
Heat Shock Proteins: Connectors between Heart and Kidney


 , ,
, ,  and
and
Abstract
:1. Heat Shock Proteins: Definition and Function
2. Participation of HSPs in Cardiorenal Diseases
2.1. HSPs and Heart
2.2. Cardiorenal Syndrome
2.3. Role of HSPs in Cardiorenal Syndrome
3. Final Considerations
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACD | α-crystallin domain |
| ACS | acute coronary syndrome |
| ADHF | acute decompensated heart failure |
| AF | atrial fibrillation |
| AKI | acute kidney injury |
| Apaf-1 | apoptosis protease-activating factor 1 |
| APC | antigen presenting cell |
| CAV | cardiac allograft vasculopathy |
| CKD | chronic kidney disease |
| CRS | cardiorenal syndrome |
| CVD | cardiovascular diseases |
| DAMPs | damage-associated molecular patterns |
| eNOS | endothelial nitric oxide synthase |
| ESKD | end-stage kidney disease |
| GFR | glomerular filtration rate |
| GSK3β | glycogen synthase kinase 3-β |
| HD | hemodialysis |
| HSPs | Heat Shock Proteins |
| ICAM-1 | Intercellular adhesion molecule 1 |
| IL | interleukin |
| IRI | ischemia and reperfusion injury |
| JNK | c-Jun N-terminal kinase |
| LDL | low-density lipoproteins |
| MCP-1 | monocyte chemoattractant protein-1 |
| MKBP | myotonic dystrophy kinase-binding protein |
| NF-kB | nuclear factor kappa B |
| NO | nitric oxide |
| PAMPs | pathogen-associated molecular patterns |
| peritoneal dialysis fluid | |
| PDS | peritoneal dialysis solution |
| PIKK | PI3K-related protein kinase |
| RISC | RNA-induced silencing complex |
| ROS | reactive oxygen species |
| SCD | sudden cardiac death |
| sHSPs | small HSPs |
| TGF-β | transforming growth factor-β |
| TLRs | toll-like receptors |
| TNF-α | tumor necrosis factor-alpha |
| VSCM | vascular smooth muscle cells |
| WHO | World Health Organization |
References
- Ritossa, F. A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia 1962, 18, 571–573. [Google Scholar] [CrossRef]
- Ritossa, F. Experimental activation of specific loci in polytene chromosomes of Drosophila. Exp. Cell Res. 1964, 35, 601–607. [Google Scholar] [CrossRef]
- Currie, R.W. Protein synthesis in perfused rat hearts after in vivo hyperthermia and in vitro cold ischemia. Biochem. Cell Biol. 1988, 66, 13–19. [Google Scholar] [CrossRef]
- Currie, R.W.; Karmazyn, M.; Kloc, M.; Mailer, K. Heat-shock response is associated with enhanced postischemic ventricular recovery. Circ. Res. 1988, 63, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Currie, R.W. Effects of ischemia and perfusion temperature on the synthesis of stress-induced (heart shock) proteins in isolated and perfused rat hearts. J. Mol. Cell. Cardiol. 1987, 19, 795–808. [Google Scholar] [CrossRef]
- Li, Z.; Srivastava, P. Heat-shock proteins. Curr. Protoc. Immunol. 2004, Appendix 1, Appendix 1T. [Google Scholar] [CrossRef]
- Chen, Y.; Voegeli, T.S.; Liu, P.P.; Noble, E.G.; Currie, R.W. Heat Shock Paradox and a New Role of Heat Shock Proteins and their Receptors as Anti-Inflammation Targets. Inflamm. Allergy-Drug Targets 2007, 6, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Snoeckx, L.H.E.H.; Cornelussen, R.N.; Van Nieuwenhoven, F.A.; Reneman, R.S.; Van der Vusse, G.J. Heat Shock Proteins and Cardiovascular Pathophysiology. Physiol. Rev. 2001, 81, 1461–1497. [Google Scholar] [CrossRef]
- O’Neill, S.; Harrison, E.M.; Ross, J.A.; Wigmore, S.J.; Hughes, J. Heat-Shock Proteins and Acute Ischaemic Kidney Injury. Nephron Exp. Nephrol. 2014, 126, 167–174. [Google Scholar] [CrossRef]
- Polla, B.S.; Bachelet, M.; Elia, G.; Santoro, M.G. Stress Proteins in Inflammationa. Ann. N. Y. Acad. Sci. 1998, 851, 75–85. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Webster, J.M.; Darling, A.L.; Uversky, V.N.; Blair, L.J. Small Heat Shock Proteins, Big Impact on Protein Aggregation in Neurodegenerative Disease. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [Green Version]
- Dasari, S.; Alexander, M.P.; Vrana, J.A.; Theis, J.D.; Mills, J.R.; Negron, V.; Sethi, S.; Dispenzieri, A.; Highsmith, W.E.; Nasr, S.H.; et al. DnaJ Heat Shock Protein Family B Member 9 Is a Novel Biomarker for Fibrillary GN. J. Am. Soc. Nephrol. 2018, 29, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, M.; Imanaka-Yoshida, K.; Yoshida, T.; Wood, M.; Fearns, C.; Tatake, R.; Lee, D. A crucial role of mitochondrial Hsp40 in preventing dilated cardiomyopathy. Nat. Med. 2006, 12, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Brocchieri, L.; Karlin, S. Conservation among HSP60 sequences in relation to structure, function, and evolution. Protein Sci. 2008, 9, 476–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, F.U. Molecular Chaperones in the Cytosol: From Nascent Chain to Folded Protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.A.; Kolesar, J.E.; Perlman, P.S.; Butow, R.A. A function for the mitochondrial chaperonin Hsp60 in the structure and transmission of mitochondrial DNA nucleoids in Saccharomyces cerevisiae. J. Cell Biol. 2003, 163, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Pockley, A.G.; Muthana, M.; Calderwood, S.K. The dual immunoregulatory roles of stress proteins. Trends Biochem. Sci. 2008, 33, 71–79. [Google Scholar] [CrossRef]
