
Regulation of Store-Operated Ca2+ Entry by SARAF


Abstract
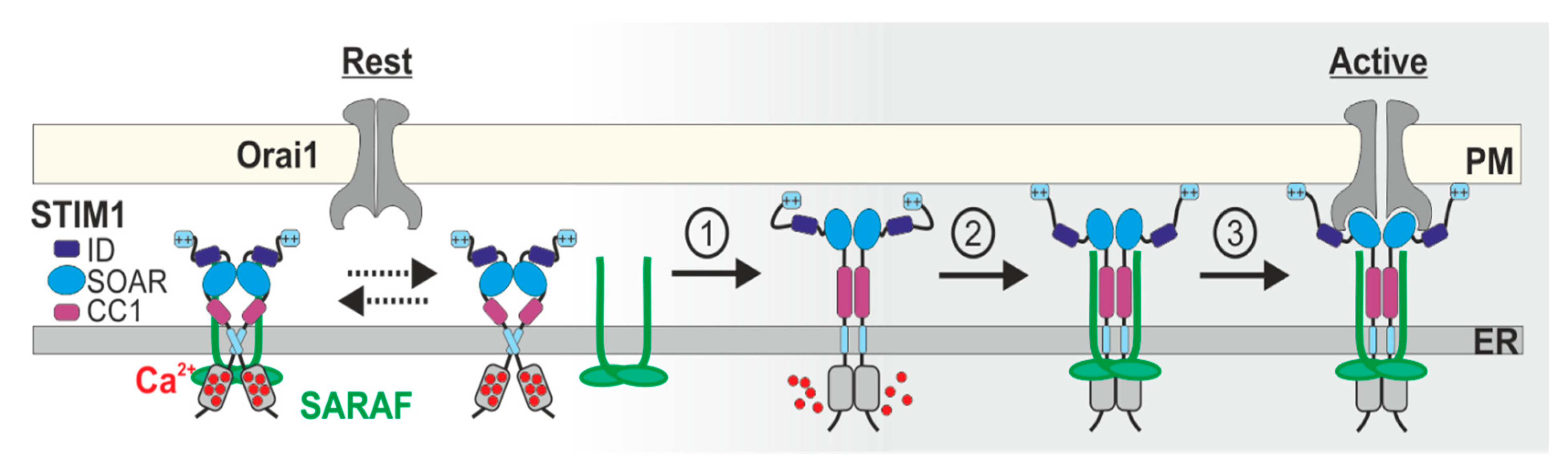
:1. Introduction
2. Slow Calcium-Dependent Inactivation—What Is the Mechanism?
3. Molecular Identification of SARAF
4. Interaction of SARAF with the CRAC Channel
5. Current Models for SCDI by SARAF
6. The Molecular Basis for Ca2+ Sensitivity of SCDI by SARAF
7. The Regulatory Function of the ER Luminal-Facing Domain of SARAF
8. The Physiological Role of SARAF
9. The Role of SARAF in Pathologies of Cardiovascular Tissues
10. The Role of SARAF in Acute Pancreatitis
11. The Role of SARAF in Smooth Muscle Remodeling
12. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Putney, J.W. A Model for Receptor-Regulated Calcium Entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Lacruz, R.S.; Feske, S. Diseases Caused by Mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [Green Version]
- Ong, H.L.; de Souza, L.B.; Ambudkar, I.S. Role of TRPC Channels in Store-Operated Calcium Entry. Adv. Exp. Med. Biol. 2016, 898, 87–109. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.-F.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-Wide RNAi Screen of Ca2+ Influx Identifies Genes That Regulate Ca2+ Release-Activated Ca2+ Channel Activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 Is a Ca2+ Sensor That Activates CRAC Channels and Migrates from the Ca2+ Store to the Plasma Membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an Essential and Conserved Component of Store-Operated Ca2+ Channel Function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Yeromin, A.V.; Zhang, S.L.; Jiang, W.; Yu, Y.; Safrina, O.; Cahalan, M.D. Molecular Identification of the CRAC Channel by Altered Ion Selectivity in a Mutant of Orai. Nature 2006, 443, 226–229. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A Mutation in Orai1 Causes Immune Deficiency by Abrogating CRAC Channel Function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 Is an Essential Pore Subunit of the CRAC Channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 Is a Plasma Membrane Protein Essential for Store-Operated Ca2+ Entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef] [Green Version]
- Hoth, M.; Niemeyer, B.A. The Neglected CRAC Proteins: Orai2, Orai3, and STIM2. Curr. Top. Membr. 2013, 71, 237–271. [Google Scholar] [CrossRef]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal Structure of the Calcium Release-Activated Calcium Channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wu, G.; Yu, Y.; Chen, X.; Ji, R.; Lu, J.; Li, X.; Zhang, X.; Yang, X.; Shen, Y. Molecular Understanding of Calcium Permeation through the Open Orai Channel. PLoS Biol. 2019, 17, e3000096. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Stanley, C.; Isacoff, E.Y. Critical Role for Orai1 C-Terminal Domain and TM4 in CRAC Channel Gating. Cell Res. 2015, 25, 963–980. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Lu, J.; Xu, P.; Xie, X.; Chen, L.; Xu, T. Mapping the Interacting Domains of STIM1 and Orai1 in Ca2+ Release-Activated Ca2+ Channel Activation. J. Biol. Chem. 2007, 282, 29448–29456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palty, R.; Isacoff, E.Y. Cooperative Binding of Stromal Interaction Molecule 1 (STIM1) to the N and C Termini of Calcium Release-Activated Calcium Modulator 1 (Orai1). J. Biol. Chem. 2016, 291, 334–341. [Google Scholar] [CrossRef] [Green Version]
- McNally, B.A.; Somasundaram, A.; Jairaman, A.; Yamashita, M.; Prakriya, M. The C- and N-Terminal STIM1 Binding Sites on Orai1 Are Required for Both Trapping and Gating CRAC Channels. J. Physiol. 2013, 591, 2833–2850. [Google Scholar] [CrossRef] [PubMed]
- Gudlur, A.; Quintana, A.; Zhou, Y.; Hirve, N.; Mahapatra, S.; Hogan, P.G. STIM1 Triggers a Gating Rearrangement at the Extracellular Mouth of the ORAI1 Channel. Nat. Commun. 2014, 5, 5164. [Google Scholar] [CrossRef] [Green Version]
- Derler, I.; Plenk, P.; Fahrner, M.; Muik, M.; Jardin, I.; Schindl, R.; Gruber, H.J.; Groschner, K.; Romanin, C. The Extended Transmembrane Orai1 N-Terminal (ETON) Region Combines Binding Interface and Gate for Orai1 Activation by STIM1*♦. J. Biol. Chem. 2013, 288, 29025–29034. [Google Scholar] [CrossRef] [Green Version]
