
Cumulative Damage: Cell Death in Posthemorrhagic Hydrocephalus of Prematurity

 ,
, 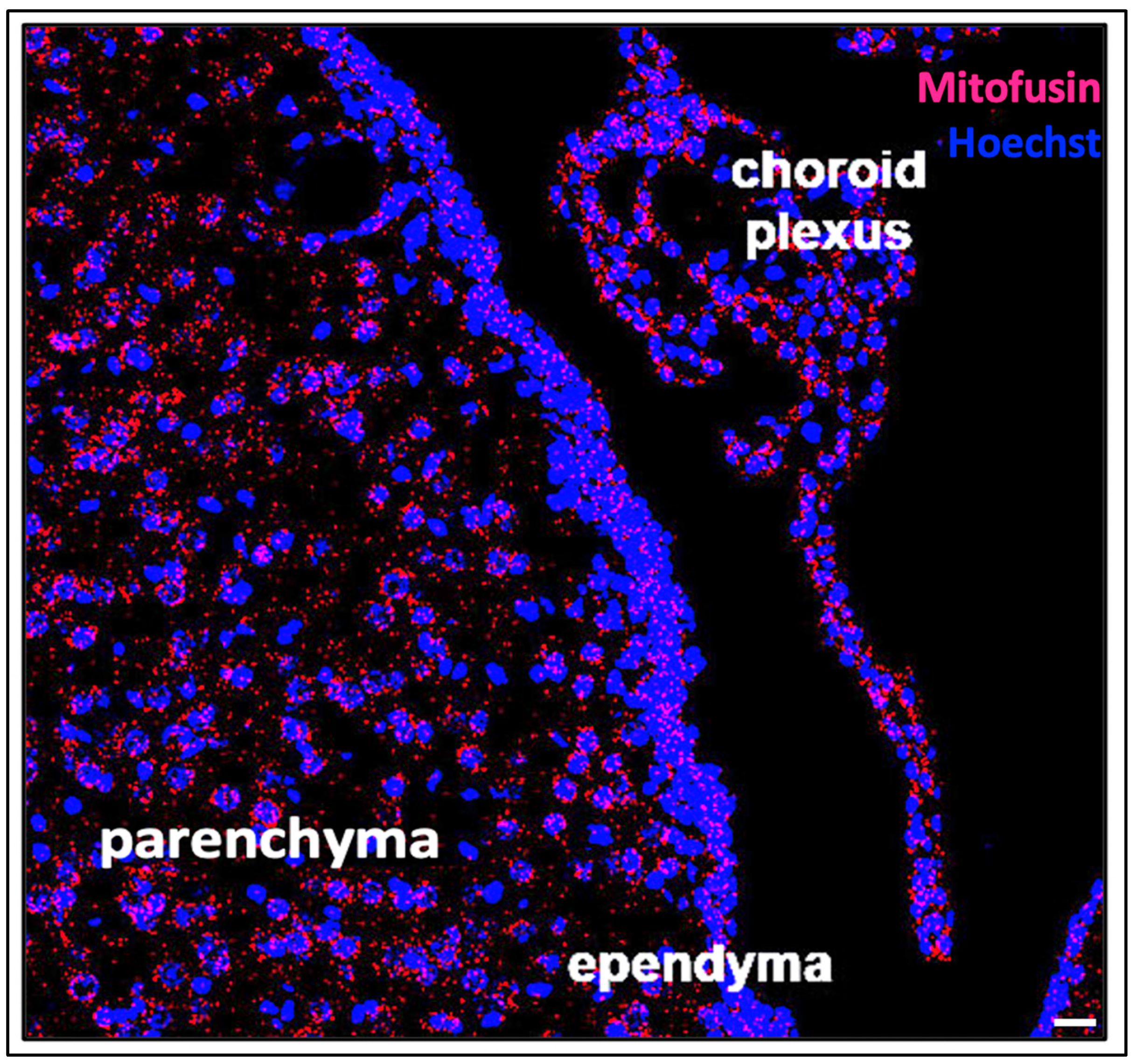{kind=link}
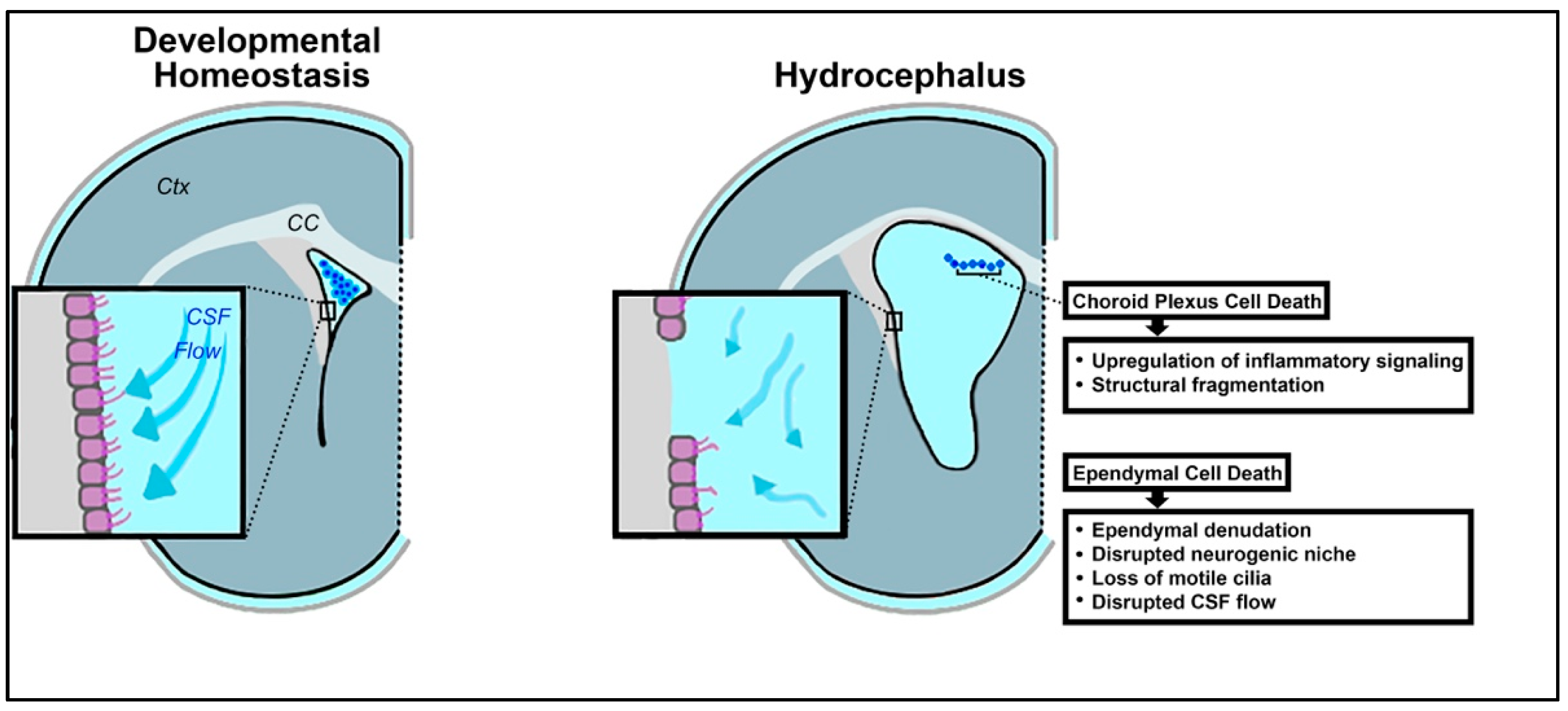{kind=link}
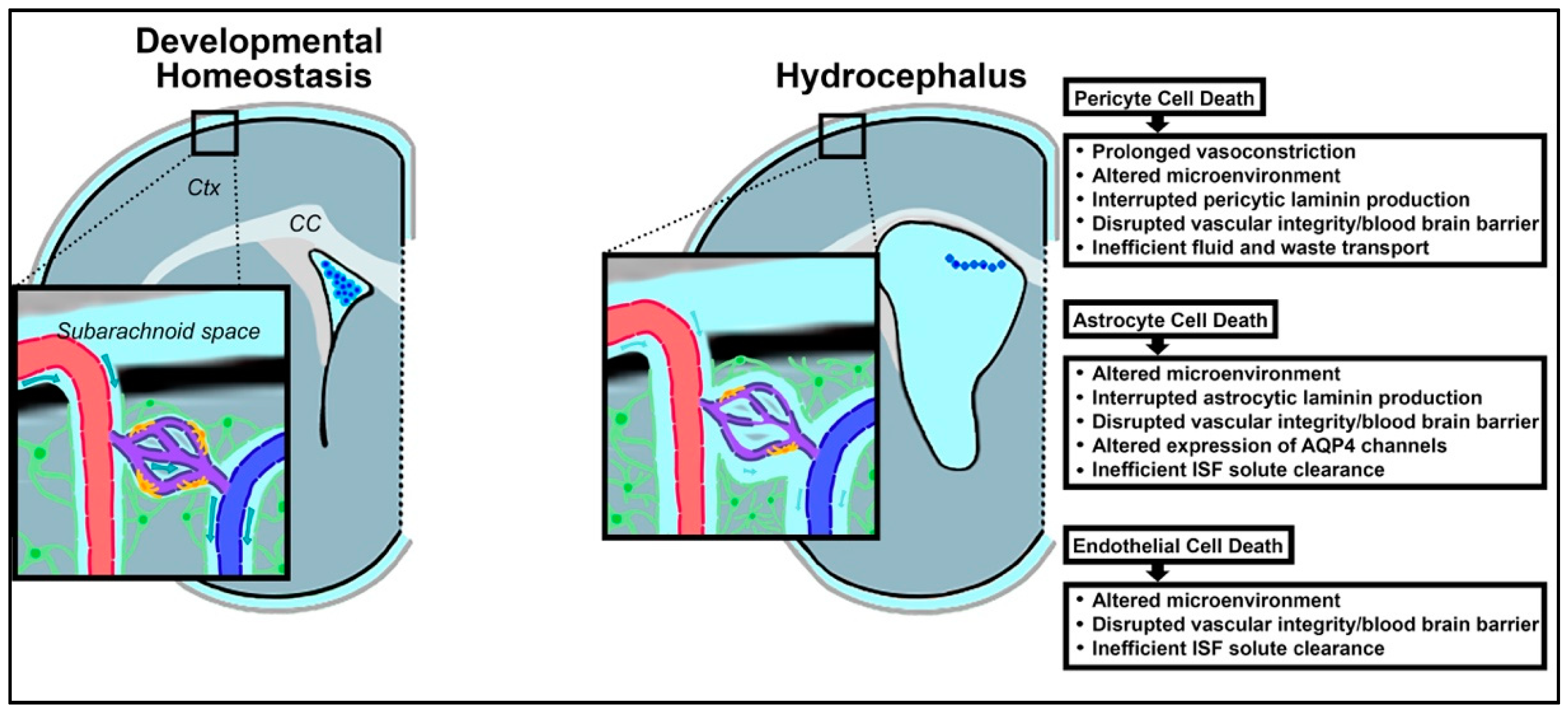{kind=link}
Abstract
:1. Introduction
1.1. A Broad Introduction to Infantile Hydrocephalus
1.2. Encephalopathy of Prematurity
1.3. Chorioamnionitis as a Driver of Dysfunction through the Maternal–Placental–Fetal Axis
1.4. Posthemorrhagic Hydrocephalus of Prematurity as a Severe Manifestation
1.5. Other Forms of Hydrocephalus
1.6. A Focus on Cell Death
2. Cell Death in the Choroid Plexus
2.1. Cellular Structure of the Choroid Plexus
2.2. Mechanisms of Choroidal Cell Death
3. Cell Death in the Ependyma
3.1. Cellular Structure of the Ependyma
3.2. Mechanisms of Ependymal Cell Death
3.2.1. Ependymal Cell Death Precipitates Hydrocephalus
3.2.2. Ependymal Cell Death Exacerbates Hydrocephalus
3.3. Downstream Impacts of Ependymal Cell Loss
4. Cell Death in the Glymphatic System
4.1. Cellular Structure of the Glymphatic System
4.2. Mechanisms of Glymphatic Cell Death
4.2.1. Pericytes
4.2.2. Astrocytes
4.2.3. Endothelial Cells
5. Cell Death via Ferroptosis
5.1. Ferroptosis Described
5.2. The Role of Ferroptosis in PHHP
6. Cell Death in White Matter
7. Cell Death in Other Neural Components
8. Discussion of Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tully, H.M.; Dobyns, W. Infantile hydrocephalus: A review of epidemiology, classification and causes. Eur. J. Med. Genet. 2014, 57, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Leinonen, V.; Vanninen, R.; Rauramaa, T. Cerebrospinal fluid circulation and hydrocephalus. Handb Clin Neurol. 2017, 145, 39–50. [Google Scholar] [CrossRef]
- Lummis, N.C.; Sánchez-Pavón, P.; Kennedy, G.; Frantz, A.J.; Kihara, Y.; Blaho, V.A.; Chun, J. LPA1/3 overactivation induces neonatal posthemorrhagic hydrocephalus through ependymal loss and ciliary dysfunction. Sci. Adv. 2019, 5, eaax2011. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.J.; Sheinberg, D.L.; Luther, E.; McCrea, H.J. Myelomeningocele-associated hydrocephalus: Nationwide analysis and systematic review. Neurosurg. Focus 2019, 47, E5. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.C.; Rattani, A.; Mekary, R.; Glancz, L.J.; Yunusa, I.; Baticulon, R.; Fieggen, G.; Wellons, J.C.; Park, K.B.; Warf, B.C. Global hydrocephalus epidemiology and incidence: Systematic review and meta-analysis [published online ahead of print, 2018 Apr 1]. J Neurosurg. 2018, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Feng, Z.; Tan, Q.; Guo, J.; Tang, J.; Tan, L.; Feng, H.; Chen, Z. Post-hemorrhagic hydrocephalus: Recent advances and new therapeutic insights. J. Neurol. Sci. 2017, 375, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S. Neonatal posthemorrhagic hydrocephalus from prematurity: Pathophysiology and current treatment concepts. J. Neurosurg. Pediatr. 2012, 9, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Shooman, D.; Portess, H.; Sparrow, O. A review of the current treatment methods for posthaemorrhagic hydrocephalus of infants. Cereb. Fluid Res. 2009, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Whitelaw, A.; Aquilina, K. Management of posthaemorrhagic ventricular dilatation. Arch. Dis. Child. Fetal Neonatal Ed. 2012, 97, F229–F233. [Google Scholar] [CrossRef]
- Riva-Cambrin, J.; Shannon, C.N.; Holubkov, R.; Whitehead, W.E.; Kulkarni, A.V.; Drake, J.; Simon, T.D.; Browd, S.R.; Kestle, J.R.W.; Wellons, J.C. Center effect and other factors influencing temporization and shunting of cerebrospinal fluid in preterm infants with intraventricular hemorrhage. J. Neurosurg. Pediatr. 2012, 9, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, S.; Kaufman, B.A.; Park, T. Outcome Analysis of Initial Neonatal Shunts: Does the Valve Make a Difference? Pediatr. Neurosurg. 2002, 37, 287–294. [Google Scholar] [CrossRef]
- Deopujari, C.E.; Karmarkar, V.; Shaikh, S. Endoscopic Third Ventriculostomy: Success and Failure. J. Korean Neurosurg. Soc. 2017, 60, 306–314. [Google Scholar] [CrossRef]
- Whitelaw, A.; Lee-Kelland, R. Repeated lumbar or ventricular punctures in newborns with intraventricular haemorrhage. Cochrane Database Syst. Rev. 2017, 2017, CD000216. [Google Scholar] [CrossRef] [PubMed]
- Vogel, T.W.; Bahuleyan, B.; Robinson, S.; Cohen, A.R. The role of endoscopic third ventriculostomy in the treatment of hydrocephalus. J. Neurosurg. Pediatr. 2013, 12, 54–61. [Google Scholar] [CrossRef]
- Riva-Cambrin, J.; Kestle, J.R.W.; Rozzelle, C.J.; Naftel, R.P.; Alvey, J.S.; Reeder, R.W.; Holubkov, R.; Browd, S.R.; Cochrane, D.; Limbrick, D.D.; et al. Predictors of success for combined endoscopic third ventriculostomy and choroid plexus cauterization in a North American setting: A Hydrocephalus Clinical Research Network study. J. Neurosurg. Pediatr. 2019, 24, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Kulkarni, A.V.; Limbrick, D.D.; Warf, B.C. Hydrocephalus in children. Lancet 2016, 387, 788–799. [Google Scholar] [CrossRef]
- Liu, L.; Oza, S.; Hogan, D.; Chu, Y.; Perin, J.; Zhu, J.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of under-5 mortality in 2000–2015: An updated systematic analysis with implications for the Sustainable Development Goals. Lancet 2016, 388, 3027–3035. [Google Scholar] [CrossRef] [Green Version]
- Blencowe, H.; Cousens, S.; Chou, D.; Oestergaard, M.; Say, L.; Moller, A.-B.; Kinney, M.; Lawn, J. The Born Too Soon Preterm Birth Action Group (see acknowledgement for full list). Born Too Soon: The global epidemiology of 15 million preterm births. Reprod. Health 2013, 10, S2. [Google Scholar] [CrossRef] [Green Version]
- Fleiss, B.; Gressens, P.; Stolp, H.B. Cortical gray matter injury in encephalopathy of prematurity: Link to neurodevelopmental disorders. Front. Neurol. 2020, 11, 575. [Google Scholar] [CrossRef]
- Marlow, N.; Wolke, D.; Bracewell, M.A.; Samara, M.; EPICure Study Group. Neurologic and Developmental Disability at Six Years of Age after Extremely Preterm Birth. N. Engl. J. Med. 2005, 352, 9–19. [Google Scholar] [CrossRef]
- O’Shea, T.; Allred, E.; Dammann, O.; Hirtz, D.; Kuban, K.; Paneth, N.; Leviton, A. The ELGAN study of the brain and related disorders in extremely low gestational age newborns. Early Hum. Dev. 2009, 85, 719–725. [Google Scholar] [CrossRef] [Green Version]
- Pierrat, V.; Marchand-Martin, L.; Arnaud, C.; Kaminski, M.; Resche-Rigon, M.; Lebeaux, C.; Bodeau-Livinec, F.; Morgan, A.; Goffinet, F.; Marret, S.; et al. Neurodevelopmental outcome at 2 years for preterm children born at 22 to 34 weeks’ gestation in France in 2011: EPIPAGE-2 cohort study. BMJ 2017, 358, j3448. [Google Scholar] [CrossRef] [Green Version]
- Gunn, A.J.; Thoresen, M. Neonatal encephalopathy and hypoxic–ischemic encephalopathy. Handb. Clin. Neurol. 2019, 162, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Jantzie, L.L.; Robinson, S. Preclinical Models of Encephalopathy of Prematurity. Dev. Neurosci. 2015, 37, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Volpe, J.J. Encephalopathy of prematurity includes neuronal abnormalities. Pediatrics 2005, 116, 221–225. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, T.M.; Shah, B.; Allred, E.N.; Fichorova, R.N.; Kuban, K.; Dammann, O.; Leviton, A. Inflammation-initiating illnesses, inflammation-related proteins, and cognitive impairment in extremely preterm infants. Brain Behav. Immun. 2013, 29, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Orchinik, L.J.; Taylor, H.G.; Espy, K.A.; Minich, N.; Klein, N.; Sheffield, T.; Hack, M. Cognitive Outcomes for Extremely Preterm/Extremely Low Birth Weight Children in Kindergarten. J. Int. Neuropsychol. Soc. 2011, 17, 1067–1079. [Google Scholar] [CrossRef] [Green Version]
- Ortinau, C.; Neil, J. The neuroanatomy of prematurity: Normal brain development and the impact of preterm birth. Clin. Anat. 2015, 28, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Galinsky, R.; Polglase, G.; Hooper, S.B.; Black, M.J.; Moss, T. The Consequences of Chorioamnionitis: Preterm Birth and Effects on Development. J. Pregnancy 2013, 2013, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.-C.; Chang, J.-H.; Lin, H.-Y.; Cheng, P.-J.; Su, B.-H. Intrauterine inflammation, infection, or both (Triple I): A new concept for chorioamnionitis. Pediatr. Neonatol. 2018, 59, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Su, B.-H. Histological Chorioamnionitis and Neonatal Outcome in Preterm Infants. Pediatr. Neonatol. 2014, 55, 154–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastek, J.A.; Weber, A.L.; McShea, M.A.; Ryan, M.E.; Elovitz, M.A. Prenatal inflammation is associated with adverse neonatal outcomes. Am. J. Obstet. Gynecol. 2014, 210, 450.e1–450.e10. [Google Scholar] [CrossRef]