- Habich, C.; Burkart, V. Heat shock protein 60: Regulatory role on innate immune cells. Cell. Mol. Life Sci. 2007, 64, 742–751. [Google Scholar] [CrossRef]
- Flaherty, K.M.; DeLuca-Flaherty, C.; McKay, D.B. Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature 1990, 346, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Asea, A.; Rehli, M.; Kabingu, E.; Boch, J.A.; Baré, O.; Auron, P.E.; Stevenson, M.A.; Calderwood, S.K. Novel Signal Transduction Pathway Utilized by Extracellular HSP70. J. Biol. Chem. 2002, 277, 15028–15034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triantafilou, M.; Triantafilou, K. Heat-shock protein 70 and heat-shock protein 90 associate with Toll-like receptor 4 in response to bacterial lipopolysaccharide. Biochem. Soc. Trans. 2004, 32, 636–639. [Google Scholar] [CrossRef] [Green Version]
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat. Cell Biol. 2000, 2, 469–475. [Google Scholar] [CrossRef]
- Vostakolaei, M.A.; Hatami-Baroogh, L.; Babaei, G.; Molavi, O.; Kordi, S.; Abdolalizadeh, J. Hsp70 in cancer: A double agent in the battle between survival and death. J. Cell. Physiol. 2021, 236, 3420–3444. [Google Scholar] [CrossRef]
- Arya, R.; Mallik, M.; Lakhotia, S.C. Heat shock genes–integrating cell survival and death. J. Biosci. 2007, 32, 595–610. [Google Scholar] [CrossRef]
- Nayak Rao, S. The role of heat shock proteins in kidney disease. J. Transl. Intern. Med. 2016, 4, 114–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asea, A. Chaperokine-induced signal transduction pathways. Exerc. Immunol. Rev. 2003, 9, 25–33. [Google Scholar]
- Matzinger, P. Tolerance, Danger, and the Extended Family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Srivastava, P. Interaction of heat shock proteins with peptides and antigen presenting cells: Chaperoning of the innate and adaptive immune responses. Annu. Rev. Immunol. 2002, 20, 395–425. [Google Scholar] [CrossRef]
- Mohamed, B.A.; Barakat, A.Z.; Zimmermann, W.-H.; Bittner, R.E.; Mühlfeld, C.; Hünlich, M.; Engel, W.; Maier, L.S.; Adham, I.M. Targeted disruption of Hspa4 gene leads to cardiac hypertrophy and fibrosis. J. Mol. Cell. Cardiol. 2012, 53, 459–468. [Google Scholar] [CrossRef]
- Polier, S.; Dragovic, Z.; Hartl, F.U.; Bracher, A. Structural Basis for the Cooperation of Hsp70 and Hsp110 Chaperones in Protein Folding. Cell 2008, 133, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Biebl, M.M.; Buchner, J. Structure, Function, and Regulation of the Hsp90 Machinery. Cold Spring Harb. Perspect. Biol. 2019, 11, a034017. [Google Scholar] [CrossRef] [Green Version]
- Karagöz, G.E.; Rüdiger, S.G.D. Hsp90 interaction with clients. Trends Biochem. Sci. 2015, 40, 117–125. [Google Scholar] [CrossRef]
- Kim, Y.; Alarcon, S.; Lee, S.; Lee, M.-J.; Giaccone, G.; Neckers, L.; Trepel, J. Update on Hsp90 Inhibitors in Clinical Trial. Curr. Top. Med. Chem. 2009, 9, 1479–1492. [Google Scholar] [CrossRef]
- Vos, M.J.; Hageman, J.; Carra, S.; Kampinga, H.H. Structural and Functional Diversities between Members of the Human HSPB, HSPH, HSPA, and DNAJ Chaperone Families. Biochemistry 2008, 47, 7001–7011. [Google Scholar] [CrossRef] [PubMed]
- Currie, R.W. Synthesis of stress-induced protein in isolated and perfused rat hearts. Biochem. Cell Biol. 1986, 64, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Welch, W.J. Mammalian stress response: Cell physiology, structure/function of stress proteins, and implications for medicine and disease. Physiol. Rev. 1992, 72, 1063–1081. [Google Scholar] [CrossRef] [PubMed]
- Latchman, D. Heat shock proteins and cardiac protection. Cardiovasc. Res. 2001, 51, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Dillmann, W.H.; Mehta, H.B.; Barrieux, A.; Guth, B.D.; Neeley, W.E.; Ross, J. Ischemia of the dog heart induces the appearance of a cardiac mRNA coding for a protein with migration characteristics similar to heat-shock/stress protein 71. Circ. Res. 1986, 59, 110–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranek, M.J.; Stachowski, M.J.; Kirk, J.A.; Willis, M.S. The role of heat shock proteins and co-chaperones in heart failure. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160530. [Google Scholar] [CrossRef] [Green Version]
- De Jong, P.R.; Schadenberg, A.W.L.; Jansen, N.J.G.; Prakken, B.J. Hsp70 and cardiac surgery: Molecular chaperone and inflammatory regulator with compartmentalized effects. Cell Stress Chaperones 2009, 14, 117–131. [Google Scholar] [CrossRef] [Green Version]
