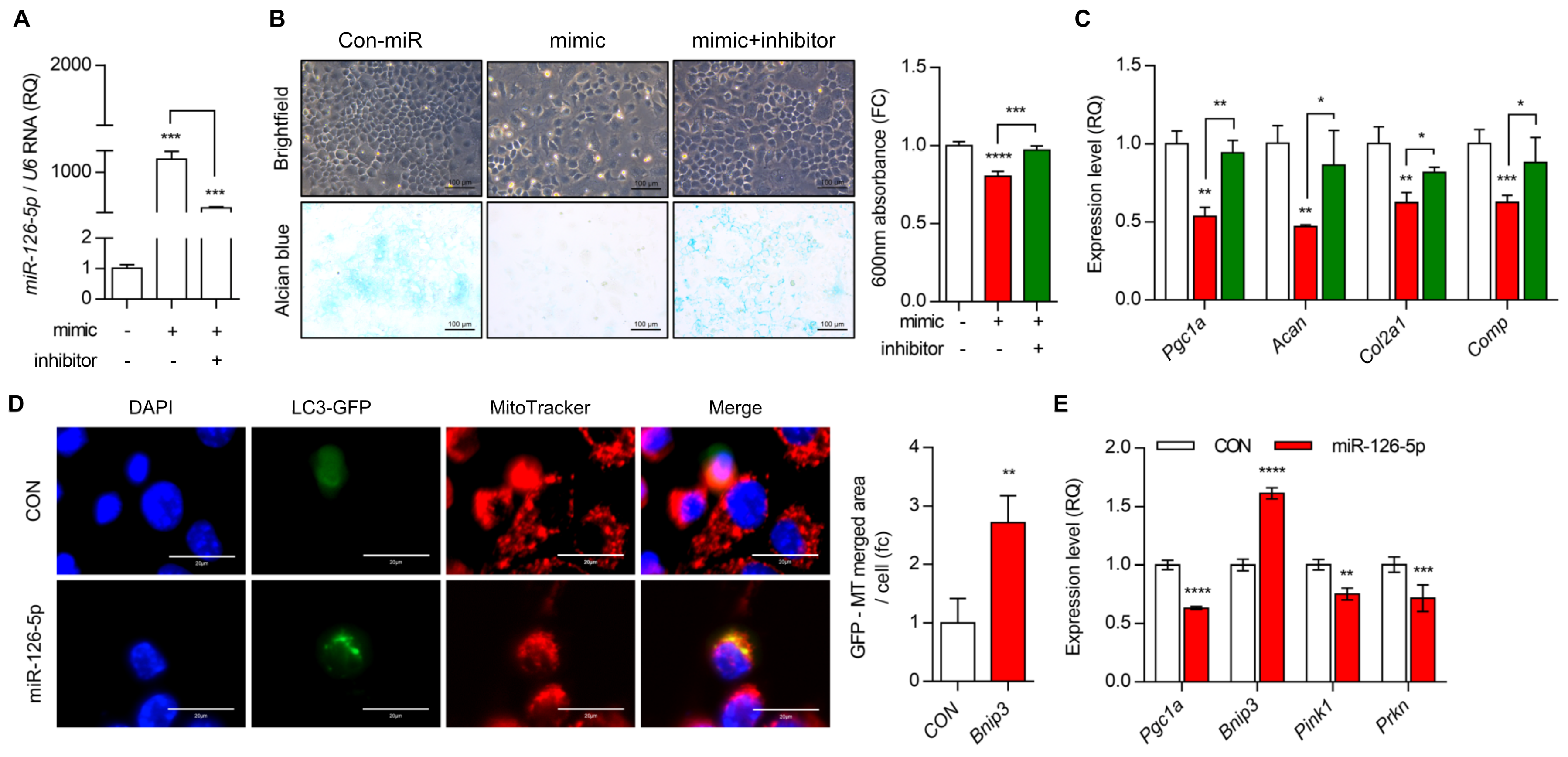
3.3. miR-126-5p Is Key Regulator for PGC1a during OA Pathogenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arden, N.; Nevitt, M.C. Osteoarthritis: Epidemiology. Best Pract. Res. Clin. Rheumatol. 2006, 20, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; He, C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y.; Hirose, J.; Yamabe, S.; Okamoto, N.; Okada, T.; Oyadomari, S.; Mizuta, H. Endoplasmic reticulum stress-induced apoptosis contributes to articular cartilage degeneration via C/EBP homologous protein. Osteoarthr. Cartil. 2014, 22, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.J.; Xiong, Z.; Lu, X.; Ye, M.; Han, X.; Jiang, R. ATF6 upregulates XBP1S and inhibits ER stress-mediated apoptosis in osteoarthritis cartilage. Cell Signal. 2014, 26, 332–342. [Google Scholar] [CrossRef]
- Kim, D.; Song, J.; Kang, Y.; Park, S.; Kim, Y.I.; Kwak, S.; Lim, D.; Park, R.; Chun, C.H.; Choe, S.K.; et al. Fis1 depletion in osteoarthritis impairs chondrocyte survival and peroxisomal and lysosomal function. J. Mol. Med. 2016, 94, 1373–1384. [Google Scholar] [CrossRef]
- Song, J.; Baek, I.J.; Chun, C.H.; Jin, E.J. Dysregulation of the NUDT7-PGAM1 axis is responsible for chondrocyte death during osteoarthritis pathogenesis. Nat. Commun. 2018, 9, 3427. [Google Scholar] [CrossRef]
- Blanco, F.J.; Rego, I.; Ruiz-Romero, C. The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 161–169. [Google Scholar] [CrossRef]
- Gavriilidis, C.; Miwa, S.; von Zglinicki, T.; Taylor, R.W.; Young, D.A. Mitochondrial dysfunction in osteoarthritis is associated with down-regulation of superoxide dismutase 2. Arthritis Rheum. 2013, 65, 378–387. [Google Scholar] [CrossRef]
- Lopez de Figueroa, P.; Lotz, M.K.; Blanco, F.J.; Carames, B. Autophagy activation and protection from mitochondrial dysfunction in human chondrocytes. Arthritis Rheumatol. 2015, 67, 966–976. [Google Scholar] [CrossRef]
- Lane, R.K.; Hilsabeck, T.; Rea, S.L. The role of mitochondrial dysfunction in age-related diseases. Biochim. Biophys. Acta BBA Bioenerg. 2015, 1847, 1387–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Armada, M.J.; Carames, B.; Martin, M.A.; Cillero-Pastor, B.; Lires-Dean, M.; Fuentes-Boquete, I.; Arenas, J.; Blanco, F.J. Mitochondrial activity is modulated by TNFα and IL-1β in normal human chondrocyte cells. Osteoarthr. Cartil. 2006, 14, 1011–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.A.; Lee, Y.J.; Seong, S.C.; Choe, K.W.; Song, Y.W. Apoptotic chondrocyte death in human osteoarthritis. J. Rheumatol. 2000, 27, 455–462. [Google Scholar] [PubMed]
- Rego, I.; Fernandez-Moreno, M.; Fernandez-Lopez, C.; Gomez-Reino, J.J.; Gonzalez, A.; Arenas, J.; Blanco, F.J. Role of European mitochondrial DNA haplogroups in the prevalence of hip osteoarthritis in Galicia, Northern Spain. Ann. Rheum. Dis. 2010, 69, 210–213. [Google Scholar] [CrossRef]