- Yoon, B.H.; Romero, R.; Bin Moon, J.; Shim, S.-S.; Kim, M.; Kim, G.; Jun, J.K. Clinical significance of intra-amniotic inflammation in patients with preterm labor and intact membranes. Am. J. Obstet. Gynecol. 2001, 185, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Blume, H.K.; Li, C.I.; Loch, C.M.; Koepsell, T.D. Intrapartum fever and chorioamnionitis as risks for encephalopathy in term newborns: A case-control study. Dev. Med. Child Neurol. 2008, 50, 19–24. [Google Scholar] [CrossRef]
- Seong, H.S.; Lee, S.E.; Kang, J.H.; Romero, R.; Yoon, B.H. The frequency of microbial invasion of the amniotic cavity and histologic chorioamnionitis in women at term with intact membranes in the presence or absence of labor. Am. J. Obstet. Gynecol. 2008, 199, 375.e1–375.e5. [Google Scholar] [CrossRef] [Green Version]
- Tita, A.T.; Andrews, W.W. Diagnosis and Management of Clinical Chorioamnionitis. Clin. Perinatol. 2010, 37, 339–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, R.S.; Duff, P. Progress in pathogenesis and management of clinical intraamniotic infection. Am. J. Obstet. Gynecol. 1991, 164, 1317–1326. [Google Scholar] [CrossRef]
- Moscuzza, F.; Belcari, F.; Nardini, V.; Bartoli, A.; Domenici, C.; Cuttano, A.; Ghirri, P.; Boldrini, A. Correlation between placental histopathology and fetal/neonatal outcome: Chorioamnionitis and funisitis are associated to intraventricular haemorrage and retinopathy of prematurity in preterm newborns. Gynecol. Endocrinol. 2010, 27, 319–323. [Google Scholar] [CrossRef]
- Salas, A.; Faye-Petersen, O.M.; Sims, B.; Peralta-Carcelen, M.; Reilly, S.D.; McGwin, G.; Carlo, W.A.; Ambalavanan, N. Histological Characteristics of the Fetal Inflammatory Response Associated with Neurodevelopmental Impairment and Death in Extremely Preterm Infants. J. Pediatr. 2013, 163, 652–657.e2. [Google Scholar] [CrossRef] [Green Version]
- Arayici, S.; Simsek, G.K.; Öncel, M.Y.; Eras, Z.; Canpolat, F.E.; Oguz, S.S.; Uras, N.; Zergeroglu, S.; Dilmen, U. The effect of histological chorioamnionitis on the short-term outcome of preterm infants ≤32 weeks: A single-center study. J. Matern. Fetal Neonatal Med. 2014, 27, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.; Conteh, F.S.; Oppong, A.Y.; Yellowhair, T.R.; Newville, J.C.; El Demerdash, N.; Shrock, C.L.; Maxwell, J.R.; Jett, S.; Northington, F.J.; et al. Extended Combined Neonatal Treatment With Erythropoietin Plus Melatonin Prevents Posthemorrhagic Hydrocephalus of Prematurity in Rats. Front. Cell. Neurosci. 2018, 12, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorner, R.A.; Burton, V.J.; Allen, M.C.; Robinson, S.; Soares, B.P. Preterm neuroimaging and neurodevelopmental outcome: A focus on intraventricular hemorrhage, post-hemorrhagic hydrocephalus, and associated brain injury. J. Perinatol. 2018, 38, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Du Plessis, A.J. The Role of Systemic Hemodynamic Disturbances in Prematurity-Related Brain Injury. J. Child Neurol. 2009, 24, 1127–1140. [Google Scholar] [CrossRef] [Green Version]
- Bangma, J.T.; Hartwell, H.; Santos, H.P., Jr.; O’Shea, T.M.; Fry, R.C. Placental programming, perinatal inflammation, and neurodevelopment impairment among those born extremely preterm. Pediatr. Res. 2021, 89, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Alan, N.; Manjila, S.; Minich, N.; Bass, N.; Cohen, A.R.; Walsh, M.; Robinson, S. Reduced ventricular shunt rate in very preterm infants with severe intraventricular hemorrhage: An institutional experience. J. Neurosurg. Pediatr. 2012, 10, 357–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Bigio, M.R.; Di Curzio, D.L. Nonsurgical therapy for hydrocephalus: A comprehensive and critical review. Fluids Barriers CNS 2015, 13, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, A.L. The Impact of Prematurity on Social and Emotional Development. Clin. Perinatol. 2018, 45, 547–555. [Google Scholar] [CrossRef]
- Agajany, N.; Gigi, M.; Ross, J.; Roth, J.; Eshel, R.; Constantini, S.; Bassan, H. The impact of neonatal posthemorrhagic hydrocephalus of prematurity on family function at preschool age. Early Hum. Dev. 2019, 137, 104827. [Google Scholar] [CrossRef]
- Dorner, R.A.; Boss, R.D.; Burton, V.J.; Raja, K.; Robinson, S.; Lemmon, M.E. Isolated and On Guard: Preparing Neonatal Intensive Care Unit Families for Life with Hydrocephalus. Am. J. Perinatol. 2021. [Google Scholar] [CrossRef]
- Estey, C.M. Congenital Hydrocephalus. Veter Clin. N. Am. Small Anim. Pr. 2016, 46, 217–229. [Google Scholar] [CrossRef] [PubMed]
- McKnight, I.; Hart, C.; Park, I.-H.; Shim, J.W. Genes causing congenital hydrocephalus: Their chromosomal characteristics of telomere proximity and DNA compositions. Exp. Neurol. 2021, 335, 113523. [Google Scholar] [CrossRef]
- Malagón-Valdez, J. Hidrocefalia congénita [Congenital hydrocephalus]. Rev. Neurol. 2006, 42 (Suppl. 3), S39–S44. [Google Scholar]
- Rajshekhar, V. Management of hydrocephalus in patients with tuberculous meningitis. Neurol. India 2009, 57, 368–374. [Google Scholar] [CrossRef] [Green Version]
- Huo, L.; Fan, Y.; Jiang, C.; Gao, J.; Yin, M.; Wang, H.; Yang, F.; Cao, Q. Clinical Features of and Risk Factors for Hydrocephalus in Childhood Bacterial Meningitis. J. Child Neurol. 2018, 34, 11–16. [Google Scholar] [CrossRef]
- Chiang, S.S.; Khan, F.A.; Milstein, M.B.; Tolman, A.W.; Benedetti, A.; Starke, J.R.; Becerra, M.C. Treatment outcomes of childhood tuberculous meningitis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 947–957. [Google Scholar] [CrossRef]
- Yellowhair, T.R.; Oppong, A.Y.; Maxwell, J.R. The Unifying Effects of Maternal-Placental-Fetal Axis Dysregulation on Neurode-velopment Following Infectious and Toxic In Utero Insults. Med. Res. Arch. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.K.; Arman, D.; Roy, H.A.; Khan, R.K.; Afreen, S.; Hossain, M.A.; Ziauddin, M.; Ekramullah, S.M.; Rahman, M.; Yusuf, A.; et al. Status of ToRCH positivity among the children presented with congenital Hydrocephalus. Bangladesh J. Neurosurg. 2020, 9, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Waldorf, K.M.A.; McAdams, R.M. Influence of infection during pregnancy on fetal development. Reproduction 2013, 146, R151–R162. [Google Scholar] [CrossRef] [Green Version]
- Muir, R.; Wang, S.; Warf, B.C. Global surgery for pediatric hydrocephalus in the developing world: A review of the history, challenges, and future directions. Neurosurg. Focus 2016, 41, E11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragheb, M.; Shah, A.H.; Jernigan, S.; Koru-Sengul, T.; Ragheb, J. Epidemiology of pediatric hydrocephalus in Haiti: Analysis of a surgical case series. J. Neurosurg. Pediatr. 2019, 23, 568–576. [Google Scholar] [CrossRef]
- Dandy, W.E.; Blackfan, K.D. Internal hydrocephalus: An experimental, clinical and pathological study. Am. J. Dis. Child. 1914, VIII, 406. [Google Scholar] [CrossRef] [Green Version]
- Beyerl, B.; Black, P.M. Posttraumatic Hydrocephalus. Neurosurgery 1984, 15, 257–261. [Google Scholar] [CrossRef]
- Robinson, S.; Northington, F.J.; Jantzie, L.L. A time for cocktails and inclusion. Neural Regen. Res. 2018, 13, 987–988. [Google Scholar] [CrossRef]
- Paredes, I.; Himmels, P.; de Almodóvar, C.R. Neurovascular Communication during CNS Development. Dev. Cell 2018, 45, 10–32. [Google Scholar] [CrossRef] [Green Version]
- Belousov, A.B.; Fontes, J.D.; Freitas-Andrade, M.; Naus, C.C. Gap junctions and hemichannels: Communicating cell death in neurodevelopment and disease. BMC Cell Biol. 2017, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Miura, M. Programmed Cell Death in Neurodevelopment. Dev. Cell 2015, 32, 478–490. [Google Scholar] [CrossRef] [Green Version]