- St. Rammos, K.; George, J.; Koullias, G.J.; Hassan, M.O.; Argyrakis, N.P.; Voucharas, C.G.; Scarupa, S.J.; Cowte, T.G. Low preoperative HSP70 atrial myocardial levels correlate significantly with high incidence of postoperative atrial fibrillation after cardiac surgery. Cardiovasc. Surg. 2002, 10, 228–232. [Google Scholar] [CrossRef]
- Mandal, K.; Torsney, E.; Poloniecki, J.; Camm, A.J.; Xu, Q.; Jahangiri, M. Association of High Intracellular, But Not Serum, Heat Shock Protein 70 With Postoperative Atrial Fibrillation. Ann. Thorac. Surg. 2005, 79, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Afzal, A.R.; Mandal, K.; Nyamweya, S.; Foteinos, G.; Poloniecki, J.; Camm, A.J.; Jahangiri, M.; Xu, Q. Association of Met439Thr Substitution in Heat Shock Protein 70 Gene with Postoperative Atrial Fibrillation and Serum HSP70 Protein Levels. Cardiology 2008, 110, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Currie, R.W.; Karmazyn, M. Improved post-ischemic ventricular recovery in the absence of changes in energy metabolism in working rat hearts following heat-shock. J. Mol. Cell. Cardiol. 1990, 22, 631–636. [Google Scholar] [CrossRef]
- Karmazyn, M.; Mailer, K.; Currie, R.W. Acquisition and decay of heat-shock-enhanced postischemic ventricular recovery. Am. J. Physiol. Circ. Physiol. 1990, 259, H424–H431. [Google Scholar] [CrossRef] [PubMed]
- Dybdahl, B.; Wahba, A.; Lien, E.; Flo, T.H.; Waage, A.; Qureshi, N.; Sellevold, O.F.M.; Espevik, T.; Sundan, A. Inflammatory response after open heart surgery: Release of heat-shock protein 70 and signaling through toll-like receptor-4. Circulation 2002, 105, 685–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.L.; Lun, M.; Schworer, C.M.; Blasick, T.M.; Masker, K.K.; Jones, J.B.; Carey, D.J. Heat shock protein expression is highly sensitive to ischemia-reperfusion injury in rat kidneys. Ann. Clin. Lab. Sci. 2008, 38, 57–64. [Google Scholar]
- Cai, W.-F.; Zhang, X.-W.; Yan, H.-M.; Ma, Y.-G.; Wang, X.-X.; Yan, J.; Xin, B.-M.; Lv, X.-X.; Wang, Q.-Q.; Wang, Z.-Y.; et al. Intracellular or extracellular heat shock protein 70 differentially regulates cardiac remodelling in pressure overload mice. Cardiovasc. Res. 2010, 88, 140–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutter, J.J.; Mestril, R.; Tam, E.K.W.; Sievers, R.E.; Dillmann, W.H.; Wolfe, C.L. Overexpression of Heat Shock Protein 72 in Transgenic Mice Decreases Infarct Size In Vivo. Circulation 1996, 94, 1408–1411. [Google Scholar] [CrossRef]
- Radford, N.B.; Fina, M.; Benjamin, I.J.; Moreadith, R.W.; Graves, K.H.; Zhao, P.; Gavva, S.; Wiethoff, A.; Sherry, A.D.; Malloy, C.R.; et al. Cardioprotective effects of 70-kDa heat shock protein in transgenic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 2339–2342. [Google Scholar] [CrossRef] [Green Version]
- Naka K, K.; Vezyraki, P.; Kalaitzakis, A.; Zerikiotis, S.; Michalis, L.; Angelidis, C. Hsp70 regulates the doxorubicin-mediated heart failure in Hsp70-transgenic mice. Cell Stress Chaperones 2014, 19, 853–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, B.C.; Sapra, G.; Patterson, N.L.; Cemerlang, N.; Kiriazis, H.; Ueyama, T.; Febbraio, M.A.; McMullen, J.R. Long-Term Overexpression of Hsp70 Does Not Protect against Cardiac Dysfunction and Adverse Remodeling in a MURC Transgenic Mouse Model with Chronic Heart Failure and Atrial Fibrillation. PLoS ONE 2015, 10, e0145173. [Google Scholar] [CrossRef]
- Su, X.; Sykes, J.B.; Ao, L.; Raeburn, C.D.; Fullerton, D.A.; Meng, X. Extracellular heat shock cognate protein 70 induces cardiac functional tolerance to endotoxin: Differential effect on TNF-α and ICAM-1 levels in heart tissue. Cytokine 2010, 51, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xu, Z.; Zhou, L.; Chen, Y.; He, M.; Cheng, L.; Hu, F.B.; Tanguay, R.M.; Wu, T. Plasma levels of Hsp70 and anti-Hsp70 antibody predict risk of acute coronary syndrome. Cell Stress Chaperones 2010, 15, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornej, J.; Reinhardt, C.; Kosiuk, J.; Arya, A.; Hindricks, G.; Adams, V.; Husser, D.; Bollmann, A. Response of circulating heat shock protein 70 and anti-heat shock protein 70 antibodies to catheter ablation of atrial fibrillation. J. Transl. Med. 2013, 11, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Marion, D.M.S.; Lanters, E.A.H.; Ramos, K.S.; Li, J.; Wiersma, M.; Baks-te Bulte, L.; Muskens, A.J.Q.M.; Boersma, E.; de Groot, N.M.S.; Brundel, B.J.J.M. Evaluating Serum Heat Shock Protein Levels as Novel Biomarkers for Atrial Fibrillation. Cells 2020, 9, 2105. [Google Scholar] [CrossRef]
- De Almeida Santos-Junior, V.; Lollo, P.C.B.; Cantero, M.A.; Moura, C.S.; Amaya-Farfan, J.; Morato, P.N. Heat shock proteins: Protection and potential biomarkers for ischemic injury of cardiomyocytes after surgery. Braz. J. Cardiovasc. Surg. 2018, 33, 291–302. [Google Scholar] [CrossRef]