- Lis, A.; Zierler, S.; Peinelt, C.; Fleig, A.; Penner, R. A Single Lysine in the N-Terminal Region of Store-Operated Channels Is Critical for STIM1-Mediated Gating. J. Gen. Physiol. 2010, 136, 673–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Zhou, M.-H.; Hu, C.; Kuo, E.; Peng, X.; Hu, J.; Kuo, L.; Zhang, S.L. Differential Roles of the C and N Termini of Orai1 Protein in Interacting with Stromal Interaction Molecule 1 (STIM1) for Ca2+ Release-Activated Ca2+ (CRAC) Channel Activation. J. Biol. Chem. 2013, 288, 11263–11272. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E.; Meyer, T. STIM Is a Ca2+ Sensor Essential for Ca2+-Store-Depletion-Triggered Ca2+ Influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudlur, A.; Zeraik, A.E.; Hirve, N.; Rajanikanth, V.; Bobkov, A.A.; Ma, G.; Zheng, S.; Wang, Y.; Zhou, Y.; Komives, E.A.; et al. Calcium Sensing by the STIM1 ER-Luminal Domain. Nat. Commun. 2018, 9, 4536. [Google Scholar] [CrossRef] [PubMed]
- Luik, R.M.; Wang, B.; Prakriya, M.; Wu, M.M.; Lewis, R.S. Oligomerization of STIM1 Couples ER Calcium Depletion to CRAC Channel Activation. Nature 2008, 454, 538–542. [Google Scholar] [CrossRef]
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P.; et al. Inside-out Ca2+ Signalling Prompted by STIM1 Conformational Switch. Nat. Commun. 2015, 6, 7826. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Srinivasan, P.; Razavi, S.; Seymour, S.; Meraner, P.; Gudlur, A.; Stathopulos, P.B.; Ikura, M.; Rao, A.; Hogan, P.G. Initial Activation of STIM1, the Regulator of Store-Operated Calcium Entry. Nat. Struct. Mol. Biol. 2013, 20, 973–981. [Google Scholar] [CrossRef] [Green Version]
- Hirve, N.; Rajanikanth, V.; Hogan, P.G.; Gudlur, A. Coiled-Coil Formation Conveys a STIM1 Signal from ER Lumen to Cytoplasm. Cell Rep. 2018, 22, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Fahrner, M.; Muik, M.; Schindl, R.; Butorac, C.; Stathopulos, P.; Zheng, L.; Jardin, I.; Ikura, M.; Romanin, C. A Coiled-Coil Clamp Controls Both Conformation and Clustering of Stromal Interaction Molecule 1 (STIM1). J. Biol. Chem. 2014, 289, 33231–33244. [Google Scholar] [CrossRef] [Green Version]
- Fahrner, M.; Stadlbauer, M.; Muik, M.; Rathner, P.; Stathopulos, P.; Ikura, M.; Müller, N.; Romanin, C. A Dual Mechanism Promotes Switching of the Stormorken STIM1 R304W Mutant into the Activated State. Nat. Commun. 2018, 9, 825. [Google Scholar] [CrossRef]
- Muik, M.; Fahrner, M.; Derler, I.; Schindl, R.; Bergsmann, J.; Frischauf, I.; Groschner, K.; Romanin, C. A Cytosolic Homomerization and a Modulatory Domain within STIM1 C Terminus Determine Coupling to ORAI1 Channels. J. Biol. Chem. 2009, 284, 8421–8426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muik, M.; Fahrner, M.; Schindl, R.; Stathopulos, P.; Frischauf, I.; Derler, I.; Plenk, P.; Lackner, B.; Groschner, K.; Ikura, M.; et al. STIM1 Couples to ORAI1 via an Intramolecular Transition into an Extended Conformation. EMBO J. 2011, 30, 1678–1689. [Google Scholar] [CrossRef] [PubMed]
- Muik, M.; Frischauf, I.; Derler, I.; Fahrner, M.; Bergsmann, J.; Eder, P.; Schindl, R.; Hesch, C.; Polzinger, B.; Fritsch, R.; et al. Dynamic Coupling of the Putative Coiled-Coil Domain of ORAI1 with STIM1 Mediates ORAI1 Channel Activation. J. Biol. Chem. 2008, 283, 8014–8022. [Google Scholar] [CrossRef] [Green Version]
- Korzeniowski, M.K.; Manjarrés, I.M.; Varnai, P.; Balla, T. Activation of STIM1-Orai1 Involves an Intramolecular Switching Mechanism. Sci. Signal. 2010, 3, ra82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.-J.; Worley, P.F.; Muallem, S. SOAR and the Polybasic STIM1 Domains Gate and Regulate Orai Channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 Clusters and Activates CRAC Channels via Direct Binding of a Cytosolic Domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, A.; Ahuja, M.; Maléth, J.; Moreno, C.M.; Yuan, J.P.; Kim, M.S.; Muallem, S. The STIM1 CTID Domain Determines Access of SARAF to SOAR to Regulate Orai1 Channel Function. J. Cell Biol. 2013, 202, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Lee, M.; Jeong, S.J.; Qin, X.; Lee, A.R.; Park, H.; Park, C.Y. IDstim Helps STIM1 Keep Inactive via Intramolecular Binding to the Coiled-Coil Domain in a Resting State. J. Cell Sci. 2019. [Google Scholar] [CrossRef]
- Luik, R.M.; Wu, M.M.; Buchanan, J.; Lewis, R.S. The Elementary Unit of Store-Operated Ca2+ Entry: Local Activation of CRAC Channels by STIM1 at ER-Plasma Membrane Junctions. J. Cell Biol. 2006, 174, 815–825. [Google Scholar] [CrossRef]