- Thion, M.S.; Ginhoux, F.; Garel, S. Microglia and early brain development: An intimate journey. Science 2018, 362, 185–189. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, B.B.; Bhutani, A.; Stary, C.M. Adult neurogenesis from reprogrammed astrocytes. Neural Regen. Res. 2020, 15, 973–979. [Google Scholar] [CrossRef]
- Ismail, F.Y.; Fatemi, A.; Johnston, M.V. Cerebral plasticity: Windows of opportunity in the developing brain. Eur. J. Paediatr. Neurol. 2017, 21, 23–48. [Google Scholar] [CrossRef]
- Lun, M.P.; Monuki, E.S.; Lehtinen, M.K. Development and functions of the choroid plexus–cerebrospinal fluid system. Nat. Rev. Neurosci. 2015, 16, 445–457. [Google Scholar] [CrossRef]
- Hunter, N.L.; Dymecki, S.M. Molecularly and temporally separable lineages form the hindbrain roof plate and contribute differentially to the choroid plexus. Development 2007, 134, 3449–3460. [Google Scholar] [CrossRef] [Green Version]
- O’Rahilly, R.; Müller, F. The meninges in human development. J. Neuropathol. Exp. Neurol. 1986, 45, 588–608. [Google Scholar] [CrossRef]
- Sakka, L.; Coll, G.; Chazal, J. Anatomy and physiology of cerebrospinal fluid. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2011, 128, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Delgehyr, N.; Meunier, A.; Faucourt, M.; Grau, M.B.; Strehl, L.; Janke, C.; Spassky, N. Ependymal cell differentiation, from monociliated to multiciliated cells. Micropatterning Cell Biol. Part B 2015, 127, 19–35. [Google Scholar] [CrossRef]
- Munk, A.S.; Wang, W.; Bèchet, N.B.; Eltanahy, A.M.; Cheng, A.X.; Sigurðsson, B.; Benraiss, A.; Mäe, M.A.; Kress, B.T.; Kelley, D.; et al. PDGF-B Is Required for Development of the Glymphatic System. Cell Rep. 2019, 26, 2955–2969.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehtinen, M.K.; Bjornsson, C.S.; Dymecki, S.M.; Gilbertson, R.J.; Holtzman, D.M.; Monuki, E.S. The Choroid Plexus and Cerebrospinal Fluid: Emerging Roles in Development. J. Neurosci. 2013, 33, 17553–17559. [Google Scholar] [CrossRef]
- Damkier, H.H.; Brown, P.D.; Praetorius, J. Cerebrospinal Fluid Secretion by the Choroid Plexus. Physiol. Rev. 2013, 93, 1847–1892. [Google Scholar] [CrossRef] [Green Version]
- Benarroch, E.E. Choroid plexus--CSF system: Recent developments and clinical correlations. Neurology 2016, 86, 286–296. [Google Scholar] [CrossRef]
- Solár, P.; Zamani, A.; Kubíčková, L.; Dubový, P.; Joukal, M. Choroid plexus and the blood–cerebrospinal fluid barrier in disease. Fluids Barriers CNS 2020, 17, 1–29. [Google Scholar] [CrossRef]
- Hutton, D.; Fadelalla, M.G.; Kanodia, A.K.; Hossain-Ibrahim, K. Choroid plexus and CSF: An updated review. Br. J. Neurosurg. 2021, 1–9, 1–9. [Google Scholar] [CrossRef]
- Liddelow, S.A. Development of the choroid plexus and blood-CSF barrier. Front. Neurosci. 2015, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Hochstetler, A.E.; Smith, H.M.; Preston, D.C.; Reed, M.M.; Territo, P.R.; Shim, J.W.; Fulkerson, D.; Blazer-Yost, B.L. TRPV4 antagonists ameliorate ventriculomegaly in a rat model of hydrocephalus. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Simpson, S.; Preston, D.; Schwerk, C.; Schroten, H.; Blazer-Yost, B. Cytokine and inflammatory mediator effects on TRPV4 function in choroid plexus epithelial cells. Am. J. Physiol. Physiol. 2019, 317, C881–C893. [Google Scholar] [CrossRef] [PubMed]
- Dutta, B.; Arya, R.K.; Goswami, R.; Alharbi, M.O.; Sharma, S.; Rahaman, S.O. Role of macrophage TRPV4 in inflammation. Lab. Investig. 2019, 100, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Grace, M.S.; Bonvini, S.; Belvisi, M.G.; McIntyre, P. Modulation of the TRPV4 ion channel as a therapeutic target for disease. Pharmacol. Ther. 2017, 177, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Kanju, P.; Chen, Y.; Lee, W.; Yeo, M.; Lee, S.H.; Romac, J.; Shahid, R.; Fan, P.; Gooden, D.M.; Simon, S.A.; et al. Small molecule dual-inhibitors of TRPV4 and TRPA1 for attenuation of inflammation and pain. Sci. Rep. 2016, 6, 26894. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, L.; An, D.; Xu, W.; Wu, C.; Sha, S.; Li, Y.; Zhu, Y.; Chen, A.; Du, Y.; et al. TRPV4-induced inflammatory response is involved in neuronal death in pilocarpine model of temporal lobe epilepsy in mice. Cell Death Dis. 2019, 10, 386. [Google Scholar] [CrossRef]
- Preston, D.; Simpson, S.; Halm, D.; Hochstetler, A.; Schwerk, C.; Schroten, H.; Blazer-Yost, B.L. Activation of TRPV4 stimulates transepithelial ion flux in a porcine choroid plexus cell line. Am. J. Physiol. Physiol. 2018, 315, C357–C366. [Google Scholar] [CrossRef]
- Stridh, L.; Ek, C.J.; Wang, X.; Nilsson, H.; Mallard, C. Regulation of Toll-Like Receptors in the Choroid Plexus in the Immature Brain After Systemic Inflammatory Stimuli. Transl. Stroke Res. 2013, 4, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Shipley, F.B.; Shannon, M.L.; Alturkistani, O.; Dani, N.; Webb, M.D.; Sugden, A.U.; Andermann, M.L.; Lehtinen, M.K. Inflammation of the Embryonic Choroid Plexus Barrier following Maternal Immune Activation. Dev. Cell. 2020, 55, 617–628. [Google Scholar] [CrossRef]
- Dziegielewska, K.M.; Ek, J.; Habgood, M.D.; Saunders, N.R. Development of the choroid plexus. Microsc. Res. Tech. 2001, 52, 5–20. [Google Scholar] [CrossRef]
- Aziz, A.A.; Coleman, L.; Morokoff, A.; Maixner, W. Diffuse choroid plexus hyperplasia: An under-diagnosed cause of hydrocephalus in children? Pediatr. Radiol. 2005, 35, 815–818. [Google Scholar] [CrossRef]
- Cardia, E.; Molina, D.; Abbate, F.; Mastroeni, P.; Stassi, G. Morphological modifications of the choroid plexus in a rodent model of acute ventriculitis induced by gram-negative liquoral sepsis. Possible implications in the pathophysiology of hypersecretory hydrocephalus. Childs Nerv. Syst. 1995, 11, 511–516. [Google Scholar] [CrossRef]
- Strahle, J.; Garton, H.J.L.; Maher, C.O.; Muraszko, K.M.; Keep, R.; Xi, G. Mechanisms of Hydrocephalus After Neonatal and Adult Intraventricular Hemorrhage. Transl. Stroke Res. 2012, 3, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Karimy, J.K.; Zhang, J.; Kurland, D.B.; Theriault, B.C.; Duran, D.; Stokum, J.A.; Furey, C.G.; Zhou, X.; Mansuri, M.S.; Montejo, J.; et al. Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat. Med. 2017, 23, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Karimy, J.K.; Duran, D.; Hu, J.K.; Gavankar, C.; Gaillard, J.R.; Bayri, Y.; Rice, H.; DiLuna, M.L.; Gerzanich, V.; Simard, J.M.; et al. Cerebrospinal fluid hypersecretion in pediatric hydrocephalus. Neurosurg. Focus 2016, 41, E10. [Google Scholar] [CrossRef] [Green Version]
- Ferrand-Drake, M. Cell death in the choroid plexus following transient forebrain global ischemia in the rat. Microsc. Res. Tech. 2001, 52, 130–136. [Google Scholar] [CrossRef]
- Schwerk, C.; Rybarczyk, K.; Essmann, F.; Seibt, A.; Mölleken, M.-L.; Zeni, P.; Schroten, H.; Tenenbaum, T. TNF Induces Choroid Plexus Epithelial Cell Barrier Alterations by Apoptotic and Nonapoptotic Mechanisms. J. Biomed. Biotechnol. 2010, 2010, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Hao, X.; Li, J.; Hua, Y.; Keep, R.F.; Xi, G. Effects of minocycline on epiplexus macrophage activation, choroid plexus injury and hydrocephalus development in spontaneous hypertensive rats. J. Cereb. Blood Flow Metab. 2019, 39, 1936–1948. [Google Scholar] [CrossRef]
- Gram, M.; Sveinsdottir, S.; Cinthio, M.; Sveinsdottir, K.; Hansson, S.R.; Mörgelin, M.; Åkerström, B.; Ley, D.H. Extracellular hemoglobin-mediator of inflammation and cell death in the choroid plexus following preterm intraventricular hemorrhage. J. Neuroinflammation 2014, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Sveinsdottir, S.; Gram, M.; Cinthio, M.; Sveinsdottir, K.; Mörgelin, M.; Ley, D. Altered Expression of Aquaporin 1 and 5 in the Choroid Plexus following Preterm Intraventricular Hemorrhage. Dev. Neurosci. 2014, 36, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Keep, R.; Hoff, J.T. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006, 5, 53–63. [Google Scholar] [CrossRef]
- Kovacs, G.G. Cellular reactions of the central nervous system. Handb. Clin. Neurol. 2017, 145, 13–23. [Google Scholar] [CrossRef]