- Hu, Y.F.; Yeh, H.I.; Tsao, H.M.; Tai, C.T.; Lin, Y.J.; Chang, S.L.; Lo, L.W.; Tuan, T.C.; Suenari, K.; Li, C.H.; et al. Electrophysiological correlation and prognostic impact of heat shock protein 27 in atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2012, 5, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenek, P.; Kmecova, J.; Kucerova, D.; Bajuszova, Z.; Musil, P.; Gazova, A.; Ochodnicky, P.; Klimas, J.; Kyselovic, J. Isoproterenol-induced heart failure in the rat is associated with nitric oxide-dependent functional alterations of cardiac function. Eur. J. Heart Fail. 2009, 11, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Ke, X.; Chen, J.; Peng, L.; Zhang, W.; Yang, Y.; Liao, X.; Mo, L.; Guo, R.; Feng, J.; Hu, C.; et al. Heat shock protein 90/Akt pathway participates in the cardioprotective effect of exogenous hydrogen sulfide against high glucose-induced injury to H9c2 cells. Int. J. Mol. Med. 2017, 39, 1001–1010. [Google Scholar] [CrossRef]
- Tu, R.-H.; Li, Q.-J.; Huang, Z.; He, Y.; Meng, J.-J.; Zheng, H.-L.; Zeng, Z.-Y.; Zhong, G.-Q. Novel Functional Role of Heat Shock Protein 90 in Mitochondrial Connexin 43-Mediated Hypoxic Postconditioning. Cell. Physiol. Biochem. 2017, 44, 982–997. [Google Scholar] [CrossRef] [Green Version]
- Businaro, R.; Profumo, E.; Tagliani, A.; Buttari, B.; Leone, S.; D’Amati, G.; Ippoliti, F.; Leopizzi, M.; D’Arcangelo, D.; Capoano, R.; et al. Heat-shock protein 90: A novel autoantigen in human carotid atherosclerosis. Atherosclerosis 2009, 207, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Madrigal-Matute, J.; Martin-Ventura, J.L.; Blanco-Colio, L.M.; Egido, J.; Michel, J.-B.; Meilhac, O. Heat-shock proteins in cardiovascular disease. Adv. Clin. Chem. 2011, 54, 1–43. [Google Scholar] [CrossRef]
- Tamura, S.; Marunouchi, T.; Tanonaka, K. Heat-shock protein 90 modulates cardiac ventricular hypertrophy via activation of MAPK pathway. J. Mol. Cell. Cardiol. 2019, 127, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Datta, R.; Bansal, T.; Rana, S.; Datta, K.; Datta Chaudhuri, R.; Chawla-Sarkar, M.; Sarkar, S. Myocyte-Derived Hsp90 Modulates Collagen Upregulation via Biphasic Activation of STAT-3 in Fibroblasts during Cardiac Hypertrophy. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- García, R.; Merino, D.; Gómez, J.M.; Nistal, J.F.; Hurlé, M.A.; Cortajarena, A.L.; Villar, A.V. Extracellular heat shock protein 90 binding to TGFβ receptor I participates in TGFβ-mediated collagen production in myocardial fibroblasts. Cell. Signal. 2016, 28, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Yellon, D.M.; Sivaraman, V.; Das, D.; Boi-Doku, C.; Arjun, S.; Zheng, Y.; A Riquelme, J.; Kearney, J.; Sharma, V.; et al. Plasma Exosomes Protect the Myocardium From Ischemia-Reperfusion Injury. J. Am. Coll. Cardiol. 2015, 65, 1525–1536. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Kim, M.; Min, H.-K.; Lee, Y.-U.; Kwon, D.-H.; Lee, M.; Lee, S.; Kook, T.; Joung, H.; Nam, K.-I.; et al. Inhibition of heat shock protein 70 blocks the development of cardiac hypertrophy by modulating the phosphorylation of histone deacetylase 2. Cardiovasc. Res. 2019, 115, 1850–1860. [Google Scholar] [CrossRef]
- Marunouchi, T.; Nishiumi, C.; Iinuma, S.; Yano, E.; Tanonaka, K. Effects of Hsp90 inhibitor on the RIP1-RIP3-MLKL pathway during the development of heart failure in mice. Eur. J. Pharmacol. 2021, 898, 173987. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Bao, H.; Jin, C.; Zhou, J.; Hua, F.; Li, K.; Lv, X.; Cui, B.; Hu, Z.; Zhang, X. Targeting Extracellular Heat Shock Protein 70 Ameliorates Doxorubicin-Induced Heart Failure Through Resolution of Toll-Like Receptor 2–Mediated Myocardial Inflammation. J. Am. Heart Assoc. 2019, 8. [Google Scholar] [CrossRef]
- Bright, R. Cases and Observations Illustrative of Renal Disease, Accompanied with the Secretion of Albuminous Urine. Med.Chir. Rev. 1836, 25, 23–35. [Google Scholar]
- Ronco, C.; McCullough, P.; Anker, S.D.; Anand, I.; Aspromonte, N.; Bagshaw, S.M.; Bellomo, R.; Berl, T.; Bobek, I.; Cruz, D.N.; et al. Cardio-renal syndromes: Report from the consensus conference of the Acute Dialysis Quality Initiative. Eur. Heart J. 2010, 31, 703–711. [Google Scholar] [CrossRef]
- Ronco, C.; Di Lullo, L. Cardiorenal syndrome. Heart Fail. Clin. 2014, 10, 251–280. [Google Scholar] [CrossRef] [PubMed]
- Savira, F.; Magaye, R.; Liew, D.; Reid, C.; Kelly, D.J.; Kompa, A.R.; Sangaralingham, S.J.; Burnett, J.C.; Kaye, D.; Wang, B.H. Cardiorenal syndrome: Multi-organ dysfunction involving the heart, kidney and vasculature. Br. J. Pharmacol. 2020, 177, 2906–2922. [Google Scholar] [CrossRef]
- Kumar, U.; Wettersten, N.; Garimella, P.S. Cardiorenal Syndrome: Pathophysiology. Cardiol. Clin. 2019, 37, 251–265. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 15 June 2021).