- Blanco, F.J.; Rego-Perez, I. Mitochondria and mitophagy: Biosensors for cartilage degradation and osteoarthritis. Osteoarthr. Cartil. 2018, 26, 989–991. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Li, Z.; Cao, Y.; Cui, Y.; Yang, X.; Meng, Z.; Wang, R. Effect of chondrocyte mitochondrial dysfunction on cartilage degeneration: A possible pathway for osteoarthritis pathology at the subcellular level. Mol. Med. Rep. 2019, 20, 3308–3316. [Google Scholar] [CrossRef] [Green Version]
- Maneiro, E.; Martin, M.A.; de Andres, M.C.; Lopez-Armada, M.J.; Fernandez-Sueiro, J.L.; del Hoyo, P.; Galdo, F.; Arenas, J.; Blanco, F.J. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum. 2003, 48, 700–708. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, X.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor γ coactivator 1α. Arthritis Rheumatol. 2015, 67, 2141–2153. [Google Scholar] [CrossRef]
- Blanco, F.J.; Fernandez-Moreno, M. Mitochondrial biogenesis: A potential therapeutic target for osteoarthritis. Osteoarthr. Cartil. 2020, 28, 1003–1006. [Google Scholar] [CrossRef]
- Lotz, M.K.; Carames, B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat. Rev. Rheumatol. 2011, 7, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.S.; Zhang, F.J.; Zeng, C.; Luo, W.; Xiao, W.F.; Gao, S.G.; Lei, G.H. Autophagy in osteoarthritis. Jt. Bone Spine 2016, 83, 143–148. [Google Scholar] [CrossRef]
- Carames, B.; Olmer, M.; Kiosses, W.B.; Lotz, M.K. The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol. 2015, 67, 1568–1576. [Google Scholar] [CrossRef]
- Zhang, Y.; Vasheghani, F.; Li, Y.H.; Blati, M.; Simeone, K.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Pelletier, J.P.; et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 2015, 74, 1432–1440. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.T. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 23–34. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125 Pt 4, 795–799. [Google Scholar] [CrossRef] [Green Version]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Um, J.H.; Yun, J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 2017, 50, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Park, H.; Shin, N.; Kwon, H.H.; Yin, Y.; Hwang, J.A.; Song, H.J.; Kim, J.; Kim, D.W.; Beom, J. Pink1-Mediated Chondrocytic Mitophagy Contributes to Cartilage Degeneration in Osteoarthritis. J. Clin. Med. 2019, 8, 1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, G.E.; Lee, H.J.; Chae, C.W.; Cho, J.H.; Jung, Y.H.; Kim, J.S.; Kim, S.Y.; Lim, J.R.; Han, H.J. BNIP3L/NIX-mediated mitophagy protects against glucocorticoid-induced synapse defects. Nat. Commun. 2021, 12, 487. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.E.; Johnson, L.R.; Kang, H.H.; Sun, J.C. BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 2015, 43, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampert, M.A.; Orogo, A.M.; Najor, R.H.; Hammerling, B.C.; Leon, L.J.; Wang, B.J.; Kim, T.; Sussman, M.A.; Gustafsson, Å.B. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 2019, 15, 1182–1198. [Google Scholar] [CrossRef]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [Green Version]