- Del Bigio, M.R. The ependyma: A protective barrier between brain and cerebrospinal fluid. Glia 1995, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Spassky, N.; Merkle, F.; Flames, N.; Tramontin, A.D.; García-Verdugo, J.M.; Alvarez-Buylla, A. Adult Ependymal Cells Are Postmitotic and Are Derived from Radial Glial Cells during Embryogenesis. J. Neurosci. 2005, 25, 10–18. [Google Scholar] [CrossRef]
- Del Bigio, M.R. Ependymal cells: Biology and pathology. Acta Neuropathol. 2010, 119, 55–73. [Google Scholar] [CrossRef]
- Bruni, J.E. Ependymal development, proliferation, and functions: A review. Microsc. Res. Tech. 1998, 41, 2–13. [Google Scholar] [CrossRef]
- McAllister, J.P. Pathophysiology of congenital and neonatal hydrocephalus. Semin. Fetal Neonatal Med. 2012, 17, 285–294. [Google Scholar] [CrossRef]
- Coletti, A.M.; Singh, D.; Kumar, S.; Shafin, T.N.; Briody, P.J.; Babbitt, B.F.; Pan, D.; Norton, E.S.; Brown, E.C.; Kahle, K.T.; et al. Characterization of the ventricular-subventricular stem cell niche during human brain development. Development 2018, 145, dev.170100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez, G.O.; Daclin, M.; Shihavuddin, A.; Lansade, P.; Fortoul, A.; Faucourt, M.; Clavreul, S.; Lalioti, M.-E.; Taraviras, S.; Hippenmeyer, S.; et al. Adult Neural Stem Cells and Multiciliated Ependymal Cells Share a Common Lineage Regulated by the Geminin Family Members. Neuron 2019, 102, 159–172.e7. [Google Scholar] [CrossRef] [Green Version]
- Spassky, N.; Meunier, A. The development and functions of multiciliated epithelia. Nat. Rev. Mol. Cell Biol. 2017, 18, 423–436. [Google Scholar] [CrossRef]
- Kokovay, E.; Wang, Y.; Kusek, G.; Wurster, R.; Lederman, P.; Lowry, N.; Shen, Q.; Temple, S. VCAM1 Is Essential to Maintain the Structure of the SVZ Niche and Acts as an Environmental Sensor to Regulate SVZ Lineage Progression. Cell Stem Cell 2012, 11, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.-L.; Chen, G.; Zhang, S.; Zheng, J.; Wu, J.; Bai, Q.-R.; Wang, Y.; Li, J.; Wang, H.; Feng, H.; et al. Persistent Expression of VCAM1 in Radial Glial Cells Is Required for the Embryonic Origin of Postnatal Neural Stem Cells. Neuron 2017, 95, 309–325.e6. [Google Scholar] [CrossRef] [Green Version]
- Mirzadeh, Z.; Merkle, F.; Soriano-Navarro, M.; García-Verdugo, J.M.; Alvarez-Buylla, A. Neural Stem Cells Confer Unique Pinwheel Architecture to the Ventricular Surface in Neurogenic Regions of the Adult Brain. Cell Stem Cell 2008, 3, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.W.; Sandlund, J.; Han, C.H.; Hameed, M.Q.; Connors, S.; Klagsbrun, M.; Madsen, J.R.; Irwin, N. VEGF, which is elevated in the CSF of patients with hydrocephalus, causes ventriculomegaly and ependymal changes in rats. Exp. Neurol. 2013, 247, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Meunier, A.; Sawamoto, K.; Spassky, N. Chapter 42- Ependyma, Choroid. In Patterning and Cell Type Specification in the Developing CNS and PNS; Rubenstein, J.L.R., Rakic, P., Eds.; Academic Press: Amsterdam, The Netherlands, 2013; pp. 819–833. [Google Scholar] [CrossRef]
- Narita, K.; Takeda, S. Cilia in the choroid plexus: Their roles in hydrocephalus and beyond. Front. Cell. Neurosci. 2015, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Zappaterra, M.W.; Lehtinen, M.K. The cerebrospinal fluid: Regulator of neurogenesis, behavior, and beyond. Cell. Mol. Life Sci. 2012, 69, 2863–2878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guirao, B.; Meunier, A.; Mortaud, S.; Aguilar, A.; Corsi, J.-M.; Strehl, L.; Hirota, Y.; Desoeuvre, A.; Boutin, C.; Han, Y.-G.; et al. Coupling between hydrodynamic forces and planar cell polarity orients mammalian motile cilia. Nat. Cell Biol. 2010, 12, 341–350. [Google Scholar] [CrossRef]
- Mirzadeh, Z.; Han, Y.-G.; Soriano-Navarro, M.; García-Verdugo, J.M.; Alvarez-Buylla, A. Cilia Organize Ependymal Planar Polarity. J. Neurosci. 2010, 30, 2600–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tissir, F.; Qu, Y.; Montcouquiol, M.; Zhou, L.; Komatsu, K.; Shi, D.; Fujimori, T.; LaBeau, J.; Tyteca, D.; Courtoy, P.J.; et al. Lack of cadherins Celsr2 and Celsr3 impairs ependymal ciliogenesis, leading to fatal hydrocephalus. Nat. Neurosci. 2010, 13, 700–707. [Google Scholar] [CrossRef]
- Hirota, Y.; Meunier, A.; Huang, S.; Shimozawa, T.; Yamada, O.; Kida, Y.; Inoue, M.; Ito, T.; Kato, H.; Sakaguchi, M.; et al. Planar polarity of multiciliated ependymal cells involves the anterior migration of basal bodies regulated by non-muscle myosin II. Development 2010, 137, 3037–3046. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.; Kozlowski, G.; Sheridan, M. Scanning Electron Microscopy in the Ultrastructural Analysis of the Mammalian Cerebral Ventricular System. Adv. Clin. Chem. 1974, 37, 349–388. [Google Scholar] [CrossRef]
- Faubel, R.; Westendorf, C.; Bodenschatz, E.; Eichele, G. Cilia-based flow network in the brain ventricles. Science 2016, 353, 176–178. [Google Scholar] [CrossRef]
- Eichele, G.; Bodenschatz, E.; Ditte, Z.; Günther, A.-K.; Kapoor, S.; Wang, Y.; Westendorf, C. Cilia-driven flows in the brain third ventricle. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siyahhan, B.; Knobloch, V.; de Zelicourt, D.; Asgari, M.; Daners, M.S.; Poulikakos, D.; Kurtcuoglu, V. Flow induced by ependymal cilia dominates near-wall cerebrospinal fluid dynamics in the lateral ventricles. J. R. Soc. Interface 2014, 11, 20131189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worthington, W.C.; Cathcart, R.S.; Cooper, P.; Goldring, I.; Klein, M. Ependymal Cilia: Distribution and Activity in the Adult Human Brain. Science 1963, 139, 221–222. [Google Scholar] [CrossRef]
- Pellicciotta, N.; Hamilton, E.; Kotar, J.; Faucourt, M.; Delgehyr, N.; Spassky, N.; Cicuta, P. Entrainment of mammalian motile cilia in the brain with hydrodynamic forces. Proc. Natl. Acad. Sci. USA 2020, 117, 8315–8325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibañez-Tallon, I.; Pagenstecher, A.; Fliegauf, M.; Olbrich, H.; Kispert, A.; Ketelsen, U.-P.; North, A.; Heintz, N.; Omran, H. Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum. Mol. Genet. 2004, 13, 2133–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawamoto, K.; Wichterle, H.; Gonzalez-Perez, O.; Cholfin, J.A.; Yamada, M.; Spassky, N.; Murcia, N.S.; Garcia-Verdugo, J.M.; Marin, O.; Rubenstein, J.L.R.; et al. New Neurons Follow the Flow of Cerebrospinal Fluid in the Adult Brain. Science 2006, 311, 629–632. [Google Scholar] [CrossRef]
- Lehtinen, M.K.; Walsh, C.A. Neurogenesis at the Brain–Cerebrospinal Fluid Interface. Annu. Rev. Cell Dev. Biol. 2011, 27, 653–679. [Google Scholar] [CrossRef] [Green Version]
- Petrik, D.; Myoga, M.H.; Grade, S.; Gerkau, N.J.; Pusch, M.; Rose, C.R.; Grothe, B.; Götz, M. Epithelial Sodium Channel Regulates Adult Neural Stem Cell Proliferation in a Flow-Dependent Manner. Cell Stem. Cell 2018, 22, 865–878.e8. [Google Scholar] [CrossRef] [Green Version]
- Ringers, C.; Olstad, E.W.; Jurisch-Yaksi, N. The role of motile cilia in the development and physiology of the nervous system. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190156. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Berbari, N.F.; Yoder, B.K. Chapter 13 Ciliary Dysfunction in Developmental Abnormalities and Diseases. Curr. Top. Dev. Biol. 2008, 85, 371–427. [Google Scholar] [CrossRef]
- Lechtreck, K.-F.; Delmotte, P.; Robinson, M.L.; Sanderson, M.J.; Witman, G.B. Mutations in Hydin impair ciliary motility in mice. J. Cell Biol. 2008, 180, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Sapiro, R.; Kostetskii, I.; Olds-Clarke, P.; Gerton, G.; Radice, G.L.; Strauss, J.F. Male Infertility, Impaired Sperm Motility, and Hydrocephalus in Mice Deficient in Sperm-Associated Antigen 6. Mol. Cell. Biol. 2002, 22, 6298–6305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallmeier, J.; Nielsen, K.G.; Kuehni, C.E.; Lucas, J.S.; Leigh, M.W.; Zariwala, M.A.; Omran, H. Motile ciliopathies. Nat. Rev. Dis. Prim. 2020, 6, 1–29. [Google Scholar] [CrossRef]
- Banizs, B.; Pike, M.M.; Millican, C.L.; Ferguson, W.B.; Komlosi, P.; Sheetz, J.; Bell, P.D.; Schwiebert, E.M.; Yoder, B.K. Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development 2005, 132, 5329–5339. [Google Scholar] [CrossRef] [Green Version]
- Davy, B.E. Congenital hydrocephalus in hy3 mice is caused by a frameshift mutation in Hydin, a large novel gene. Hum. Mol. Genet. 2003, 12, 1163–1170. [Google Scholar] [CrossRef]