- Bagshaw, S.M.; Hoste, E.A.; Braam, B.; Briguori, C.; Kellum, J.A.; McCullough, P.A.; Ronco, C. Cardiorenal Syndrome Type 3: Pathophysiologic and Epidemiologic Considerations. Contrib. Nephrol. 2013, 182, 137–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trentin-Sonoda, M.; Da Silva, R.C.; Kmit, F.V.; Abrahão, M.V.; Cahli, G.M.; Brasil, G.V.; Muzi-Filho, H.; Silva, P.A.; Tovar-Moll, F.F.; Vieyra, A.; et al. Knockout of toll-like receptors 2 and 4 prevents renal ischemia-reperfusion-induced cardiac hypertrophy in mice. PLoS ONE 2015, 10, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Milanesi, S.; Garibaldi, S.; Saio, M.; Ghigliotti, G.; Picciotto, D.; Ameri, P.; Garibotto, G.; Barisione, C.; Verzola, D. Indoxyl Sulfate Induces Renal Fibroblast Activation through a Targetable Heat Shock Protein 90-Dependent Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 1–11. [Google Scholar] [CrossRef]
- Musial, K.; Szprynger, K.; Szczepańska, M.; Zwolińska, D. The Heat Shock Protein Profile in Children with Chronic Kidney Disease. Perit. Dial. Int. J. Int. Soc. Perit. Dial. 2010, 30, 227–232. [Google Scholar] [CrossRef]
- Amador-Martínez, I.; Pérez-Villalva, R.; Uribe, N.; Cortés-González, C.; Bobadilla, N.A.; Barrera-Chimal, J. Reduced endothelial nitric oxide synthase activation contributes to cardiovascular injury during chronic kidney disease progression. Am. J. Physiol. Physiol. 2019, 317, F275–F285. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Chimal, J.; Perez-Villalva, R.; Ortega, J.A.; Uribe, N.; Gamba, G.; Cortes-Gonzalez, C.; Bobadilla, N.A. Intra-renal transfection of heat shock protein 90 alpha or beta (Hsp90 or Hsp90 ) protects against ischemia/reperfusion injury. Nephrol. Dial. Transplant. 2014, 29, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, V.; Mejía-Vilet, J.M.; Hernández, D.; Gamba, G.; Bobadilla, N.A. Radicicol, a heat shock protein 90 inhibitor, reduces glomerular filtration rate. Am. J. Physiol. Physiol. 2008, 295, F1044–F1051. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; Fernandez-Garcia, C.E.; Gomez-Guerrero, C.; Lopez-Franco, O.; Muñoz-Garcia, B.; Egido, J.; Blanco-Colio, L.M.; Martin-Ventura, J.L. HSP90 inhibition by 17-DMAG attenuates oxidative stress in experimental atherosclerosis. Cardiovasc. Res. 2012, 95, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Lazaro, I.; Oguiza, A.; Recio, C.; Mallavia, B.; Madrigal-Matute, J.; Blanco, J.; Egido, J.; Martin-Ventura, J.-L.; Gomez-Guerrero, C. Targeting HSP90 Ameliorates Nephropathy and Atherosclerosis Through Suppression of NF-κB and STAT Signaling Pathways in Diabetic Mice. Diabetes 2015, 64, 3600–3613. [Google Scholar] [CrossRef] [Green Version]
- Thakar, C.V.; Christianson, A.; Himmelfarb, J.; Leonard, A.C. Acute Kidney Injury Episodes and Chronic Kidney Disease Risk in Diabetes Mellitus. Clin. J. Am. Soc. Nephrol. 2011, 6, 2567–2572. [Google Scholar] [CrossRef] [PubMed]
- Pinier, C.; Gatault, P.; François, M.; Barbet, C.; Longuet, H.; Rabot, N.; Noble, J.; Bailly, E.; Buchler, M.; Sautenet, B.; et al. Renal function at the time of nephrology referral but not dialysis initiation as a risk for death in patients with diabetes mellitus. Clin. Kidney J. 2018, 11, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gall, J.M.; Bonegio, R.G.B.; Havasi, A.; Hunt, C.R.; Sherman, M.Y.; Schwartz, J.H.; Borkan, S.C. Induction of heat shock protein 70 inhibits ischemic renal injury. Kidney Int. 2011, 79, 861–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinstein, D.L.; Galea, E.; Aquino, D.A.; Li, G.C.; Xu, H.; Reis, D.J. Heat Shock Protein 70 Suppresses Astroglial-inducible Nitric-oxide Synthase Expression by Decreasing NFκB Activation. J. Biol. Chem. 1996, 271, 17724–17732. [Google Scholar] [CrossRef] [Green Version]
- Yoo, C.-G.; Lee, S.; Lee, C.-T.; Kim, Y.W.; Han, S.K.; Shim, Y.-S. Anti-Inflammatory Effect of Heat Shock Protein Induction Is Related to Stabilization of IκBα Through Preventing IκB Kinase Activation in Respiratory Epithelial Cells. J. Immunol. 2000, 164, 5416–5423. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Xiao, W.; Wang, H.; Calderwood, S.K.; Xiao, X. The Anti-inflammatory Effects of Heat Shock Protein 72 Involve Inhibition of High-Mobility-Group Box 1 Release and Proinflammatory Function in Macrophages. J. Immunol. 2007, 179, 1236–1244. [Google Scholar] [CrossRef] [Green Version]
- Ran, R.; Lu, A.; Zhang, L.; Tang, Y.; Zhu, H.; Xu, H.; Feng, Y.; Han, C.; Zhou, G.; Rigby, A.C.; et al. Hsp70 promotes TNF-mediated apoptosis by binding IKK and impairing NF- B survival signaling. Genes Dev. 2004, 18, 1466–1481. [Google Scholar] [CrossRef] [Green Version]
- Maddock, A.L.; Westenfelder, C. Urea induces the heat shock response in human neuroblastoma cells. J. Am. Soc. Nephrol. 1996, 7, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Neuhofer, W.; Lugmayr, K.; Fraek, M.-L.; Beck, F.-X. Regulated Overexpression of Heat Shock Protein 72 Protects Madin-Darby Canine Kidney Cells from the Detrimental Effects of High Urea Concentrations. J. Am. Soc. Nephrol. 2001, 12, 2565–2571. [Google Scholar] [CrossRef]