- Wu, M.M.; Buchanan, J.; Luik, R.M.; Lewis, R.S. Ca2+ Store Depletion Causes STIM1 to Accumulate in ER Regions Closely Associated with the Plasma Membrane. J. Cell Biol. 2006, 174, 803–813. [Google Scholar] [CrossRef]
- Xu, P.; Lu, J.; Li, Z.; Yu, X.; Chen, L.; Xu, T. Aggregation of STIM1 underneath the Plasma Membrane Induces Clustering of Orai1. Biochem. Biophys. Res. Commun. 2006, 350, 969–976. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium—A Life and Death Signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Jousset, H.; Frieden, M.; Demaurex, N. STIM1 Knockdown Reveals That Store-Operated Ca2+ Channels Located Close to Sarco/Endoplasmic Ca2+ ATPases (SERCA) Pumps Silently Refill the Endoplasmic Reticulum. J. Biol. Chem. 2007, 282, 11456–11464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malli, R.; Naghdi, S.; Romanin, C.; Graier, W.F. Cytosolic Ca2+ Prevents the Subplasmalemmal Clustering of STIM1: An Intrinsic Mechanism to Avoid Ca2+ Overload. J. Cell Sci. 2008, 121, 3133–3139. [Google Scholar] [CrossRef] [Green Version]
- Smyth, J.T.; Dehaven, W.I.; Bird, G.S.; Putney, J.W. Ca2+-Store-Dependent and -Independent Reversal of Stim1 Localization and Function. J. Cell Sci. 2008, 121, 762–772. [Google Scholar] [CrossRef] [Green Version]
- Bird, G.S.; Hwang, S.-Y.; Smyth, J.T.; Fukushima, M.; Boyles, R.R.; Putney, J.W. STIM1 Is a Calcium Sensor Specialized for Digital Signaling. Curr. Biol. 2009, 19, 1724–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derler, I.; Fahrner, M.; Muik, M.; Lackner, B.; Schindl, R.; Groschner, K.; Romanin, C. A Ca2+ Release-Activated Ca2+ (CRAC) Modulatory Domain (CMD) within STIM1 Mediates Fast Ca2+-Dependent Inactivation of ORAI1 Channels. J. Biol. Chem. 2009, 284, 24933–24938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, F.M.; Yen, M.; Lewis, R.S. Orai1 Pore Residues Control CRAC Channel Inactivation Independently of Calmodulin. J. Gen. Physiol. 2016, 147, 137–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zweifach, A.; Lewis, R.S. Rapid Inactivation of Depletion-Activated Calcium Current (ICRAC) Due to Local Calcium Feedback. J. Gen. Physiol. 1995, 105, 209–226. [Google Scholar] [CrossRef] [Green Version]
- Mullins, F.M.; Lewis, R.S. The Inactivation Domain of STIM1 Is Functionally Coupled with the Orai1 Pore to Enable Ca2+-Dependent Inactivation. J. Gen. Physiol. 2016, 147, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.P.; Yuan, J.P.; Zeng, W.; So, I.; Worley, P.F.; Muallem, S. Molecular Determinants of Fast Ca2+-Dependent Inactivation and Gating of the Orai Channels. Proc. Natl. Acad. Sci. USA 2009, 106, 14687–14692. [Google Scholar] [CrossRef] [Green Version]
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF Inactivates the Store Operated Calcium Entry Machinery to Prevent Excess Calcium Refilling. Cell 2012, 149, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Zweifach, A.; Lewis, R.S. Slow Calcium-Dependent Inactivation of Depletion-Activated Calcium Current. Store-Dependent and -Independent Mechanisms. J. Biol. Chem. 1995, 270, 14445–14451. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.B. Slow Feedback Inhibition of Calcium Release-Activated Calcium Current by Calcium Entry. J. Biol. Chem. 1998, 273, 14925–14932. [Google Scholar] [CrossRef] [Green Version]
- Carreras-Sureda, A.; Cantero-Recasens, G.; Rubio-Moscardo, F.; Kiefer, K.; Peinelt, C.; Niemeyer, B.A.; Valverde, M.A.; Vicente, R. ORMDL3 Modulates Store-Operated Calcium Entry and Lymphocyte Activation. Hum. Mol. Genet. 2013, 22, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wu, G.; Yang, Y.; Fu, S.; Liu, X.; Kang, H.; Yang, X.; Su, X.-C.; Shen, Y. Calmodulin Dissociates the STIM1-Orai1 Complex and STIM1 Oligomers. Nat. Commun. 2017, 8, 1042. [Google Scholar] [CrossRef] [PubMed]
- Nesin, V.; Wiley, G.; Kousi, M.; Ong, E.-C.; Lehmann, T.; Nicholl, D.J.; Suri, M.; Shahrizaila, N.; Katsanis, N.; Gaffney, P.M.; et al. Activating Mutations in STIM1 and ORAI1 Cause Overlapping Syndromes of Tubular Myopathy and Congenital Miosis. Proc. Natl. Acad. Sci. USA 2014, 111, 4197–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louzao, M.C.; Ribeiro, C.M.; Bird, G.S.; Putney, J.W. Cell Type-Specific Modes of Feedback Regulation of Capacitative Calcium Entry. J. Biol. Chem. 1996, 271, 14807–14813. [Google Scholar] [CrossRef] [Green Version]