- Ohata, S.; Herranz-Pérez, V.; Nakatani, J.; Boletta, A.; García-Verdugo, J.M.; Álvarez-Buylla, A. Mechanosensory Genes Pkd1 and Pkd2 Contribute to the Planar Polarization of Brain Ventricular Epithelium. J. Neurosci. 2015, 35, 11153–11168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohata, S.; Alvarez-Buylla, A. Planar Organization of Multiciliated Ependymal (E1) Cells in the Brain Ventricular Epithelium. Trends Neurosci. 2016, 39, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Antony, D.; Becker-Heck, A.; Zariwala, M.A.; Schmidts, M.; Onoufriadis, A.; Forouhan, M.; Wilson, R.; Taylor-Cox, T.; Dewar, A.; Jackson, C.; et al. Mutations inCCDC39andCCDC40are the Major Cause of Primary Ciliary Dyskinesia with Axonemal Disorganization and Absent Inner Dynein Arms. Hum. Mutat. 2013, 34, 462–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelhamed, Z.; Vuong, S.M.; Hill, L.; Shula, C.; Timms, A.; Beier, D.; Campbell, K.; Mangano, F.T.; Stottmann, R.W.; Goto, J. A mutation in Ccdc39 causes neonatal hydrocephalus with abnormal motile cilia development in mice. Development 2018, 145, dev154500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarnat, H.B. Ependymal Reactions to Injury. A Review. J. Neuropathol. Exp. Neurol. 1995, 54, 1–15. [Google Scholar] [CrossRef]
- Ferland, R.; Batiz, L.F.; Neal, J.; Lian, G.; Bundock, E.; Lu, J.; Hsiao, Y.-C.; Diamond, R.; Mei, D.; Banham, A.; et al. Disruption of neural progenitors along the ventricular and subventricular zones in periventricular heterotopia. Hum. Mol. Genet. 2008, 18, 497–516. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, A.J.; Tome, M.; Paez, P.; Wagner, C.; Rodriguez, S.; Fernández-Llebrez, P.; Rodríguez, E.M.; Pérez-Fígares, J.M. A Programmed Ependymal Denudation Precedes Congenital Hydrocephalus in thehyhMutant Mouse. J. Neuropathol. Exp. Neurol. 2001, 60, 1105–1119. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, A.J.; García-Verdugo, J.M.; González, C.A.; Bátiz, L.F.; Rodríguez-Pérez, L.M.; Páez, P.; Soriano-Navarro, M.; Roales-Buján, R.; Rivera, P.; Rodríguez, S.; et al. Disruption of the Neurogenic Niche in the Subventricular Zone of Postnatal Hydrocephalic hyh Mice. J. Neuropathol. Exp. Neurol. 2009, 68, 1006–1020. [Google Scholar] [CrossRef] [Green Version]
- Sival, D.A.; Guerra, M.; Dunnen, W.F.A.D.; Batiz, L.F.; Alvial, G.; Castañeyra-Perdomo, A.; Rodríguez, E.M. Neuroependymal Denudation is in Progress in Full-term Human Foetal Spina Bifida Aperta. Brain Pathol. 2011, 21, 163–179. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Wang, J.; Tseng, I.-C.; Huang, T.; Zhao, Y.; Zheng, Q.; Gao, Y.; Luo, H.; Zhang, X.; et al. SNX27 Deletion Causes Hydrocephalus by Impairing Ependymal Cell Differentiation and Ciliogenesis. J. Neurosci. 2016, 36, 12586–12597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, Y.; Sakane, F.; Hashimoto, K. N-cadherin-based adherens junction regulates the maintenance, proliferation, and differentiation of neural progenitor cells during development. Cell Adhes. Migr. 2015, 9, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Roales-Bujan, R.; Paez-Gonzalez, P.; Guerra, M.; Rodríguez, S.; Vío, K.; Ho-Plagaro, A.; Bonilla, M.G.; Rodriguez-Perez, L.M.; Dominguez-Pinos, M.D.; Rodríguez, E.-M.; et al. Astrocytes acquire morphological and functional characteristics of ependymal cells following disruption of ependyma in hydrocephalus. Acta Neuropathol. 2012, 124, 531–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, C.; González, C.A.; Alvial, G.; Flores, C.A.; Rodríguez, E.M.; Bátiz, L.F. Disruption of CDH2/N-Cadherin–Based Adherens Junctions Leads to Apoptosis of Ependymal Cells and Denudation of Brain Ventricular Walls. J. Neuropathol. Exp. Neurol. 2013, 72, 846–860. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, K.J.; Malakhov, M.P.; Hetherington, C.J.; Zhou, L.; Little, M.-T.; Malakhova, O.A.; Sipe, J.C.; Orkin, S.H.; Zhang, D.-E. Dysregulation of protein modification by ISG15 results in brain cell injury. Genes Dev. 2002, 16, 2207–2212. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.-Y.; Tang, F.-L.; Lee, D.; Zhao, Y.; Song, H.; Zhu, X.-J.; Mei, L.; Xiong, W.-C. Ependymal Vps35 Promotes Ependymal Cell Differentiation and Survival, Suppresses Microglial Activation, and Prevents Neonatal Hydrocephalus. J. Neurosci. 2020, 40, 3862–3879. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, M.; Sato, R.; Yamashita, T. Loss of p73 in ependymal cells during the perinatal period leads to aqueductal stenosis. Sci. Rep. 2017, 7, 12007. [Google Scholar] [CrossRef]
- Jackson, P.K.; Attardi, L.D. p73 and FoxJ1: Programming Multiciliated Epithelia. Trends Cell Biol. 2016, 26, 239–240. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.; Mays, D.J.; Beeler, S.; Rosenbluth, J.M.; Boyd, K.L.; Guasch, G.L.S.; Shaver, T.M.; Tang, L.J.; Liu, Q.; Shyr, Y.; et al. p73 Is Required for Multiciliogenesis and Regulates the Foxj1-Associated Gene Network. Cell Rep. 2016, 14, 2289–2300. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Maruo, T.; Majima, T.; Ishizaki, H.; Tanaka-Okamoto, M.; Miyoshi, J.; Mandai, K.; Takai, Y. Genetic Deletion of Afadin Causes Hydrocephalus by Destruction of Adherens Junctions in Radial Glial and Ependymal Cells in the Midbrain. PLoS ONE 2013, 8, e80356. [Google Scholar] [CrossRef]
- Pozniak, C.D.; Barnabé-Heider, F.; Rymar, V.V.; Lee, A.F.; Sadikot, A.F.; Miller, F.D. p73 Is Required for Survival and Maintenance of CNS Neurons. J. Neurosci. 2002, 22, 9800–9809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkes, P.; Sharpe, A.; et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nat. Cell Biol. 2000, 404, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Tozluoğlu, M.; Karaca, E.; Haliloglu, T.; Nussinov, R. Cataloging and organizing p73 interactions in cell cycle arrest and apoptosis. Nucleic Acids Res. 2008, 36, 5033–5049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandai, K.; Rikitake, Y.; Shimono, Y.; Takai, Y. Afadin/AF-6 and Canoe: Roles in cell adhesion and beyond. Prog. Mol. Biol. Transl. Sci. 2013, 116, 433–454. [Google Scholar] [CrossRef]
- Mack, P.F. Intracranial haemorrhage: Therapeutic interventions and anaesthetic management. Br. J. Anaesth. 2014, 113, ii17–ii25. [Google Scholar] [CrossRef] [Green Version]
- Ahdab-Barmada, M.; Moossy, J.; Preble, O.T.; Youngner, J.S. Hydrocephalus in Weanling Mice Induced by a Temperature-sensitive Mutant of Vesicular Stomatitis Virus. J. Neuropathol. Exp. Neurol. 1982, 41, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Zhang, H.; Fu, C.; Liu, X.; Chen, B.; Dang, Y.; Chen, H.; Liu, L. Prolonged hydrocephalus induced by intraventricular hemorrhage in rats is reduced by curcumin therapy. Neurosci. Lett. 2017, 637, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Mayfrank, L.; Kim, Y.; Kissler, J.; Delsing, P.; Gilsbach, J.M.; Schröder, J.M.; Weis, J. Morphological changes following experimental intraventricular haemorrhage and intraventricular fibrinolytic treatment with recombinant tissue plasminogen activator. Acta Neuropathol. 2000, 100, 561–567. [Google Scholar] [CrossRef]
- Nathoo, N.; Jalal, H.; Natah, S.S.; Zhang, Q.; Wu, Y.; Dunn, J.F. Hypoxia and Inflammation-Induced Disruptions of the Blood-Brain and Blood-Cerebrospinal Fluid Barriers Assessed Using a Novel T1-Based MRI Method. Acta Neurochir Suppl. 2016, 121, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Schubert, J.J.; Veronese, M.; Marchitelli, L.; Bodini, B.; Tonietto, M.; Stankoff, B.; Brooks, D.; Bertoldo, A.; Edison, P.; Turkheimer, F.E. Dynamic 11C-PiB PET Shows Cerebrospinal Fluid Flow Alterations in Alzheimer Disease and Multiple Sclerosis. J. Nucl. Med. 2019, 60, 1452–1460. [Google Scholar] [CrossRef] [Green Version]
- Lisanti, C.J.; Asbach, P.; Bradley, W.G., Jr. The ependymal “Dot-Dash” sign: An MR imaging finding of early multiple sclerosis. AJNR Am. J. Neuroradiol. 2005, 26, 2033–2036. [Google Scholar]
- Hatrock, D.; Caporicci-Dinucci, N.; Stratton, J.A. Ependymal cells and multiple sclerosis: Proposing a relationship. Neural Regen. Res. 2020, 15, 263–264. [Google Scholar] [CrossRef]