- Marzec, L.; Zdrojewski, Z.; Liberek, T.; Bryl, E.; Chmielewski, M.; Witkowski, J.M.; Rutkowski, B. Expression of Hsp72 protein in chronic kidney disease patients. Scand. J. Urol. Nephrol. 2009, 43, 400–408. [Google Scholar] [CrossRef]
- Lebherz-Eichinger, D.; Ankersmit, H.J.; Hacker, S.; Hetz, H.; Kimberger, O.; Schmidt, E.M.; Reiter, T.; Hörl, W.H.; Haas, M.; Krenn, C.G.; et al. HSP27 and HSP70 serum and urine levels in patients suffering from chronic kidney disease. Clin. Chim. Acta 2012, 413, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Bangen, P.; Edemir, B.; Hillebrand, U.; Pavenstadt, H.; Heidenreich, S.; Lang, D. The HSP72 stress response of monocytes from patients on haemodialysis is impaired. Nephrol. Dial. Transplant. 2009, 24, 2838–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aufricht, C.; Endemann, M.; Bidmon, B.; Arbeiter, K.; Mueller, T.; Regele, H.; Herkner, K.; Eickelberg, O. Peritoneal Dialysis Fluids Induce the Stress Response in Human Mesothelial Cells. Perit. Dial. Int. J. Int. Soc. Perit. Dial. 2001, 21, 1–5. [Google Scholar] [CrossRef]
- Arbeiter, K.; Bidmon, B.; Endemann, M.; Ruffingshofer, D.; Mueller, T.; Regele, H.; Eickelberg, O.; Aufricht, C. Induction of Mesothelial HSP-72 upon In vivo Exposure to Peritoneal Dialysis Fluid. J. Int. Soc. Perit. Dial. 2003, 23, 499–501. [Google Scholar] [CrossRef]
- Bender, T.O.; Böhm, M.; Kratochwill, K.; Lederhuber, H.; Endemann, M.; Bidmon, B.; Aufricht, C. HSP-Mediated Cytoprotection of Mesothelial Cells in Experimental Acute Peritoneal Dialysis. Perit. Dial. Int. J. Int. Soc. Perit. Dial. 2010, 30, 294–299. [Google Scholar] [CrossRef]
- Lu, T.-S.; Lim, K.; Molostvov, G.; Yang, Y.-C.; Yiao, S.-Y.; Zehnder, D.; Hsiao, L.-L. Induction of intracellular heat-shock protein 72 prevents the development of vascular smooth muscle cell calcification. Cardiovasc. Res. 2012, 96, 524–532. [Google Scholar] [CrossRef] [Green Version]
- London, G.M. Arterial media calcification in end-stage renal disease: Impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transplant. 2003, 18, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Detrano, R.; Guerci, A.D.; Carr, J.J.; Bild, D.E.; Burke, G.; Folsom, A.R.; Liu, K.; Shea, S.; Szklo, M.; Bluemke, D.A.; et al. Coronary Calcium as a Predictor of Coronary Events in Four Racial or Ethnic Groups. N. Engl. J. Med. 2008, 358, 1336–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Quyyumi, A.A.; Wu, H.; Csako, G.; Rott, D.; Zalles-Ganley, A.; Ogunmakinwa, J.; Halcox, J.; Epstein, S.E. Increased Serum Levels of Heat Shock Protein 70 Are Associated With Low Risk of Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Fu, X. Insights into How Small Heat Shock Proteins Bind a Great Diversity of Substrate Proteins: A Super-Transformer Model. In The Big Book on Small Heat Shock Proteins; Springer: Berlin/Heidelberg, Germany, 2015; pp. 101–117. [Google Scholar]
- Arrigo, A.-P.; Virot, S.; Chaufour, S.; Firdaus, W.; Kretz-Remy, C.; Diaz-Latoud, C. Hsp27 Consolidates Intracellular Redox Homeostasis by Upholding Glutathione in Its Reduced Form and by Decreasing Iron Intracellular Levels. Antioxid. Redox Signal. 2005, 7, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Jaroszyński, A.; Jaroszyńska, A.; Zaborowski, T.; Drelich-Zbroja, A.; Zapolski, T.; Dąbrowski, W. Serum heat shock protein 27 levels predict cardiac mortality in hemodialysis patients. BMC Nephrol. 2018, 19, 359. [Google Scholar] [CrossRef]
- Ghayour-Mobarhan, M.; Saber, H.; Ferns, G.A.A. The potential role of heat shock protein 27 in cardiovascular disease. Clin. Chim. Acta 2012, 413, 15–24. [Google Scholar] [CrossRef]
- Rayner, K.; Chen, Y.-X.; Siebert, T.; O’Brien, E.R. Heat Shock Protein 27: Clue to Understanding Estrogen-Mediated Atheroprotection? Trends Cardiovasc. Med. 2010, 20, 53–57. [Google Scholar] [CrossRef]
- Keezer, S.M.; Ivie, S.E.; Krutzsch, H.C.; Tandle, A.; Libutti, S.K.; Roberts, D.D. Angiogenesis inhibitors target the endothelial cell cytoskeleton through altered regulation of heat shock protein 27 and cofilin. Cancer Res. 2003, 63, 6405–6412. [Google Scholar] [PubMed]