- Esteban-Martinez, L.; Sierra-Filardi, E.; McGreal, R.S.; Salazar-Roa, M.; Mariño, G.; Seco, E.; Durand, S.; Enot, D.; Graña, O.; Malumbres, M.; et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 2017, 36, 1688–1706. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Zhuang, H.; Chen, H.G.; Chen, Y.; Tian, W.; Wu, W.; Li, Y.; Wang, S.; Zhang, L.; et al. MicroRNA-137 is a novel hypoxia-responsive microRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX. J Biol. Chem. 2014, 289, 10691–10701. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Glasson, S.S.; Blanchet, T.J.; Morris, E.A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr. Cartil. 2007, 15, 1061–1069. [Google Scholar] [CrossRef] [Green Version]
- Culley, K.L.; Dragomir, C.L.; Chang, J.; Wondimu, E.B.; Coico, J.; Plumb, D.A.; Otero, M.; Goldring, M.B. Mouse models of osteoarthritis: Surgical model of posttraumatic osteoarthritis induced by destabilization of the medial meniscus. Methods Mol. Biol. 2015, 1226, 143–173. [Google Scholar] [CrossRef]
- Cheng, N.T.; Meng, H.; Ma, L.F.; Zhang, L.; Yu, H.M.; Wang, Z.Z.; Guo, A. Role of autophagy in the progression of osteoarthritis: The autophagy inhibitor, 3-methyladenine, aggravates the severity of experimental osteoarthritis. Int. J. Mol. Med. 2017, 39, 1224–1232. [Google Scholar] [CrossRef] [Green Version]
- Luo, P.; Gao, F.; Niu, D.; Sun, X.; Song, Q.; Guo, C.; Liang, Y.; Sun, W. The Role of Autophagy in Chondrocyte Metabolism and Osteoarthritis: A Comprehensive Research Review. BioMed Res. Int. 2019, 2019, 5171602. [Google Scholar] [CrossRef]
- Carames, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010, 62, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Pellegrino, M.W.; Haynes, C.M. Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol. 2015, 13, 22. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.Y.; Khan, N.M.; Ahmad, I.; Haqqi, T.M. Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthr. Cartil. 2018, 26, 1087–1097. [Google Scholar] [CrossRef]
- Su, X.J.; Zheng, C.; Hu, Q.Q.; Du, H.Y.; Liao, R.Z.; Zhang, M.T. Bimetallic cooperative effect on O-O bond formation: Copper polypyridyl complexes as water oxidation catalyst. Dalton Trans. 2018, 47, 8670–8675. [Google Scholar] [CrossRef] [PubMed]
- Vaamonde-Garcia, C.; Riveiro-Naveira, R.R.; Valcarcel-Ares, M.N.; Hermida-Carballo, L.; Blanco, F.J.; Lopez-Armada, M.J. Mitochondrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheum. 2012, 64, 2927–2936. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melser, S.; Chatelain, E.H.; Lavie, J.; Mahfouf, W.; Jose, C.; Obre, E.; Goorden, S.; Priault, M.; Elgersma, Y.; Rezvani, H.R.; et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab. 2013, 17, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Qi, W.; Chen, G.; Feng, D.; Liu, J.; Ma, B.; Zhou, C.; Mu, C.; Zhang, W.; Chen, Q.; et al. Mitochondrial outer-membrane E3 ligase MUL1 ubiquitinates ULK1 and regulates selenite-induced mitophagy. Autophagy 2015, 11, 1216–1229. [Google Scholar] [CrossRef] [Green Version]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Bingol, B.; Sheng, M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic. Biol. Med. 2016, 100, 210–222. [Google Scholar] [CrossRef]