- Shah, P.T.; Stratton, J.; Stykel, M.; Abbasi, S.; Sharma, S.; Mayr, K.; Koblinger, K.; Whelan, P.; Biernaskie, J. Single-Cell Transcriptomics and Fate Mapping of Ependymal Cells Reveals an Absence of Neural Stem Cell Function. Cell 2018, 173, 1045–1057.e9. [Google Scholar] [CrossRef] [Green Version]
- Fukumizu, M.; Takashima, S.; Becker, L.E. Neonatal posthemorrhagic hydrocephalus: Neuropathologic and immunohistochemical studies. Pediatr. Neurol. 1995, 13, 230–234. [Google Scholar] [CrossRef]
- Bruni, J.; Delbigio, M.; Clattenburg, R. Ependyma: Normal and pathological. A review of the literature. Brain Res. Rev. 1985, 9, 1–19. [Google Scholar] [CrossRef]
- Pang, D.; Sclabassi, R.J.; Horton, J.A. Lysis of Intraventricular Blood Clot with Urokinase in a Canine Model: Part 3. Effects of intraventricular urokinase on clot lysis and posthemorrhagic hydrocephalus. Neurosurg. 1986, 19, 553–572. [Google Scholar] [CrossRef]
- Page, R.B.; Leure-Dupree, A.E. Ependymal Alterations in Hydrocephalus. In Neurobiology of Cerebrospinal Fluid; Wood, J.H., Ed.; Plenum Press: New York, NY, USA, 1983; pp. 789–820. [Google Scholar] [CrossRef]
- Abdi, K.; Lai, C.-H.; Paez-Gonzalez, P.; Lay, M.; Pyun, J.; Kuo, C.T. Uncovering inherent cellular plasticity of multiciliated ependyma leading to ventricular wall transformation and hydrocephalus. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Carlén, M.; Meletis, K.; Göritz, C.; Darsalia, V.; Evergren, E.; Tanigaki, K.; Amendola, M.; Barnabé-Heider, F.; Yeung, M.S.Y.; Naldini, L.; et al. Forebrain ependymal cells are Notch-dependent and generate neuroblasts and astrocytes after stroke. Nat. Neurosci. 2009, 12, 259–267. [Google Scholar] [CrossRef]
- Sarnat, H.B. Role of human fetal ependyma. Pediatr. Neurol. 1992, 8, 163–178. [Google Scholar] [CrossRef]
- Lavezzi, A.M.; Corna, M.F.; Matturri, L. Ependymal alterations in sudden intrauterine unexplained death and sudden infant death syndrome: Possible primary consequence of prenatal exposure to cigarette smoking. Neural Dev. 2010, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Moral, Y.; Robertson, N.J.; Goni-de-Cerio, F.; Alonso-Alconada, D. Hipoxia-isquemia neonatal: Bases celulares y moleculares del daño cerebral y modulacion terapeutica de la neurogenesis [Neonatal hypoxia-ischemia: Cellular and molecular brain damage and therapeutic modulation of neurogenesis]. Rev. Neurol. 2019, 68, 23–36. [Google Scholar]
- Domínguez-Pinos, M.D.; Páez, P.; Jiménez, A.-J.; Weil, B.; Arráez, M.-A.; Pérez-Fígares, J.-M.; Rodríguez, E.-M. Ependymal Denudation and Alterations of the Subventricular Zone Occur in Human Fetuses With a Moderate Communicating Hydrocephalus. J. Neuropathol. Exp. Neurol. 2005, 64, 595–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.M.; Kumar, R.; McAllister, J.; Krause, G.S. Gene expression analysis of the development of congenital hydrocephalus in the H-Tx rat. Brain Res. 2006, 1075, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Deren, K.E.; Packer, M.; Forsyth, J.; Milash, B.; Abdullah, O.M.; Hsu, E.W.; McAllister, J.P. Reactive astrocytosis, microgliosis and inflammation in rats with neonatal hydrocephalus. Exp. Neurol. 2010, 226, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Sarnat, H.B. Histochemistry and immunocytochemistry of the developing ependyma and choroid plexus. Microsc. Res. Tech. 1998, 41, 14–28. [Google Scholar] [CrossRef]
- Kostović, I.; Sedmak, G.; Judaš, M. Neural histology and neurogenesis of the human fetal and infant brain. NeuroImage 2019, 188, 743–773. [Google Scholar] [CrossRef] [PubMed]
- Whitelaw, A. Intraventricular haemorrhage and posthaemorrhagic hydrocephalus: Pathogenesis, prevention and future interventions. Semin. Neonatol. 2001, 6, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Ihrie, R.; Álvarez-Buylla, A. Lake-Front Property: A Unique Germinal Niche by the Lateral Ventricles of the Adult Brain. Neuron 2011, 70, 674–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, R.C.; Wu, H.; Zandian, M.; Danielpour, M.; Kabos, P.; Yu, J.S.; Sun, Y.E. Neural progenitors populate the cerebrospinal fluid of preterm patients with hydrocephalus. J. Pediatr. 2006, 148, 337–340.e3. [Google Scholar] [CrossRef]
- Emmert, A.S.; Iwasawa, E.; Shula, C.; Schultz, P.; Lindquist, D.; Dunn, R.S.; Fugate, E.M.; Hu, Y.-C.; Mangano, F.T.; Goto, J. Impaired neural differentiation and glymphatic CSF flow in the Ccdc39 rat model of neonatal hydrocephalus: Genetic interaction with L1cam. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louveau, A.; Smirnov, I.; Keyes, T.; Eccles, J.; Rouhani, S.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.H.; Prince, E.A. Basic Vascular Neuroanatomy of the Brain and Spine: What the General Interventional Radiologist Needs to Know. Semin. Interv. Radiol. 2013, 30, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Klebe, D.; McBride, D.; Krafft, P.R.; Flores, J.J.; Tang, J.; Zhang, J.H. Posthemorrhagic hydrocephalus development after germinal matrix hemorrhage: Established mechanisms and proposed pathways. J. Neurosci. Res. 2020, 98, 105–120. [Google Scholar] [CrossRef] [Green Version]
- Iliff, J.J.; Nedergaard, M. Is There a Cerebral Lymphatic System? Stroke 2013, 44, S93–S95. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Nagelhus, E.A.; Ottersen, O.P. Physiological Roles of Aquaporin-4 in Brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, E.T.; Inman, C.B.; Weller, R.O. Interrelationships of the pia mater and the perivascular (Virchow-Robin) spaces in the human cerebrum. J. Anat. 1990, 170, 111–123. [Google Scholar] [PubMed]
- Johnston, M.; Zakharov, A.; Papaiconomou, C.; Salmasi, G.; Armstrong, D. Evidence of connections between cerebrospinal fluid and nasal lymphatic vessels in humans, non-human primates and other mammalian species. Cereb. Fluid Res. 2004, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plog, B.A.; Nedergaard, M. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 379–394. [Google Scholar] [CrossRef] [Green Version]
- Benveniste, H.; Heerdt, P.M.; Fontes, M.; Rothman, D.L.; Volkow, N.D. Glymphatic System Function in Relation to Anesthesia and Sleep States. Anesthesia Analg. 2019, 128, 747–758. [Google Scholar] [CrossRef]
- Ba Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef]
- Gaberel, T.; Gakuba, C.; Goulay, R.; De Lizarrondo, S.M.; Hanouz, J.-L.; Emery, E.; Touzé, E.; Vivien, D.; Gauberti, M. Impaired Glymphatic Perfusion After Strokes Revealed by Contrast-Enhanced MRI: A new target for fibrinolysis? Stroke 2014, 45, 3092–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [Green Version]
- Bennett, H.C.; Kim, Y. Pericytes Across the Lifetime in the Central Nervous System. Front. Cell. Neurosci. 2021, 15. [Google Scholar] [CrossRef]
- Greif, D.M.; Eichmann, A. Vascular biology: Brain vessels squeezed to death. Nat. Cell Biol. 2014, 508, 50–51. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nat. Cell Biol. 2010, 468, 562–566. [Google Scholar] [CrossRef] [Green Version]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood–brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, M.; Glees, P. The fine structure of human cerebral perivascular pericytes and juxtavascular phagocytes: Their possible role in hydrocephalic edema resolution. J. Fur Hirnforsch. 1990, 31, 237–249. [Google Scholar]
- Gautam, J.; Zhang, X.; Yao, Y. The role of pericytic laminin in blood brain barrier integrity maintenance. Sci. Rep. 2016, 6, 36450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Norris, E.H.; Strickland, S. The cellular origin of laminin determines its role in blood pressure regulation. Cell. Mol. Life Sci. 2014, 72, 999–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Edelman, D.A.; Jiang, Y.; Tyburski, J.; Wilson, R.F.; Steffes, C. Pericytes and Their Role in Microvasculature Homeostasis. J. Surg. Res. 2006, 135, 305–311. [Google Scholar] [CrossRef]
- Kloner, R.A.; King, K.; Harrington, M. No-reflow phenomenon in the heart and brain. Am. J. Physiol. Circ. Physiol. 2018, 315, H550–H562. [Google Scholar] [CrossRef]