- De, A.K.; Kodys, K.M.; Yeh, B.S.; Miller-Graziano, C. Exaggerated Human Monocyte IL-10 Concomitant to Minimal TNF-α Induction by Heat-Shock Protein 27 (Hsp27) Suggests Hsp27 Is Primarily an Antiinflammatory Stimulus. J. Immunol. 2000, 165, 3951–3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.K.; Park, E.-C.; Bae, S.W.; Park, M.Y.; Kim, S.W.; Yoo, H.S.; Tudev, M.; Ko, Y.H.; Choi, Y.-H.; Kim, S.; et al. Expression of Heat Shock Protein 27 in Human Atherosclerotic Plaques and Increased Plasma Level of Heat Shock Protein 27 in Patients With Acute Coronary Syndrome. Circulation 2006, 114, 886–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, A.I.; Wait, R.; Mitchell, A.G.; Banner, N.R.; Dunn, M.J.; Rose, M.L. Heat Shock Protein 27 Is Associated With Freedom From Graft Vasculopathy After Human Cardiac Transplantation. Circ. Res. 2005, 97, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohke, T.; Wada, A.; Isono, T.; Fujii, M.; Yamamoto, T.; Tsutamoto, T.; Horie, M. Proteomic Analysis Reveals Significant Alternations of Cardiac Small Heat Shock Protein Expression in Congestive Heart Failure. J. Card. Fail. 2006, 12, 77–84. [Google Scholar] [CrossRef]
- Yamboliev, I.A.; Hedges, J.C.; Mutnick, J.L.-M.; Adam, L.P.; Gerthoffer, W.T. Evidence for modulation of smooth muscle force by the p38 MAP kinase/HSP27 pathway. Am. J. Physiol. Circ. Physiol. 2000, 278, H1899–H1907. [Google Scholar] [CrossRef] [Green Version]
- Vander Heide, R.S. Increased expression of HSP27 protects canine myocytes from simulated ischemia-reperfusion injury. Am. J. Physiol. Circ. Physiol. 2002, 282, H935–H941. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Cleveland, J.C.; Ao, L.; Li, J.; Zeng, Q.; Fullerton, D.A.; Meng, X. Human Myocardium Releases Heat Shock Protein 27 (HSP27) after Global Ischemia: The Proinflammatory Effect of Extracellular HSP27 through Toll-like Receptor (TLR)-2 and TLR4. Mol. Med. 2014, 20, 280–289. [Google Scholar] [CrossRef]
- Chen, H.; Wu, X.; Lu, X.; Zhu, L.; Wang, L.; Yang, H.; Chen, H.; Yuan, W. Phosphorylated heat shock protein 27 is involved in enhanced heart tolerance to ischemia in short-term type 1 diabetic rats1. Acta Pharmacol. Sin. 2005, 26, 806–812. [Google Scholar] [CrossRef]
- Batulan, Z.; Pulakazhi Venu, V.K.; Li, Y.; Koumbadinga, G.; Alvarez-Olmedo, D.G.; Shi, C.; O’Brien, E.R. Extracellular Release and Signaling by Heat Shock Protein 27: Role in Modifying Vascular Inflammation. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, S.; Ingman, T.G.; Wigmore, S.J.; Harrison, E.M.; Bellamy, C.O. Differential expression of heat shock proteins in healthy and diseased human renal allografts. Ann. Transplant. 2013, 18, 550–557. [Google Scholar] [CrossRef]
- Guo, Q.; Du, X.; Zhao, Y.; Zhang, D.; Yue, L.; Wang, Z. Ischemic postconditioning prevents renal ischemia reperfusion injury through the induction of heat shock proteins in rats. Mol. Med. Rep. 2014, 10, 2875–2881. [Google Scholar] [CrossRef] [Green Version]
- Roguin, N.; Greif, Z.; Schneeweiss, A.; Yahalom, M.; Hartman, C.; Saab, K.; Glusman, A.; Milgram, E.; Shasha, S. Transient mitral regurgitation in acute glomerulonephritis. Pediatr. Cardiol. 1993, 14, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Caruso Bavisotto, C.; Alberti, G.; Vitale, A.M.; Paladino, L.; Campanella, C.; Rappa, F.; Gorska, M.; Conway de Macario, E.; Cappello, F.; Macario, A.J.L.; et al. Hsp60 Post-translational Modifications: Functional and Pathological Consequences. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef]
- Lin, L.; Kim, S.C.; Wang, Y.; Gupta, S.; Davis, B.; Simon, S.I.; Torre-Amione, G.; Knowlton, A.A. HSP60 in heart failure: Abnormal distribution and role in cardiac myocyte apoptosis. Am. J. Physiol. Circ. Physiol. 2007, 293, H2238–H2247. [Google Scholar] [CrossRef] [Green Version]
- Pfister, G.; Stroh, C.M.; Perschinka, H.; Kind, M.; Knoflach, M.; Hinterdorfer, P.; Wick, G. Detection of HSP60 on the membrane surface of stressed human endothelial cells by atomic force and confocal microscopy. J. Cell Sci. 2005, 118, 1587–1594. [Google Scholar] [CrossRef] [Green Version]
- Osterloh, A.; Veit, A.; Gessner, A.; Fleischer, B.; Breloer, M. Hsp60-mediated T cell stimulation is independent of TLR4 and IL-12. Int. Immunol. 2008, 20, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; He, M.; Cheng, L.; Chen, Y.; Zhou, L.; Zeng, H.; Pockley, A.G.; Hu, F.B.; Wu, T. Elevated Heat Shock Protein 60 Levels Are Associated With Higher Risk of Coronary Heart Disease in Chinese. Circulation 2008, 118, 2687–2693. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Mandal, K.; Schett, G.; Mayr, M.; Wick, G.; Oberhollenzer, F.; Willeit, J.; Kiechl, S.; Xu, Q. Association of Serum-Soluble Heat Shock Protein 60 With Carotid Atherosclerosis. Stroke 2005, 36, 2571–2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonanad, C.; Núñez, J.; Sanchis, J.; Bodi, V.; Chaustre, F.; Chillet, M.; Miñana, G.; Forteza, M.J.; Palau, P.; Núñez, E.; et al. Serum Heat Shock Protein 60 in Acute Heart Failure: A New Biomarker? Congest. Hear. Fail. 2013, 19, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-C.; Stice, J.P.; Chen, L.; Jung, J.S.; Gupta, S.; Wang, Y.; Baumgarten, G.; Trial, J.; Knowlton, A.A. Extracellular Heat Shock Protein 60, Cardiac Myocytes, and Apoptosis. Circ. Res. 2009, 105, 1186–1195. [Google Scholar] [CrossRef] [Green Version]
