
GSK3 as a Regulator of Cytoskeleton Architecture: Consequences for Health and Disease

 , , , , and
, , , , and 
Abstract
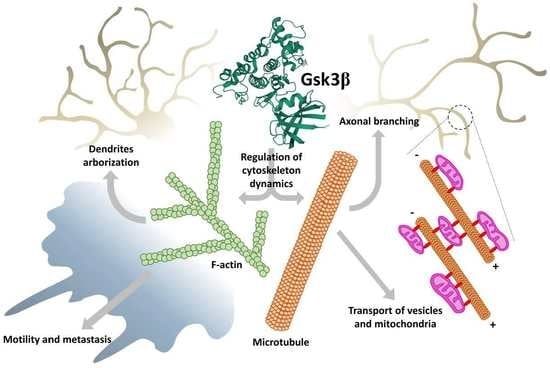
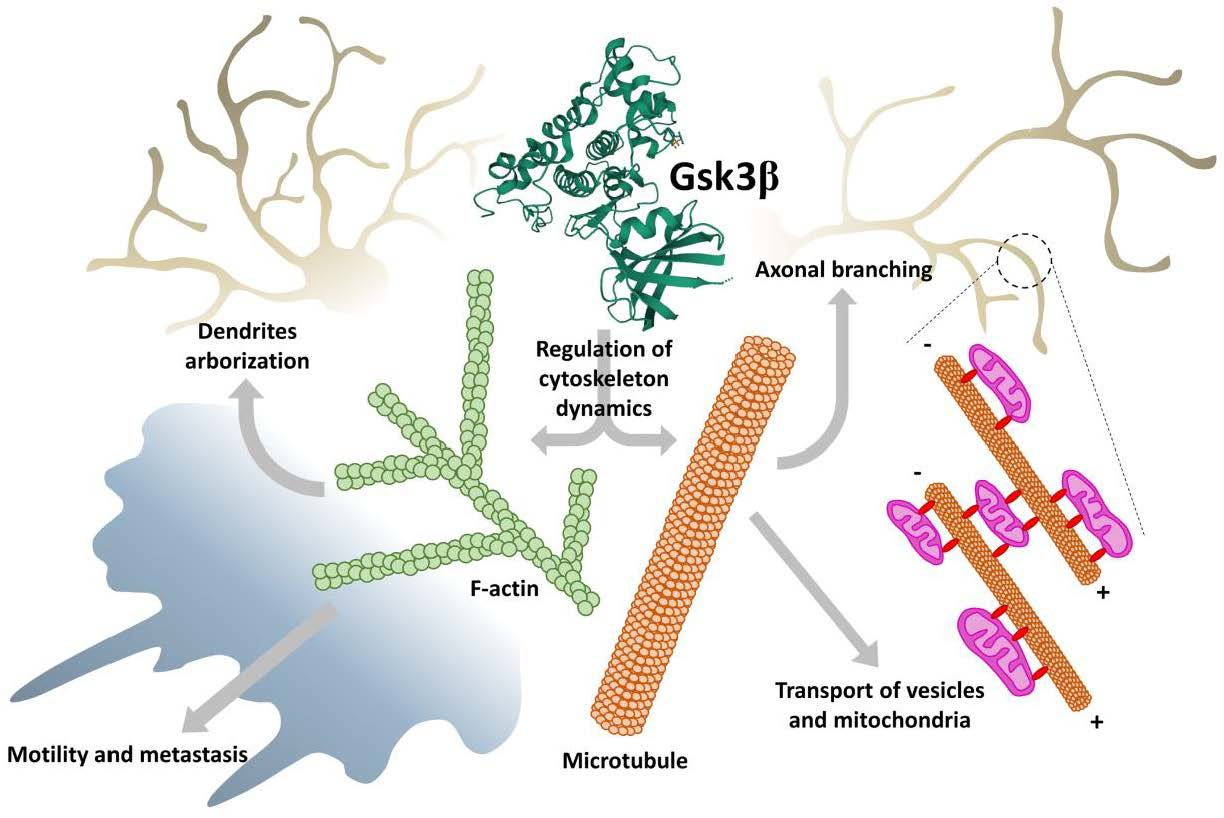
:
1. Introduction
2. GSK3–Cytoskeleton Interplay in Brain Development and Pathology
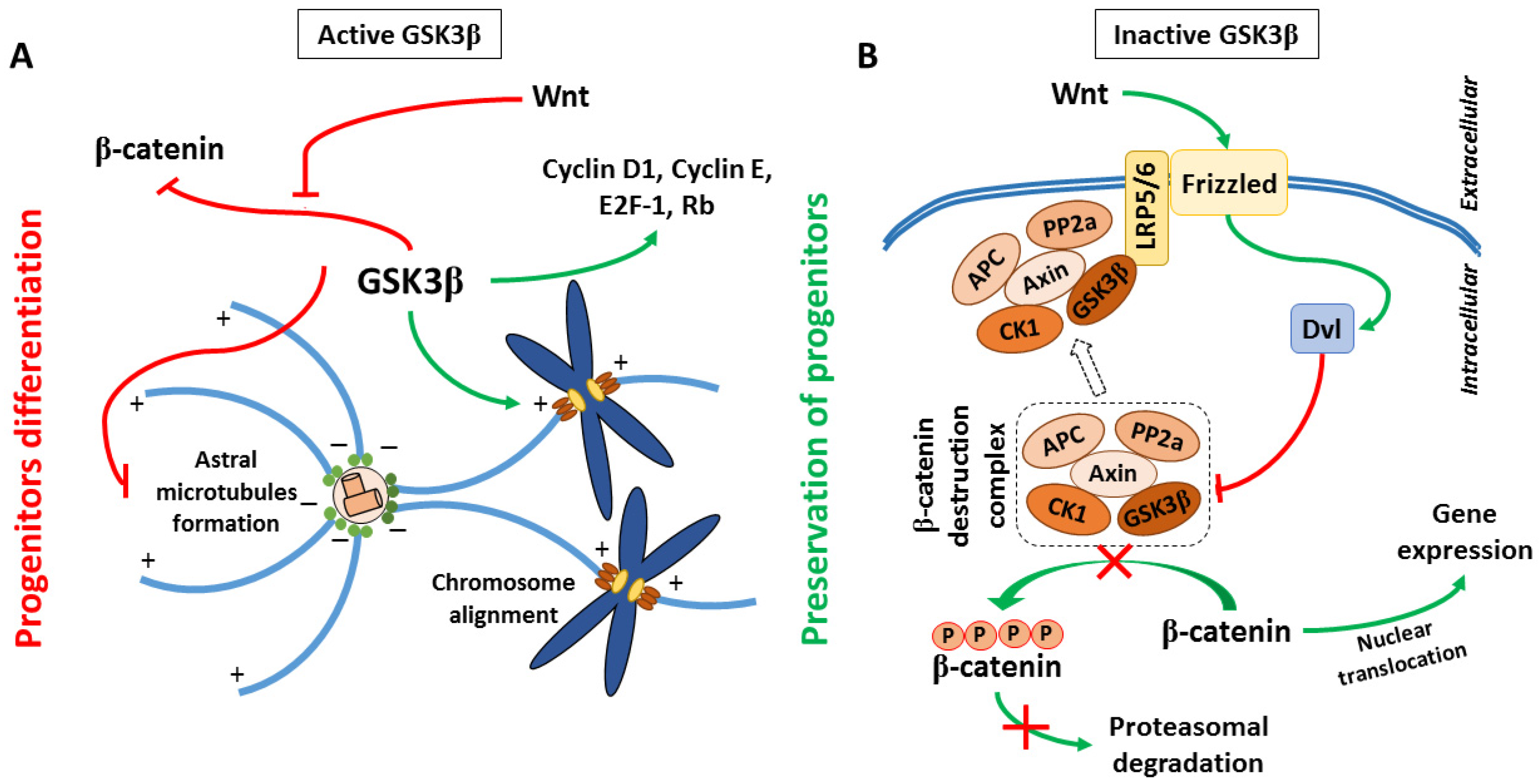
2.1. Neurogenesis
2.1.1. Proliferation
2.1.2. Migration
2.1.3. Differentiation
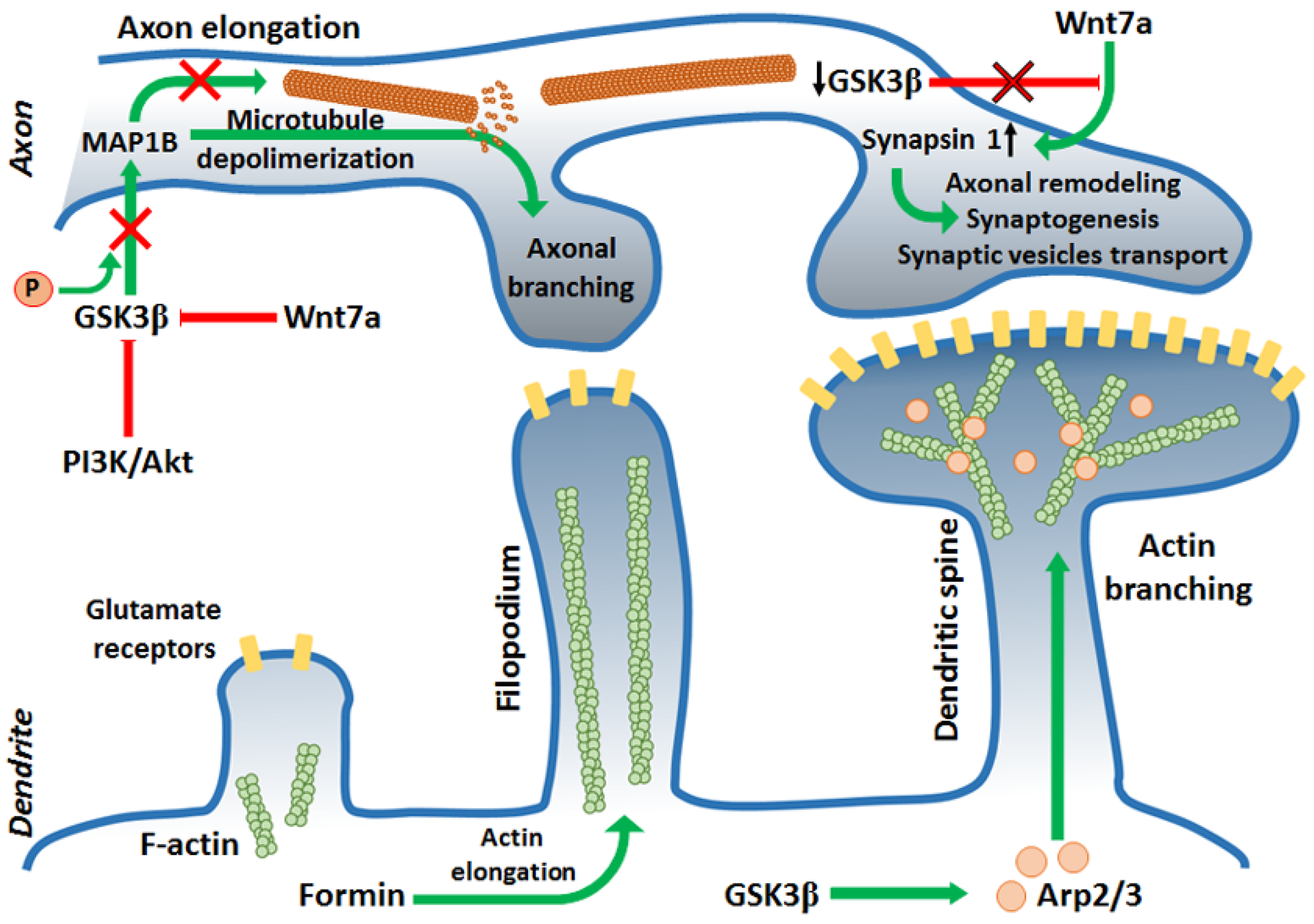
2.2. Synaptic Plasticity
2.3. Neurodegenerative Disorders and Neuronal Survival
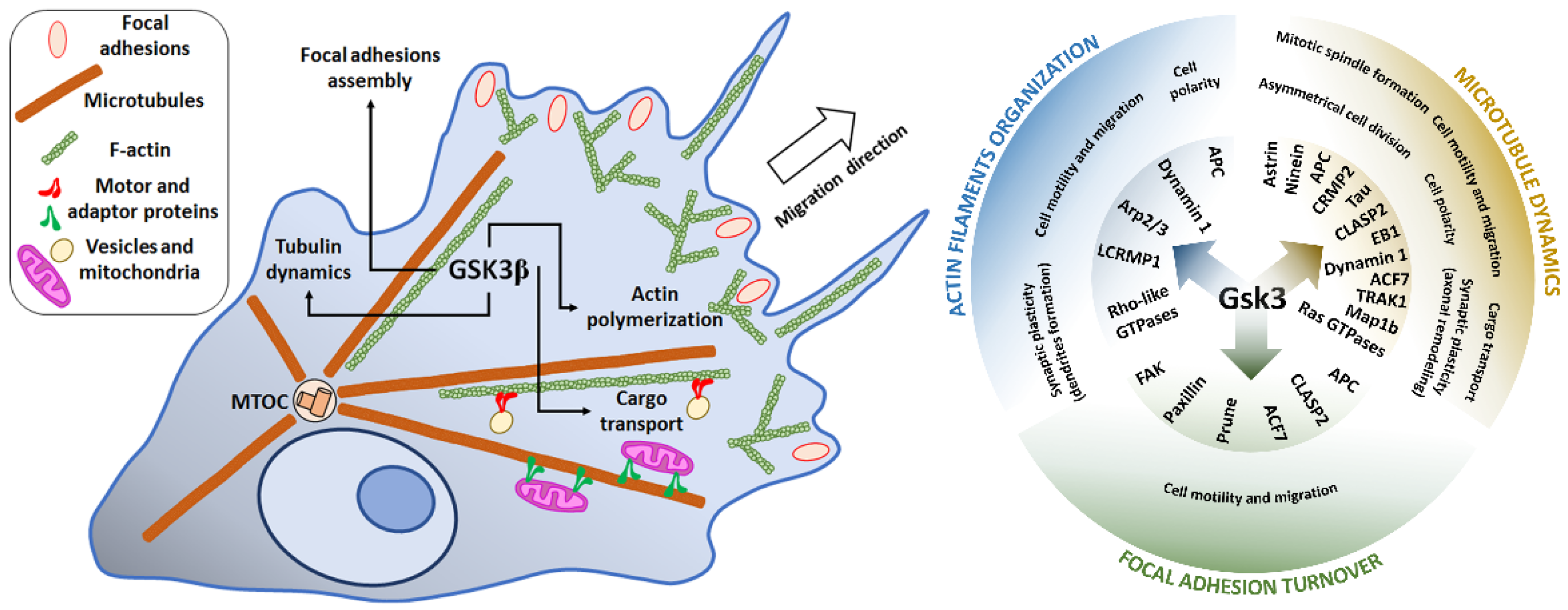
3. GSK3–Cytoskeleton Interplay in Cell Motility and Migration of Cancer Cells