- Montalvo, G.B.; Artalejo, A.R.; Gilabert, J.A. ATP from Subplasmalemmal Mitochondria Controls Ca2+-Dependent Inactivation of CRAC Channels. J. Biol. Chem. 2006, 281, 35616–35623. [Google Scholar] [CrossRef]
- Gilabert, J.A.; Parekh, A.B. Respiring Mitochondria Determine the Pattern of Activation and Inactivation of the Store-Operated Ca2+ Current I(CRAC). EMBO J. 2000, 19, 6401–6407. [Google Scholar] [CrossRef]
- Lis, A.; Peinelt, C.; Beck, A.; Parvez, S.; Monteilh-Zoller, M.; Fleig, A.; Penner, R. CRACM1, CRACM2, and CRACM3 Are Store-Operated Ca2+ Channels with Distinct Functional Properties. Curr. Biol. 2007, 17, 794–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.-W.; Bakowski, D.; Nelson, C.; Mehta, R.; Almeyda, R.; Bates, G.; Parekh, A.B. Cysteinyl Leukotriene Type I Receptor Desensitization Sustains Ca2+-Dependent Gene Expression. Nature 2012, 482, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maléth, J.; Choi, S.; Muallem, S.; Ahuja, M. Translocation between PI(4,5)P2-Poor and PI(4,5)P2-Rich Microdomains during Store Depletion Determines STIM1 Conformation and Orai1 Gating. Nat. Commun. 2014, 5, 5843. [Google Scholar] [CrossRef] [PubMed]
- Naraghi, M.; Neher, E. Linearized Buffered Ca2+ Diffusion in Microdomains and Its Implications for Calculation of [Ca2+] at the Mouth of a Calcium Channel. J. Neurosci. 1997, 17, 6961–6973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastián-Eugenio, C.E.; Bohórquez-Hernández, A.; Pacheco, J.; Sampieri, A.; Asanov, A.; Ocelotl-Oviedo, J.P.; Guerrero, A.; Darszon, A.; Vaca, L. Heterologous Calcium-Dependent Inactivation of Orai1 by Neighboring TRPV1 Channels Modulates Cell Migration and Wound Healing. Commun. Biol. 2019, 2, 88. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, X.; Mueller, G.A.; Sobhany, M.; DeRose, E.F.; Zhang, Y.; London, R.E.; Birnbaumer, L. Crystal Structure of Calmodulin Binding Domain of Orai1 in Complex with Ca2+ Calmodulin Displays a Unique Binding Mode. J. Biol. Chem. 2012, 287, 43030–43041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traxler, L.; Rathner, P.; Fahrner, M.; Stadlbauer, M.; Faschinger, F.; Charnavets, T.; Müller, N.; Romanin, C.; Hinterdorfer, P.; Gruber, H.J. Detailed Evidence for an Unparalleled Interaction Mode between Calmodulin and Orai Proteins. Angew. Chem. Int. Ed. Engl. 2017, 56, 15755–15759. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.C.; O’Connell, D.; Cahill, D.J.; Linse, S. Calmodulin Binding to the Polybasic C-Termini of STIM Proteins Involved in Store-Operated Calcium Entry. Biochemistry 2008, 47, 6089–6091. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.; Müller, H.-M.; Nickel, W.; Seedorf, M. Oligomerization and Ca2+/Calmodulin Control Binding of the ER Ca2+-Sensors STIM1 and STIM2 to Plasma Membrane Lipids. Biosci. Rep. 2013, 33. [Google Scholar] [CrossRef]
- Albarran, L.; Regodón, S.; Salido, G.M.; Lopez, J.J.; Rosado, J.A. Role of STIM1 in the Surface Expression of SARAF. Channels 2017, 11, 84–88. [Google Scholar] [CrossRef]
- Albarran, L.; Lopez, J.J.; Woodard, G.E.; Salido, G.M.; Rosado, J.A. Store-Operated Ca2+ Entry-Associated Regulatory Factor (SARAF) Plays an Important Role in the Regulation of Arachidonate-Regulated Ca2+ (ARC) Channels. J. Biol. Chem. 2016, 291, 6982–6988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albarran, L.; Lopez, J.J.; Gomez, L.J.; Salido, G.M.; Rosado, J.A. SARAF Modulates TRPC1, but Not TRPC6, Channel Function in a STIM1-Independent Manner. Biochem. J. 2016, BCJ20160348. [Google Scholar] [CrossRef]
- Lopez, E.; Frischauf, I.; Jardin, I.; Derler, I.; Muik, M.; Cantonero, C.; Salido, G.M.; Smani, T.; Rosado, J.A.; Redondo, P.C. STIM1 Phosphorylation at Y316 Modulates Its Interaction with SARAF and the Activation of SOCE and ICRAC. J. Cell. Sci. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimberlin, C.R.; Meshcheriakova, A.; Palty, R.; Raveh, A.; Karbat, I.; Reuveny, E.; Minor, D.L. SARAF Luminal Domain Structure Reveals a Novel Domain-Swapped β-Sandwich Fold Important for SOCE Modulation. J. Mol. Biol. 2019, 431, 2869–2883. [Google Scholar] [CrossRef] [PubMed]
- Albarran, L.; Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Berna-Erro, A.; Smani, T.; Camello, P.J.; Salido, G.M.; Rosado, J.A. EFHB Is a Novel Cytosolic Ca2+ Sensor That Modulates STIM1-SARAF Interaction. Cell. Physiol. Biochem. 2018, 51, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Albarran, L.; Lopez, J.J.; Amor, N.B.; Martin-Cano, F.E.; Berna-Erro, A.; Smani, T.; Salido, G.M.; Rosado, J.A. Dynamic Interaction of SARAF with STIM1 and Orai1 to Modulate Store-Operated Calcium Entry. Sci. Rep. 2016, 6, 24452. [Google Scholar] [CrossRef]