- Junho, C.V.C.; Trentin-Sonoda, M.; Alvim, J.M.; Gaisler-Silva, F.; Carneiro-Ramos, M.S. Ca2+/calmodulin-dependent kinase ii delta b is essential for cardiomyocyte hypertrophy and complement gene expression after LPS and HSP60 stimulation in vitro. Braz. J. Med. Biol. Res. 2019, 52. [Google Scholar] [CrossRef] [Green Version]
- Malik, Z.A.; Kott, K.S.; Poe, A.J.; Kuo, T.; Chen, L.; Ferrara, K.W.; Knowlton, A.A. Cardiac myocyte exosomes: Stability, HSP60, and proteomics. Am. J. Physiol. Circ. Physiol. 2013, 304, H954–H965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, C.; Wei, X.; Li, P.; Cui, Y.; Qin, Y.; Wei, X.; Jin, M.; Kohama, K.; Gao, Y. Heat shock protein 60 stimulates the migration of vascular smooth muscle cells via Toll-like receptor 4 and ERK MAPK activation. Sci. Rep. 2015, 5, 15352. [Google Scholar] [CrossRef]
- Kreutmayer, S.; Csordas, A.; Kern, J.; Maass, V.; Almanzar, G.; Offterdinger, M.; Öllinger, R.; Maass, M.; Wick, G. Chlamydia pneumoniae infection acts as an endothelial stressor with the potential to initiate the earliest heat shock protein 60-dependent inflammatory stage of atherosclerosis. Cell Stress Chaperones 2013, 18, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Xie, T.; Xue, N.; Kuang, Q.; Wei, Z.; Liang, M.; Ding, X. miR-382 Contributes to Renal Tubulointerstitial Fibrosis by Downregulating HSPD1. Oxid. Med. Cell. Longev. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aluksanasuwan, S.; Sueksakit, K.; Fong-ngern, K.; Thongboonkerd, V. Role of HSP60 (HSPD1) in diabetes-induced renal tubular dysfunction: Regulation of intracellular protein aggregation, ATP production, and oxidative stress. FASEB J. 2017, 31, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Barutta, F.; Pinach, S.; Giunti, S.; Vittone, F.; Forbes, J.M.; Chiarle, R.; Arnstein, M.; Perin, P.C.; Camussi, G.; Cooper, M.E.; et al. Heat shock protein expression in diabetic nephropathy. Am. J. Physiol. Physiol. 2008, 295, F1817–F1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Chebotareva, N.; Bobkova, I.; Shilov, E. Heat shock proteins and kidney disease: Perspectives of HSP therapy. Cell Stress Chaperones 2017, 22, 319–343. [Google Scholar] [CrossRef]
- Noh, H.; Kim, H.J.; Yu, M.R.; Kim, W.-Y.; Kim, J.; Ryu, J.H.; Kwon, S.H.; Jeon, J.S.; Han, D.C.; Ziyadeh, F. Heat shock protein 90 inhibitor attenuates renal fibrosis through degradation of transforming growth factor-β type II receptor. Lab. Investig. 2012, 92, 1583–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, M.; Minamino, T.; Toko, H.; Kayama, Y.; Zou, Y.; Sano, M.; Takaki, E.; Aoyagi, T.; Tojo, K.; Tajima, N.; et al. Upregulation of Heat Shock Transcription Factor 1 Plays a Critical Role in Adaptive Cardiac Hypertrophy. Circ. Res. 2006, 99, 1411–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, Q.; Hu, Y.; Ma, Y.; Dong, Z. Heat shock factor 1 induces crystallin-αB to protect against cisplatin nephrotoxicity. Am. J. Physiol. Physiol. 2016, 311, F94–F102. [Google Scholar] [CrossRef] [PubMed] [Green Version]



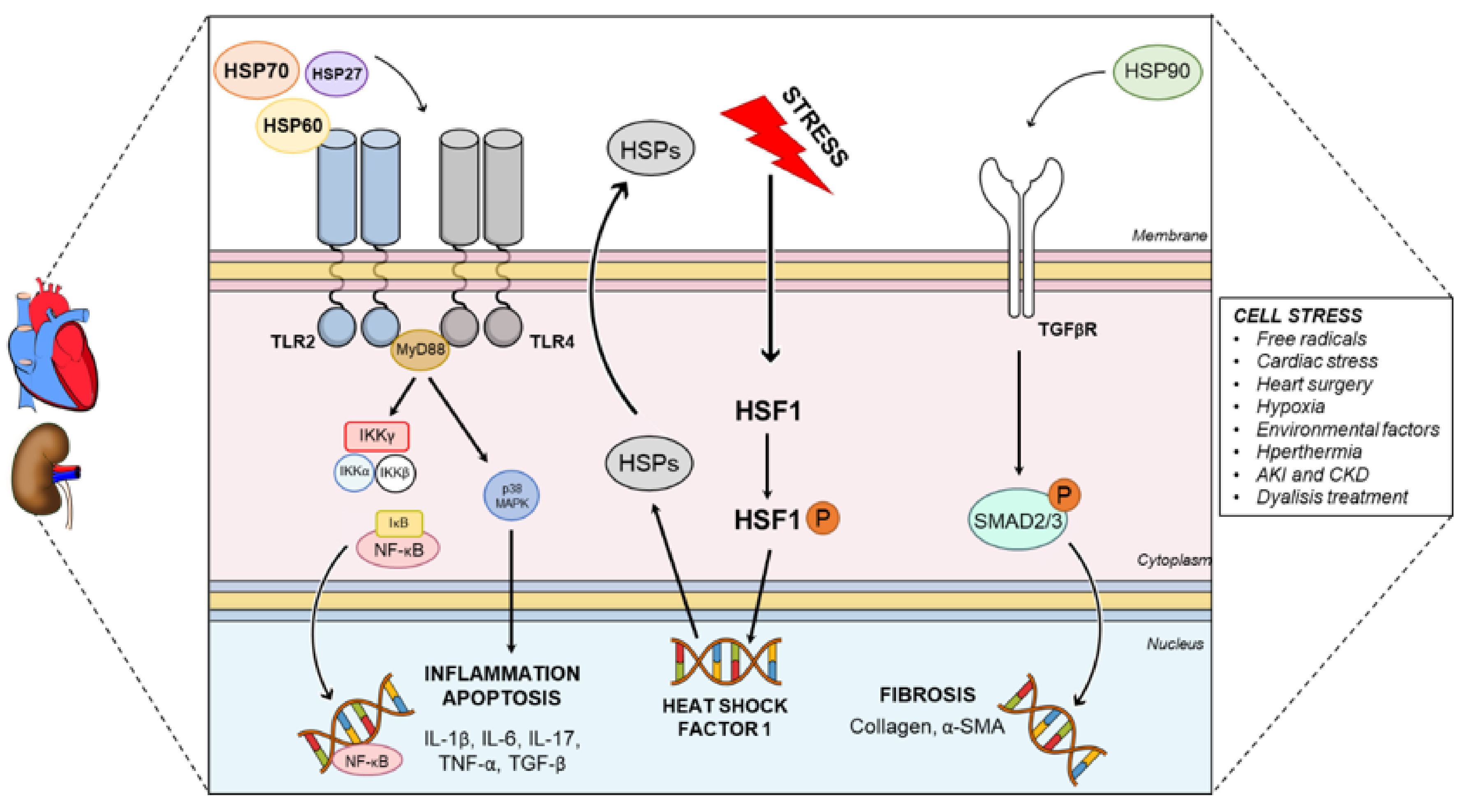{kind=link}
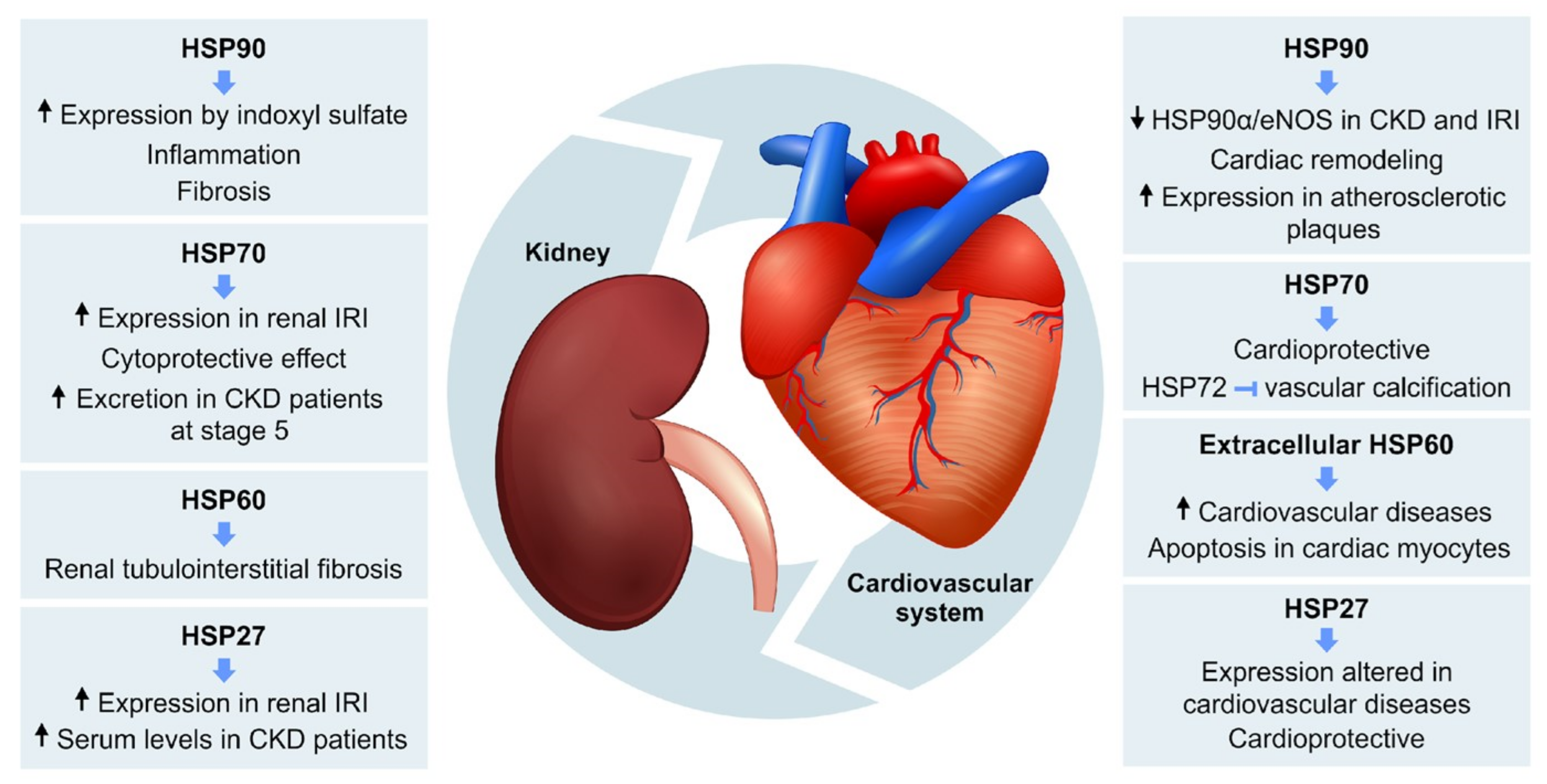{kind=link}
| Classification | CRS Types | Pathologies | HSPs |
|---|---|---|---|
| Cardio-renal | 1 | Decompensated heart failure, congestion, acute coronary injury, acute kidney injury | HSP70 |
| 2 | Chronic heart failure, coronary heart disease, chronic kidney disease | HSP60 HSP90 | |
| Reno-cardiac | 3 | Renal ischemia, acute renal failure, arrhythmia, acute heart failure, myocardial infarction, atrial fibrillation, cardiac hypertrophy | HSP27 HSP60 HSP90 |
| 4 | Chronic kidney disease, uremic toxins accumulation, diastolic dysfunction, myocardial remodeling | HSP70 HSP72 HSP90 | |
| Systemic | 5 | Sepsis, cirrhosis, diabetes | HSP60 HSP90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Junho, C.V.C.; Azevedo, C.A.B.; da Cunha, R.S.; de Yurre, A.R.; Medei, E.; Stinghen, A.E.M.; Carneiro-Ramos, M.S. Heat Shock Proteins: Connectors between Heart and Kidney. Cells 2021, 10, 1939. https://doi.org/10.3390/cells10081939
Junho CVC, Azevedo CAB, da Cunha RS, de Yurre AR, Medei E, Stinghen AEM, Carneiro-Ramos MS. Heat Shock Proteins: Connectors between Heart and Kidney. Cells. 2021; 10(8):1939. https://doi.org/10.3390/cells10081939
Chicago/Turabian StyleJunho, Carolina Victória Cruz, Carolina Amaral Bueno Azevedo, Regiane Stafim da Cunha, Ainhoa Rodriguez de Yurre, Emiliano Medei, Andréa Emilia Marques Stinghen, and Marcela Sorelli Carneiro-Ramos. 2021. "Heat Shock Proteins: Connectors between Heart and Kidney" Cells 10, no. 8: 1939. https://doi.org/10.3390/cells10081939
APA StyleJunho, C. V. C., Azevedo, C. A. B., da Cunha, R. S., de Yurre, A. R., Medei, E., Stinghen, A. E. M., & Carneiro-Ramos, M. S. (2021). Heat Shock Proteins: Connectors between Heart and Kidney. Cells, 10(8), 1939. https://doi.org/10.3390/cells10081939