- Fernández-Klett, F.; Potas, J.; Hilpert, D.; Blazej, K.; Radke, J.; Huck, J.; Engel, O.; Stenzel, W.; Genové, G.; Priller, J. Early Loss of Pericytes and Perivascular Stromal Cell-Induced Scar Formation after Stroke. J. Cereb. Blood Flow Metab. 2012, 33, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.L.A.; Michael-Titus, A.T.; Shah, D.K. Hypoxic-Ischaemic Encephalopathy and the Blood-Brain Barrier in Neonates. Dev. Neurosci. 2017, 39, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Q.; Tang, J.; Feng, H.; Zhang, J.H. The evolving roles of pericyte in early brain injury after subarachnoid hemorrhage. Brain Res. 2015, 1623, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.; Reynell, C.; Gesslein, B.; Hamilton-Whitaker, N.; Mishra, A.; Sutherland, B.; O’Farrell, F.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nat. Cell Biol. 2014, 508, 55–60. [Google Scholar] [CrossRef] [Green Version]
- O’Farrell, F.; Attwell, D. A role for pericytes in coronary no-reflow. Nat. Rev. Cardiol. 2014, 11, 427–432. [Google Scholar] [CrossRef]
- Yemisci, M.; Gürsoy-Ozdemir, Y.; Vural, A.; Can, A.; Topalkara, K.; Dalkara, T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 2009, 15, 1031–1037. [Google Scholar] [CrossRef]
- Østergaard, L.; Aamand, R.; Karabegovic, S.; Tietze, A.; Blicher, J.; Mikkelsen, I.K.; Iversen, N.K.; Secher, N.; Engedal, T.S.; Anzabi, M.; et al. The Role of the Microcirculation in Delayed Cerebral Ischemia and Chronic Degenerative Changes after Subarachnoid Hemorrhage. J. Cereb. Blood Flow Metab. 2013, 33, 1825–1837. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Rege, S.V.; Ramanathan, A.; Wang, Y.; Ahuja, A.; Lazic, D.; Tsai, P.S.; Zhao, Z.; Zhou, Y.; et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat. Neurosci. 2017, 20, 406–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Ding, F.; Deng, S.; Guo, X.; Wang, W.; Iliff, J.J.; Nedergaard, M. Focal Solute Trapping and Global Glymphatic Pathway Impairment in a Murine Model of Multiple Microinfarcts. J. Neurosci. 2017, 37, 2870–2877. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Birkelund, S.; Burkhart, A.; Stensballe, A.; Moos, T. Synthesis and deposition of basement membrane proteins by primary brain capillary endothelial cells in a murine model of the blood-brain barrier. J. Neurochem. 2016, 140, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Vandebroek, A.; Yasui, M. Regulation of AQP4 in the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 1603. [Google Scholar] [CrossRef] [Green Version]
- Sweger, E.J.; Casper, K.B.; Scearce-Levie, K.; Conklin, B.R.; McCarthy, K.D. Development of Hydrocephalus in Mice Expressing the Gi-Coupled GPCR Ro1 RASSL Receptor in Astrocytes. J. Neurosci. 2007, 27, 2309–2317. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.G.; Milhorat, T.H. Experimental Hydrocephalus. 3. Light microscopic findings in acute and subacute obstructive hydrocephalus in the monkey. J. Neurosurg. 1970, 32, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Tradtrantip, L.; Smith, A.J.; Yao, X. Aquaporin Water Channels and Hydrocephalus. Pediatr. Neurosurg. 2017, 52, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Giffard, R.G.; Swanson, R.A. Ischemia-induced programmed cell death in astrocytes. Glia 2005, 50, 299–306. [Google Scholar] [CrossRef]
- Solaroglu, I.; Gürsoy-Ozdemir, Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr. Neuropharmacol. 2018, 16, 1396–1415. [Google Scholar] [CrossRef]
- Martin, L.J.; Brambrink, A.M.; Lehmann, C.; Portera-Cailliau, C.; Koehler, R.; Rothstein, J.; Traystman, R.J. Hypoxia?ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann. Neurol. 1997, 42, 335–348. [Google Scholar] [CrossRef]
- Rossi, D.; Brambilla, L.; Valori, C.F.; Roncoroni, C.; Crugnola, A.; Yokota, T.; Bredesen, D.E.; Volterra, A. Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ. 2008, 15, 1691–1700. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, J.J.; Olabarria, M.; Chvatal, A.; Verkhratsky, A.; Rodr, J.J. Astroglia in dementia and Alzheimer’s disease. Cell Death Differ. 2008, 16, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Lukaszevicz, A.-C.; Sampaïo, N.; Guégan, C.; Benchoua, A.; Couriaud, C.; Chevalier, E.; Sola, B.; Lacombe, P.; Onténiente, B. High Sensitivity of Protoplasmic Cortical Astroglia to Focal Ischemia. J. Cereb. Blood Flow Metab. 2002, 22, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Krupinski, J.; Lopez, E.; Martí, E.; Ferrer, I. Expression of Caspases and Their Substrates in the Rat Model of Focal Cerebral Ischemia. Neurobiol. Dis. 2000, 7, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjelloun, N.; Joly, L.-M.; Palmier, B.; Plotkine, M.; Charriaut-Marlangue, C. Apoptotic mitochondrial pathway in neurones and astrocytes after neonatal hypoxia-ischaemia in the rat brain. Neuropathol. Appl. Neurobiol. 2003, 29, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Solenov, E.; Watanabe, H.; Manley, G.T.; Verkman, A.S. Sevenfold-reduced osmotic water permeability in primary astrocyte cultures from AQP-4-deficient mice, measured by a fluorescence quenching method. Am. J. Physiol. Physiol. 2004, 286, C426–C432. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.; Hsu, Y.; Schneller, B.; Hobbs, J.G.; Mehta, A.I.; Linninger, A. Hydrocephalus: The role of cerebral aquaporin-4 channels and computational modeling considerations of cerebrospinal fluid. Neurosurg. Focus 2016, 41, E8. [Google Scholar] [CrossRef] [Green Version]
- Teng, Z.; Wang, A.; Wang, P.; Wang, R.; Wang, W.; Han, H. The Effect of Aquaporin-4 Knockout on Interstitial Fluid Flow and the Structure of the Extracellular Space in the Deep Brain. Aging Dis. 2018, 9, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Bloch, O.; Manley, G.T. The role of aquaporin-4 in cerebral water transport and edema. Neurosurg. Focus 2007, 22, 1–7. [Google Scholar] [CrossRef]
- Paul, L.; Madan, M.; Rammling, M.; Chigurupati, S.; Chan, S.L.; Pattisapu, J.V. Expression of Aquaporin 1 and 4 in a Congenital Hydrocephalus Rat Model. Neurosurgery 2011, 68, 462–473. [Google Scholar] [CrossRef]
- Tourdias, T.; Dragonu, I.; Fushimi, Y.; Deloire, M.S.; Boiziau, C.; Brochet, B.; Moonen, C.; Petry, K.G.; Dousset, V. Aquaporin 4 correlates with apparent diffusion coefficient and hydrocephalus severity in the rat brain: A combined MRI–histological study. NeuroImage 2009, 47, 659–666. [Google Scholar] [CrossRef]
- Castaneyra-Ruiz, L.; González-Marrero, I.; Gonzalez-Toledo, J.M.; Castañeyra-Ruiz, A.; De Paz-Carmona, H.; Castañeyra-Perdomo, A.; Carmona-Calero, E.M. Aquaporin-4 expression in the cerebrospinal fluid in congenital human hydrocephalus. Fluids Barriers CNS 2013, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Chen, Z.-L.; Norris, E.; Strickland, S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat. Commun. 2014, 5, 3413. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.-L.; Yao, Y.; Norris, E.; Kruyer, A.; Jnocharles, O.C.; Akhmerov, A.; Strickland, S. Ablation of astrocytic laminin impairs vascular smooth muscle cell function and leads to hemorrhagic stroke. J. Cell Biol. 2013, 202, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.-O.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In vitro models of the blood–brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef] [PubMed]
- Guerra, M.; Blázquez, J.L.; Rodríguez, E.M. Blood–brain barrier and foetal-onset hydrocephalus, with a view on potential novel treatments beyond managing CSF flow. Fluids Barriers CNS 2017, 14, 1–15. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Routhe, L.J.; Moos, T. The vascular basement membrane in the healthy and pathological brain. J. Cereb. Blood Flow Metab. 2017, 37, 3300–3317. [Google Scholar] [CrossRef]
- Rossi, D.M. Astrocyte physiopathology: At the crossroads of intercellular networking, inflammation and cell death. Prog. Neurobiol. 2015, 130, 86–120. [Google Scholar] [CrossRef] [PubMed]
- Nehrkorn, K. The Role of Pericytes in Microcirculatory Dysfunction after Subarachnoid Hemorrhage. Ph.D. Thesis, Graduate School of Systemic Neurosciences, Planegg, Germany, 2016. [Google Scholar] [CrossRef]
- Friedrich, V.; Flores, R.; Sehba, F.A. Cell death starts early after subarachnoid hemorrhage. Neurosci. Lett. 2012, 512, 6–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducruet, A.F.; Zacharia, B.; Hickman, Z.; Grobelny, B.T.; Yeh, M.L.; Sosunov, S.A.; Connolly, E.S. The complement cascade as a therapeutic target in intracerebral hemorrhage. Exp. Neurol. 2009, 219, 398–403. [Google Scholar] [CrossRef] [Green Version]