- Galeano-Otero, I.; Del Toro, R.; Khatib, A.-M.; Rosado, J.A.; Ordóñez-Fernández, A.; Smani, T. SARAF and Orai1 Contribute to Endothelial Cell Activation and Angiogenesis. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Ma, G.; He, L.; Liu, S.; Xie, J.; Huang, Z.; Jing, J.; Lee, Y.-T.; Wang, R.; Luo, H.; Han, W.; et al. Optogenetic Engineering to Probe the Molecular Choreography of STIM1-Mediated Cell Signaling. Nat. Commun. 2020, 11, 1039. [Google Scholar] [CrossRef]
- Grigoriev, I.; Gouveia, S.M.; van der Vaart, B.; Demmers, J.; Smyth, J.T.; Honnappa, S.; Splinter, D.; Steinmetz, M.O.; Putney, J.W.; Hoogenraad, C.C.; et al. STIM1 Is a MT-plus-End-Tracking Protein Involved in Remodeling of the ER. Curr. Biol. 2008, 18, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honnappa, S.; Gouveia, S.M.; Weisbrich, A.; Damberger, F.F.; Bhavesh, N.S.; Jawhari, H.; Grigoriev, I.; van Rijssel, F.J.A.; Buey, R.M.; Lawera, A.; et al. An EB1-Binding Motif Acts as a Microtubule Tip Localization Signal. Cell 2009, 138, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, J.; Fivaz, M.; Inoue, T.; Meyer, T. Live-Cell Imaging Reveals Sequential Oligomerization and Local Plasma Membrane Targeting of Stromal Interaction Molecule 1 after Ca2+ Store Depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 9301–9306. [Google Scholar] [CrossRef] [Green Version]
- Ercan, E.; Momburg, F.; Engel, U.; Temmerman, K.; Nickel, W.; Seedorf, M. A Conserved, Lipid-Mediated Sorting Mechanism of Yeast Ist2 and Mammalian STIM Proteins to the Peripheral ER. Traffic 2009, 10, 1802–1818. [Google Scholar] [CrossRef] [PubMed]
- Korzeniowski, M.K.; Popovic, M.A.; Szentpetery, Z.; Varnai, P.; Stojilkovic, S.S.; Balla, T. Dependence of STIM1/Orai1-Mediated Calcium Entry on Plasma Membrane Phosphoinositides. J. Biol. Chem. 2009, 284, 21027–21035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C.M.; Chvanov, M.; Haynes, L.P.; Petersen, O.H.; Tepikin, A.V.; Burgoyne, R.D. Role of Phosphoinositides in STIM1 Dynamics and Store-Operated Calcium Entry. Biochem. J. 2009, 425, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-J.; Chang, C.-L.; Lee, W.-R.; Liou, J. RASSF4 Controls SOCE and ER-PM Junctions through Regulation of PI(4,5)P2. J. Cell Biol. 2017, 216, 2011–2025. [Google Scholar] [CrossRef]
- Chang, C.-L.; Chen, Y.-J.; Quintanilla, C.G.; Hsieh, T.-S.; Liou, J. EB1 Binding Restricts STIM1 Translocation to ER-PM Junctions and Regulates Store-Operated Ca2+ Entry. J. Cell Biol. 2018, 217, 2047–2058. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Zhou, L.; Ma, G.; Zhang, T.; Liu, J.; Li, J.; Nguyen, N.T.; Zhang, X.; Li, W.; Nwokonko, R.; et al. Calcium Store Refilling and STIM Activation in STIM- and Orai-Deficient Cell Lines. Pflug. Arch. 2018, 470, 1555–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 Mediate CRAC Currents and Store-Operated Calcium Entry Important for Endothelial Cell Proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Dong, K.; Sun, R. STIM1 Regulates Endothelial Calcium Overload and Cytokine Upregulation During Sepsis. J. Surg. Res. 2021, 263, 236–244. [Google Scholar] [CrossRef]
- Zhang, W.; Muramatsu, A.; Matsuo, R.; Teranishi, N.; Kahara, Y.; Takahara, T.; Shibata, H.; Maki, M. The Penta-EF-Hand ALG-2 Protein Interacts with the Cytosolic Domain of the SOCE Regulator SARAF and Interferes with Ubiquitination. Int. J. Mol. Sci. 2020, 21, 6315. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, S.; Jung, H.-J.; Kim, K.-D.; Souda, P.; Whitelegge, J.; Gwack, Y. A Novel EF-Hand Protein, CRACR2A, Is a Cytosolic Ca2+ Sensor That Stabilizes CRAC Channels in T Cells. Nat. Cell Biol. 2010, 12, 436–446. [Google Scholar] [CrossRef] [Green Version]
- Mullins, F.M.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. STIM1 and Calmodulin Interact with Orai1 to Induce Ca2+-Dependent Inactivation of CRAC Channels. Proc. Natl. Acad. Sci. USA 2009, 106, 15495–15500. [Google Scholar] [CrossRef] [Green Version]