- Zhao, X.; Petursson, F.; Viollet, B.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. Peroxisome proliferator-activated receptor γ coactivator 1α and FoxO3A mediate chondroprotection by AMP-activated protein kinase. Arthritis Rheumatol. 2014, 66, 3073–3082. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Soriano, F.X.; Liesa, M.; Bach, D.; Chan, D.C.; Palacín, M.; Zorzano, A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-γ coactivator-1α, estrogen-related receptor-α, and mitofusin 2. Diabetes 2006, 55, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Vainshtein, A.; Desjardins, E.M.; Armani, A.; Sandri, M.; Hood, D.A. PGC-1α modulates denervation-induced mitophagy in skeletal muscle. Skelet. Muscle 2015, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Bernard-Marissal, N.; Moullan, N.; D’Amico, D.; Auwerx, J.; Moore, D.J.; Knott, G.; Aebischer, P.; Schneider, B.L. Parkin functionally interacts with PGC-1α to preserve mitochondria and protect dopaminergic neurons. Hum. Mol. Genet. 2017, 26, 582–598. [Google Scholar] [CrossRef] [Green Version]
- Olmos, Y.; Valle, I.; Borniquel, S.; Tierrez, A.; Soria, E.; Lamas, S.; Monsalve, M. Mutual dependence of Foxo3a and PGC-1alpha in the induction of oxidative stress genes. J. Biol. Chem. 2009, 284, 14476–14484. [Google Scholar] [CrossRef] [Green Version]
- Olmos, Y.; Sanchez-Gomez, F.J.; Wild, B.; Garcia-Quintans, N.; Cabezudo, S.; Lamas, S.; Monsalve, M. SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1alpha complex. Antioxid. Redox Signal. 2013, 19, 1507–1521. [Google Scholar] [CrossRef]
- Sanchez, A.M.; Bernardi, H.; Py, G.; Candau, R.B. Autophagy is essential to support skeletal muscle plasticity in response to endurance exercise. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R956–R969. [Google Scholar] [CrossRef]
- Zhu, Y.; Massen, S.; Terenzio, M.; Lang, V.; Chen-Lindner, S.; Eils, R.; Novak, I.; Dikic, I.; Hamacher-Brady, A.; Brady, N.R. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 2013, 288, 1099–1113. [Google Scholar] [CrossRef] [Green Version]
- Ney, P.A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta BBA Mol. Cell Res. 2015, 1853, 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.F.; Liu, S.Y.; Qi, Z.F.; Lv, Z.H.; Ding, H.R.; Zhou, W.J. Mir-145 targeting Bnip3 reduces apoptosis of chondrocytes in osteroarthritis through Notch signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8263–8272. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Song, J.; Jin, E.-J. BNIP3-Dependent Mitophagy via PGC1α Promotes Cartilage Degradation. Cells 2021, 10, 1839. https://doi.org/10.3390/cells10071839
Kim D, Song J, Jin E-J. BNIP3-Dependent Mitophagy via PGC1α Promotes Cartilage Degradation. Cells. 2021; 10(7):1839. https://doi.org/10.3390/cells10071839
Chicago/Turabian StyleKim, Deokha, Jinsoo Song, and Eun-Jung Jin. 2021. "BNIP3-Dependent Mitophagy via PGC1α Promotes Cartilage Degradation" Cells 10, no. 7: 1839. https://doi.org/10.3390/cells10071839
APA StyleKim, D., Song, J., & Jin, E. -J. (2021). BNIP3-Dependent Mitophagy via PGC1α Promotes Cartilage Degradation. Cells, 10(7), 1839. https://doi.org/10.3390/cells10071839