- Bellander, B.-M.; Singhrao, S.K.; Ohlsson, M.; Mattsson, P.; Svensson, M. Complement Activation in the Human Brain after Traumatic Head Injury. J. Neurotrauma 2001, 18, 1295–1311. [Google Scholar] [CrossRef] [PubMed]
- Thyboll, J.; Kortesmaa, J.; Cao, R.; Soininen, R.; Wang, L.; Iivanainen, A.; Sorokin, L.; Risling, M.; Cao, Y.; Tryggvason, K. Deletion of the Laminin α4 Chain Leads to Impaired Microvessel Maturation. Mol. Cell. Biol. 2002, 22, 1194–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castejón, O.J. Submicroscopic pathology of human and experimental hydrocephalic cerebral cortex. Folia Neuropathol. 2010, 48, 159–174. [Google Scholar]
- Gautam, J.; Cao, Y.; Yao, Y. Pericytic Laminin Maintains Blood-Brain Barrier Integrity in an Age-Dependent Manner. Transl. Stroke Res. 2019, 11, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Safo, M.K.; Ahmed, M.H.; Ghatge, M.S.; Boyiri, T. Hemoglobin–ligand binding: Understanding Hb function and allostery on atomic level. Biochim. Biophys Acta. 2011, 1814, 797–809. [Google Scholar] [CrossRef]
- Ahmed, M.H.; Ghatge, M.S.; Safo, M.K. Hemoglobin: Structure, Function and Allostery. Subcell Biochem. 2020, 94, 345–382. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Wu, Y.; Song, J.; Wang, Y.; Wang, X.; Culmsee, C.; Zhu, C. The Potential Role of Ferroptosis in Neonatal Brain Injury. Front. Neurosci. 2019, 13, 115. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, J.L. Iron Biology in Immune Function, Muscle Metabolism and Neuronal Functioning. J. Nutr. 2001, 131, 568S–580S. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyokuni, S.; Ito, F.; Yamashita, K.; Okazaki, Y.; Akatsuka, S. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free. Radic. Biol. Med. 2017, 108, 610–626. [Google Scholar] [CrossRef]
- Tang, S.; Xiao, X. Ferroptosis and kidney diseases. Int. Urol. Nephrol. 2019, 52, 497–503. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Savman, K.; Nilsson, U.A.; Blennow, M.; Kjellmer, I.; Whitelaw, A. Non-Protein-Bound Iron Is Elevated in Cerebrospinal Fluid from Preterm Infants with Posthemorrhagic Ventricular Dilatation. Pediatr. Res. 2001, 49, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Du, H.; Hua, Y.; Keep, R.; Strahle, J.; Xi, G. Role of Red Blood Cell Lysis and Iron in Hydrocephalus after Intraventricular Hemorrhage. J. Cereb. Blood Flow Metab. 2014, 34, 1070–1075. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Gao, C.; Hua, Y.; Keep, R.; Muraszko, K.; Xi, G. Role of Iron in Brain Injury After Intraventricular Hemorrhage. Stroke 2011, 42, 465–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free. Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Fang, H.; Wang, P.-F.; Zhou, Y.; Wang, Y.-C.; Yang, Q.-W. Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. J. Neuroinflammation 2013, 10, 27. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Gao, P.; Chen, H.; Zhou, X.; Ou, Y.; He, Y. The Role of Iron, Its Metabolism and Ferroptosis in Traumatic Brain Injury. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Gu, Y.; Hua, Y.; Keep, R.; Morgenstern, L.B.; Xi, G. Deferoxamine Reduces Intracerebral Hematoma-Induced Iron Accumulation and Neuronal Death in Piglets. Stroke 2009, 40, 2241–2243. [Google Scholar] [CrossRef]
- Li, Q.; Weiland, A.; Chen, X.; Lan, X.; Han, X.; Durham, F.; Liu, X.; Wan, J.; Ziai, W.C.; Hanley, D.F.; et al. Ultrastructural Characteristics of Neuronal Death and White Matter Injury in Mouse Brain Tissues After Intracerebral Hemorrhage: Coexistence of Ferroptosis, Autophagy, and Necrosis. Front. Neurol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Volpe, J.J.; Kinney, H.C.; Jensen, F.E.; Rosenberg, P. The developing oligodendrocyte: Key cellular target in brain injury in the premature infant. Int. J. Dev. Neurosci. 2011, 29, 423–440. [Google Scholar] [CrossRef] [Green Version]
- Lebel, C.; Deoni, S. The development of brain white matter microstructure. NeuroImage 2018, 182, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J. The Encephalopathy of Prematurity—Brain Injury and Impaired Brain Development Inextricably Intertwined. Semin. Pediatr. Neurol. 2009, 16, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Cohen, C.C.; Popovic, M.A.; Klooster, J.; Weil, M.-T.; Möbius, W.; Nave, K.-A.; Kole, M.H. Saltatory Conduction along Myelinated Axons Involves a Periaxonal Nanocircuit. Cell 2020, 180, 311–322.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nave, K.-A.; Werner, H.B. Myelination of the Nervous System: Mechanisms and Functions. Annu. Rev. Cell Dev. Biol. 2014, 30, 503–533. [Google Scholar] [CrossRef]
- Moore, S.; Meschkat, M.; Ruhwedel, T.; Trevisiol, A.; Tzvetanova, I.D.; Battefeld, A.; Kusch, K.; Kole, M.H.P.; Strenzke, N.; Möbius, W.; et al. A role of oligodendrocytes in information processing. Nat. Commun. 2020, 11, 5497. [Google Scholar] [CrossRef]
- Del Bigio, M.; Da Silva, M.C.; Drake, J.M.; Tuor, U.I. Acute and Chronic Cerebral White Matter Damage in Neonatal Hydrocephalus. Can. J. Neurol. Sci. 1994, 21, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Del Bigio, M.R. Neuropathological changes caused by hydrocephalus. Acta Neuropathol. 1993, 85, 573–585. [Google Scholar] [CrossRef]
- Schneider, J.; Miller, S.P. Preterm brain Injury: White matter injury. Handb. Clin. Neurol. 2019, 162, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, E.R.; Hristova, M. Plasticity in the Neonatal Brain following Hypoxic-Ischaemic Injury. Neural. Plast. 2016, 2016, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truttmann, A.C.; Ginet, V.; Puyal, J. Current Evidence on Cell Death in Preterm Brain Injury in Human and Preclinical Models. Front. Cell Dev. Biol. 2020, 8, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuillen, P.S.; Sheldon, R.A.; Shatz, C.J.; Ferriero, D.M. Selective Vulnerability of Subplate Neurons after Early Neonatal Hypoxia-Ischemia. J. Neurosci. 2003, 23, 3308–3315. [Google Scholar] [CrossRef] [Green Version]
- Robinson, S.; Li, Q.; DeChant, A.; Cohen, M.L. Neonatal loss of γ–aminobutyric acid pathway expression after human perinatal brain injury. J. Neurosurg. Pediatr. 2006, 104, 396–408. [Google Scholar] [CrossRef]
- Kaur, C.; Rathnasamy, G.; Ling, E.-A. The Choroid Plexus in Healthy and Diseased Brain. J. Neuropathol. Exp. Neurol. 2016, 75, 198–213. [Google Scholar] [CrossRef]
- Redzic, Z.; Preston, J.; Duncan, J.A.; Chodobski, A.; Szmydynger-Chodobska, J. The Choroid Plexus-Cerebrospinal Fluid System: From Development to Aging. Curr. Top. Dev. Biol. 2005, 71, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Gordleeva, S.; Kanakov, O.; Ivanchenko, M.; Zaikin, A.; Franceschi, C. Brain aging and garbage cleaning: Modelling the role of sleep, glymphatic system, and microglia senescence in the propagation of inflammaging. Semin. Immunopathol. 2020, 42, 647–665. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.; Yamakawa, G.R.; Shultz, S.R.; Mychasiuk, R. Is the glymphatic system the missing link between sleep impairments and neurological disorders? Examining the implications and uncertainties. Prog. Neurobiol. 2021, 198, 101917. [Google Scholar] [CrossRef]
- Hablitz, L.M.; Plá, V.; Giannetto, M.; Vinitsky, H.S.; Stæger, F.F.; Metcalfe, T.; Nguyen, R.; Benrais, A.; Nedergaard, M. Circadian control of brain glymphatic and lymphatic fluid flow. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sevensky, R.; Newville, J.C.; Tang, H.L.; Robinson, S.; Jantzie, L.L. Cumulative Damage: Cell Death in Posthemorrhagic Hydrocephalus of Prematurity. Cells 2021, 10, 1911. https://doi.org/10.3390/cells10081911
Sevensky R, Newville JC, Tang HL, Robinson S, Jantzie LL. Cumulative Damage: Cell Death in Posthemorrhagic Hydrocephalus of Prematurity. Cells. 2021; 10(8):1911. https://doi.org/10.3390/cells10081911
Chicago/Turabian StyleSevensky, Riley, Jessie C. Newville, Ho Lam Tang, Shenandoah Robinson, and Lauren L. Jantzie. 2021. "Cumulative Damage: Cell Death in Posthemorrhagic Hydrocephalus of Prematurity" Cells 10, no. 8: 1911. https://doi.org/10.3390/cells10081911
APA StyleSevensky, R., Newville, J. C., Tang, H. L., Robinson, S., & Jantzie, L. L. (2021). Cumulative Damage: Cell Death in Posthemorrhagic Hydrocephalus of Prematurity. Cells, 10(8), 1911. https://doi.org/10.3390/cells10081911