- Derler, I.; Jardin, I.; Stathopulos, P.B.; Muik, M.; Fahrner, M.; Zayats, V.; Pandey, S.K.; Poteser, M.; Lackner, B.; Absolonova, M.; et al. Cholesterol Modulates Orai1 Channel Function. Sci. Signal. 2016, 9, ra10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, J.; Dominguez, L.; Bohórquez-Hernández, A.; Asanov, A.; Vaca, L. A Cholesterol-Binding Domain in STIM1 Modulates STIM1-Orai1 Physical and Functional Interactions. Sci. Rep. 2016, 6, 29634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohórquez-Hernández, A.; Gratton, E.; Pacheco, J.; Asanov, A.; Vaca, L. Cholesterol Modulates the Cellular Localization of Orai1 Channels and Its Disposition among Membrane Domains. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2017, 1862, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Calloway, N.; Owens, T.; Corwith, K.; Rodgers, W.; Holowka, D.; Baird, B. Stimulated Association of STIM1 and Orai1 Is Regulated by the Balance of PtdIns(4,5)P2 between Distinct Membrane Pools. J. Cell Sci. 2011, 124, 2602–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosado, J.A.; Sage, S.O. Phosphoinositides Are Required for Store-Mediated Calcium Entry in Human Platelets. J. Biol. Chem. 2000, 275, 9110–9113. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Takahashi, R.; Zhang, X.X.; Kakizawa, H.; Hayashi, H.; Ohno, R. Inhibition of Agonist-Induced Ca2+ Entry in Endothelial Cells by Myosin Light-Chain Kinase Inhibitor. Biochem. Biophys. Res. Commun. 1996, 225, 777–784. [Google Scholar] [CrossRef]
- Broad, L.M.; Braun, F.J.; Lievremont, J.P.; Bird, G.S.; Kurosaki, T.; Putney, J.W. Role of the Phospholipase C-Inositol 1,4,5-Trisphosphate Pathway in Calcium Release-Activated Calcium Current and Capacitative Calcium Entry. J. Biol. Chem. 2001, 276, 15945–15952. [Google Scholar] [CrossRef] [Green Version]
- Heo, W.D.; Inoue, T.; Park, W.S.; Kim, M.L.; Park, B.O.; Wandless, T.J.; Meyer, T. PI(3,4,5)P3 and PI(4,5)P2 Lipids Target Proteins with Polybasic Clusters to the Plasma Membrane. Science 2006, 314, 1458–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.K.; Kim, O.-H.; Hur, J.; Yu, S.H.; Lamichhane, S.; Lee, J.W.; Ojha, U.; Hong, J.H.; Lee, C.S.; Cha, J.-Y.; et al. Increased Intracellular Ca2+ Concentrations Prevent Membrane Localization of PH Domains through the Formation of Ca2+-Phosphoinositides. Proc. Natl. Acad. Sci. USA 2017, 114, 11926–11931. [Google Scholar] [CrossRef] [Green Version]
- Bilkova, E.; Pleskot, R.; Rissanen, S.; Sun, S.; Czogalla, A.; Cwiklik, L.; Róg, T.; Vattulainen, I.; Cremer, P.S.; Jungwirth, P.; et al. Calcium Directly Regulates Phosphatidylinositol 4,5-Bisphosphate Headgroup Conformation and Recognition. J. Am. Chem. Soc. 2017, 139, 4019–4024. [Google Scholar] [CrossRef]
- Sarmento, M.J.; Coutinho, A.; Fedorov, A.; Prieto, M.; Fernandes, F. Ca2+ Induces PI(4,5)P2 Clusters on Lipid Bilayers at Physiological PI(4,5)P2 and Ca2+ Concentrations. Biochim. Biophys. Acta 2014, 1838, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Graber, Z.T.; Shi, Z.; Baumgart, T. Cations Induce Shape Remodeling of Negatively Charged Phospholipid Membranes. Phys. Chem. Chem. Phys. 2017, 19, 15285–15295. [Google Scholar] [CrossRef] [PubMed]
- Avezov, E.; Cross, B.C.S.; Kaminski Schierle, G.S.; Winters, M.; Harding, H.P.; Melo, E.P.; Kaminski, C.F.; Ron, D. Lifetime Imaging of a Fluorescent Protein Sensor Reveals Surprising Stability of ER Thiol Redox. J. Cell. Biol. 2013, 201, 337–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enyedi, B.; Várnai, P.; Geiszt, M. Redox State of the Endoplasmic Reticulum Is Controlled by Ero1L-Alpha and Intraluminal Calcium. Antioxid. Redox Signal. 2010, 13, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Birk, J.; Meyer, M.; Aller, I.; Hansen, H.G.; Odermatt, A.; Dick, T.P.; Meyer, A.J.; Appenzeller-Herzog, C. Endoplasmic Reticulum: Reduced and Oxidized Glutathione Revisited. J. Cell. Sci. 2013, 126, 1604–1617. [Google Scholar] [CrossRef] [Green Version]
- Kalidhindi, R.S.R.; Katragadda, R.; Beauchamp, K.L.; Pabelick, C.M.; Prakash, Y.S.; Sathish, V. Androgen Receptor-Mediated Regulation of Intracellular Calcium in Human Airway Smooth Muscle Cells. Cell Physiol. Biochem. 2019, 53, 215–228. [Google Scholar] [CrossRef]
- Berry, P.A.; Birnie, R.; Droop, A.P.; Maitland, N.J.; Collins, A.T. The Calcium Sensor STIM1 Is Regulated by Androgens in Prostate Stromal Cells. Prostate 2011, 71, 1646–1655. [Google Scholar] [CrossRef]
- Liu, G.; Honisch, S.; Liu, G.; Schmidt, S.; Alkahtani, S.; AlKahtane, A.A.; Stournaras, C.; Lang, F. Up-Regulation of Orai1 Expression and Store Operated Ca2+ Entry Following Activation of Membrane Androgen Receptors in MCF-7 Breast Tumor Cells. BMC Cancer 2015, 15, 995. [Google Scholar] [CrossRef] [Green Version]
- Romanuik, T.L.; Ueda, T.; Le, N.; Haile, S.; Yong, T.M.K.; Thomson, T.; Vessella, R.L.; Sadar, M.D. Novel Biomarkers for Prostate Cancer Including Noncoding Transcripts. Am. J. Pathol. 2009, 175, 2264–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanuik, T.L.; Wang, G.; Holt, R.A.; Jones, S.J.M.; Marra, M.A.; Sadar, M.D. Identification of Novel Androgen-Responsive Genes by Sequencing of LongSAGE Libraries. BMC Genom. 2009, 10, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, A.; Ahuja, M.; Schwartz, D.M.; Varga, A.; Swaim, W.; Kang, N.; Maleth, J.; Shin, D.M.; Muallem, S. Ca2+ Influx Channel Inhibitor SARAF Protects Mice From Acute Pancreatitis. Gastroenterology 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iżykowska, K.; Przybylski, G.K.; Gand, C.; Braun, F.C.; Grabarczyk, P.; Kuss, A.W.; Olek-Hrab, K.; Bastidas Torres, A.N.; Vermeer, M.H.; Zoutman, W.H.; et al. Genetic Rearrangements Result in Altered Gene Expression and Novel Fusion Transcripts in Sézary Syndrome. Oncotarget 2017, 8, 39627–39639. [Google Scholar] [CrossRef] [Green Version]
- Taha, S.; Aljishi, M.; Alsharoqi, I.; Bakhiet, M. Differential Upregulation of the Hypothetical Transmembrane Protein 66 (TMEM66) in Multiple Sclerosis Patients with Potential Inflammatory Response. Biomed. Rep. 2015, 3, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Twine, N.A.; Janitz, K.; Wilkins, M.R.; Janitz, M. Whole Transcriptome Sequencing Reveals Gene Expression and Splicing Differences in Brain Regions Affected by Alzheimer’s Disease. PLoS ONE 2011, 6, e16266. [Google Scholar] [CrossRef]
- Yang, J.; Li, S.; Wang, Q.; Yang, D. Transmembrane Protein 66 Attenuates Neointimal Hyperplasia after Carotid Artery Injury by SOCE Inactivation. Mol. Med. Rep. 2019. [Google Scholar] [CrossRef]
- Dai, F.; Zhang, Y.; Wang, Q.; Li, D.; Yang, Y.; Ma, S.; Yang, D. Overexpression of SARAF Ameliorates Pressure Overload-Induced Cardiac Hypertrophy Through Suppressing STIM1-Orai1 in Mice. Cell. Physiol. Biochem. 2018, 47, 817–826. [Google Scholar] [CrossRef]
- Camargo, A.; Azuaje, F. Identification of Dilated Cardiomyopathy Signature Genes through Gene Expression and Network Data Integration. Genomics 2008, 92, 404–413. [Google Scholar] [CrossRef] [Green Version]
- Sanlialp, A.; Schumacher, D.; Kiper, L.; Varma, E.; Riechert, E.; Ho, T.C.; Hofmann, C.; Kmietczyk, V.; Zimmermann, F.; Dlugosz, S.; et al. Saraf-Dependent Activation of MTORC1 Regulates Cardiac Growth. J. Mol. Cell. Cardiol. 2020. [Google Scholar] [CrossRef]
- La Russa, D.; Frisina, M.; Secondo, A.; Bagetta, G.; Amantea, D. Modulation of Cerebral Store-Operated Calcium Entry-Regulatory Factor (SARAF) and Peripheral Orai1 Following Focal Cerebral Ischemia and Preconditioning in Mice. Neuroscience 2020. [Google Scholar] [CrossRef] [PubMed]
- Bulla, M.; Gyimesi, G.; Kim, J.H.; Bhardwaj, R.; Hediger, M.A.; Frieden, M.; Demaurex, N. ORAI1 Channel Gating and Selectivity Is Differentially Altered by Natural Mutations in the First or Third Transmembrane Domain. J. Physiol. 2019, 597, 561–582. [Google Scholar] [CrossRef] [Green Version]
- Hulot, J.-S.; Fauconnier, J.; Ramanujam, D.; Chaanine, A.; Aubart, F.; Sassi, Y.; Merkle, S.; Cazorla, O.; Ouillé, A.; Dupuis, M.; et al. Critical Role for Stromal Interaction Molecule 1 in Cardiac Hypertrophy. Circulation 2011, 124, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Correll, R.N.; Goonasekera, S.A.; van Berlo, J.H.; Burr, A.R.; Accornero, F.; Zhang, H.; Makarewich, C.A.; York, A.J.; Sargent, M.A.; Chen, X.; et al. STIM1 Elevation in the Heart Results in Aberrant Ca2+ Handling and Cardiomyopathy. J. Mol. Cell Cardiol. 2015, 87, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.-H.; Zheng, H.; Si, H.; Jin, Y.; Peng, J.M.; He, L.; Zhou, Y.; Muñoz-Garay, C.; Zawieja, D.C.; Kuo, L.; et al. Stromal Interaction Molecule 1 (STIM1) and Orai1 Mediate Histamine-Evoked Calcium Entry and Nuclear Factor of Activated T-Cells (NFAT) Signaling in Human Umbilical Vein Endothelial Cells. J. Biol. Chem. 2014, 289, 29446–29456. [Google Scholar] [CrossRef] [Green Version]
- Gerasimenko, J.V.; Gerasimenko, O.V.; Petersen, O.H. The Role of Ca2+ in the Pathophysiology of Pancreatitis. J. Physiol. 2014, 592, 269–280. [Google Scholar] [CrossRef]
- Eisenhut, M.; Wallace, H. Ion Channels in Inflammation. Pflug. Arch 2011, 461, 401–421. [Google Scholar] [CrossRef]
- Wen, L.; Voronina, S.; Javed, M.A.; Awais, M.; Szatmary, P.; Latawiec, D.; Chvanov, M.; Collier, D.; Huang, W.; Barrett, J.; et al. Inhibitors of ORAI1 Prevent Cytosolic Calcium-Associated Injury of Human Pancreatic Acinar Cells and Acute Pancreatitis in 3 Mouse Models. Gastroenterology 2015, 149, 481–492.e7. [Google Scholar] [CrossRef] [Green Version]
- Gerasimenko, J.V.; Gryshchenko, O.; Ferdek, P.E.; Stapleton, E.; Hébert, T.O.G.; Bychkova, S.; Peng, S.; Begg, M.; Gerasimenko, O.V.; Petersen, O.H. Ca2+ Release-Activated Ca2+ Channel Blockade as a Potential Tool in Antipancreatitis Therapy. Proc. Natl. Acad Sci. USA 2013, 110, 13186–13191. [Google Scholar] [CrossRef] [Green Version]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 Mediate SOCE and Contribute to Apoptotic Resistance of Pancreatic Adenocarcinoma. Biochim. Biophys. Acta 2014, 1843, 2263–2269. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Hong, J.H.; Li, Q.; Shin, D.M.; Abramowitz, J.; Birnbaumer, L.; Muallem, S. Deletion of TRPC3 in Mice Reduces Store-Operated Ca2+ Influx and the Severity of Acute Pancreatitis. Gastroenterology 2009, 137, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Crottès, D.; Lin, Y.-H.T.; Peters, C.J.; Gilchrist, J.M.; Wiita, A.P.; Jan, Y.N.; Jan, L.Y. TMEM16A Controls EGF-Induced Calcium Signaling Implicated in Pancreatic Cancer Prognosis. Proc. Natl. Acad. Sci. USA 2019, 116, 13026–13035. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Zeng, W.; Kiselyov, K.; Yuan, J.P.; Dehoff, M.H.; Mikoshiba, K.; Worley, P.F.; Muallem, S. Homer 1 Mediates Store- and Inositol 1,4,5-Trisphosphate Receptor-Dependent Translocation and Retrieval of TRPC3 to the Plasma Membrane. J. Biol. Chem. 2006, 281, 32540–32549. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 Heteromultimerizes TRPC Channels to Determine Their Function as Store-Operated Channels. Nat. Cell Biol. 2007, 9, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Shim, K.-N.; Li, J.M.J.; Estrema, C.; Ornelas, T.A.; Nguyen, F.; Liu, S.; Ramamoorthy, S.L.; Ho, S.; Carethers, J.M.; et al. Molecular Mechanisms Underlying Ca2+-Mediated Motility of Human Pancreatic Duct Cells. Am. J. Physiol. Cell Physiol. 2010, 299, C1493–C1503. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, A.M.; Trebak, M. Orai Channel-Mediated Ca2+ Signals in Vascular and Airway Smooth Muscle. Am. J. Physiol. Cell Physiol. 2016, 310, C402–C413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-M.; Xu, W.-J.; Xiang, L.-L.; Ding, M.; Zhang, J.-J.; Lu, J.-Y.; Xie, B.-J.; Gao, Y.-D. Store-Operated Calcium Entry-Associated Regulatory Factor Regulates Airway Inflammation and Airway Remodeling in Asthma Mice Models. Am. J. Physiol. Lung Cell Mol. Physiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.; di Capite, J.; Singaravelu, K.; Parekh, A.B. Sustained Activation of the Tyrosine Kinase Syk by Antigen in Mast Cells Requires Local Ca2+ Influx through Ca2+ Release-Activated Ca2+ Channels. J. Biol. Chem. 2008, 283, 31348–31355. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| SCDI Properties | Jurkat T-lymphocytes | Rat Basophilic Leukemia Cells | Human Embryonic Kidney Cells |
|---|---|---|---|
| Distance from CRAC channel | ~10–50 nm [59] or >100 nm [53] | >100 nm [54,60] | ~10–50 nm [57,61] |
| Ca2+ IC50 | ? | ~0.5 µM [62] | ~0.2 µM [61] |
| Recovery from inactivation | Full current recovery [53] | No current recovery [54] | Intermediate current recovery [52,61] |
| Role of regulators | Promoted by SARAF [52] and ORMDL3 [55] | Insensitive to CaM [54] | Promoted by SARAF [37,52,63] and CaM [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dagan, I.; Palty, R. Regulation of Store-Operated Ca2+ Entry by SARAF. Cells 2021, 10, 1887. https://doi.org/10.3390/cells10081887
Dagan I, Palty R. Regulation of Store-Operated Ca2+ Entry by SARAF. Cells. 2021; 10(8):1887. https://doi.org/10.3390/cells10081887
Chicago/Turabian StyleDagan, Inbal, and Raz Palty. 2021. "Regulation of Store-Operated Ca2+ Entry by SARAF" Cells 10, no. 8: 1887. https://doi.org/10.3390/cells10081887
APA StyleDagan, I., & Palty, R. (2021). Regulation of Store-Operated Ca2+ Entry by SARAF. Cells, 10(8), 1887. https://doi.org/10.3390/cells10081887