3.1. Lamellipodia, Filopodia, and Invadopodia Formation and Dynamics
3.2. Microtubule Plus-End Tracking Proteins in Direct Cell Movement
3.3. Focal Adhesion
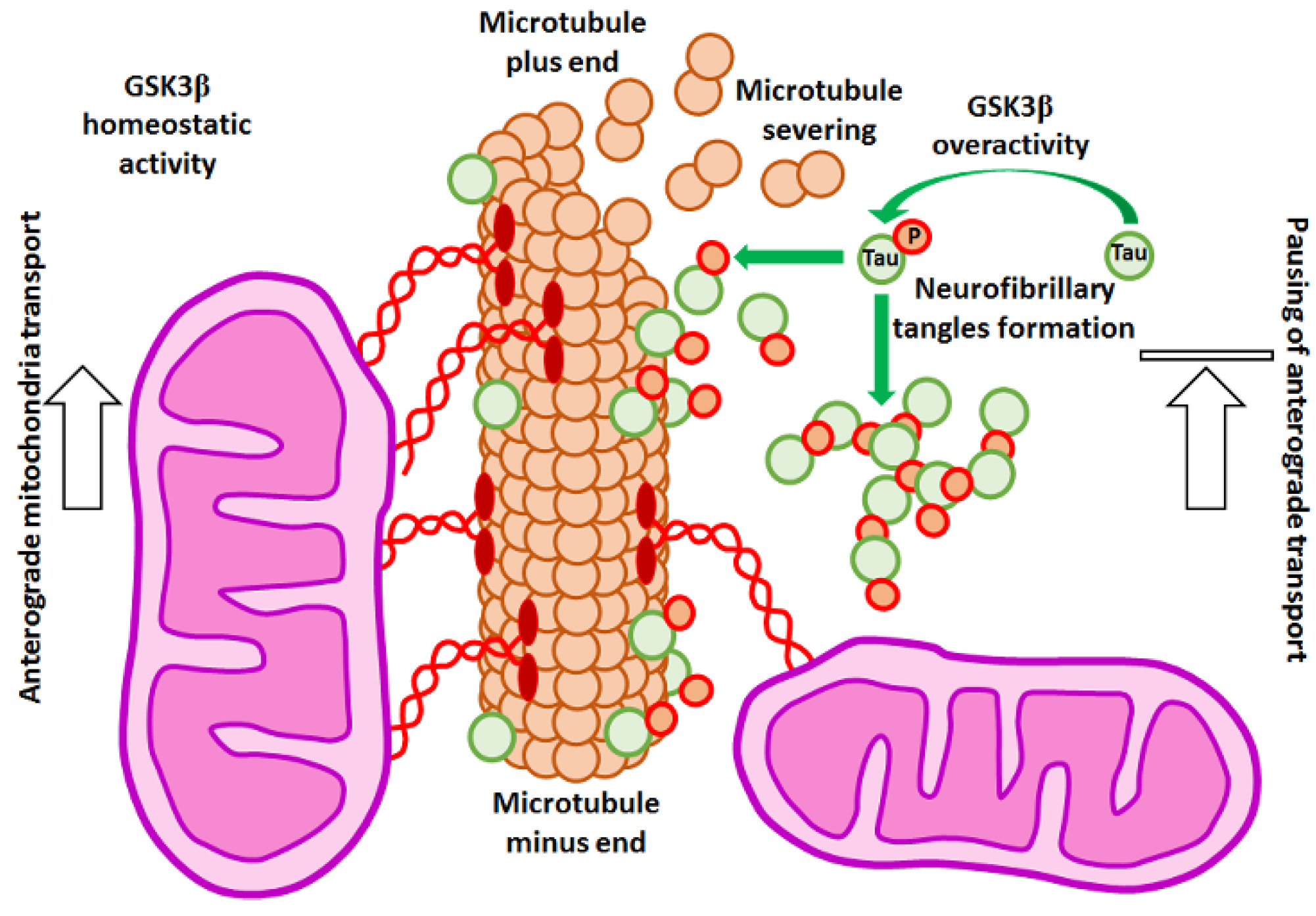
4. GSK3β–Cytoskeleton Interplay in Mitochondria Trafficking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Embi, N.; Rylatt, D.B.; Cohen, P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 1980, 107, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Woodgett, J.R. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990, 9, 2431–2438. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Lipina, T.V.; Takao, K.; van Eede, M.; Hattori, S.; Laliberté, C.; Khan, M.; Okamoto, K.; Chambers, J.W.; Fletcher, P.J.; et al. Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol. Brain. 2009, 2, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soutar, M.P.; Kim, W.Y.; Williamson, R.; Peggie, M.; Hastie, C.J.; McLauchlan, H.; Snider, W.D.; Gordon-Weeks, P.R.; Sutherland, C. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J. Neurochem. 2010, 115, 974–983. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [Green Version]
- Duda, P.; Wiśniewski, J.; Wójtowicz, T.; Wójcicka, O.; Jaśkiewicz, M.; Drulis-Fajdasz, D.; Rakus, D.; McCubrey, J.A.; Gizak, A. Targeting GSK3 signaling as a potential therapy of neurodegenerative diseases and aging. Expert Opin. Ther. Targets 2018, 22, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.V.; Andersson, U.; Andersson, S.; Knerr, L.; Bauer, U.; Sundgren-Andersson, A.K. The Conundrum of GSK3 Inhibitors: Is it the Dawn of a New Beginning? J. Alzheimers Dis. 2018, 64, S547–S554. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Rodriguez, M.; Kim, L. Glycogen synthase kinase 3 in the world of cell migration. Dev. Growth Differ. 2009, 51, 735–742. [Google Scholar] [CrossRef]
- Xu, W.; Ge, Y.; Liu, Z.; Gong, R. Glycogen synthase kinase 3β orchestrates microtubule remodeling in compensatory glomerular adaptation to podocyte depletion. J. Biol. Chem. 2015, 290, 1348–1363. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.; De Lucchini, S.; Marin, O.; Turner, D.L.; Hanks, S.K.; Villa-Moruzzi, E. Regulation of FAK Ser-722 phosphorylation and kinase activity by GSK3 and PP1 during cell spreading and migration. Biochem. J. 2005, 391, 359–370. [Google Scholar] [CrossRef]
- Luo, J. The Role of GSK3beta in the Development of the Central Nervous System. Front. Biol. 2012, 7, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, K.; Brion, J.P. Developmental Expression and Localization of Glycogen Synthase Kinase-3β in Rat Brain. J. Chem. Neuroanat. 1999, 16, 279–293. [Google Scholar] [CrossRef]
- Takahashi, M.; Tomizawa, K.; Ishiguro, K. Distribution of Tau Protein Kinase I/Glycogen Synthase Kinase-3β, Phosphatases 2A and 2B, and Phosphorylated Tau in the Developing Rat Brain. Brain Res. 2000, 857, 193–206. [Google Scholar] [CrossRef]
- Drulis-Fajdasz, D.; Rakus, D.; Wiśniewski, J.R.; McCubrey, J.A.; Gizak, A. Systematic Analysis of GSK-3 Signaling Pathways in Aging of Cerebral Tissue. Adv. Biol. Regul. 2018, 69, 35–42. [Google Scholar] [CrossRef]
- Lee, S.J.; Chung, Y.H.; Joo, K.M.; Lim, H.C.; Jeon, G.S.; Kim, D.; Lee, W.B.; Kim, Y.S.; Cha, C.I. Age-Related Changes in Glycogen Synthase Kinase 3β (GSK3β) Immunoreactivity in the Central Nervous System of Rats. Neurosci. Lett. 2006, 409, 134–139. [Google Scholar] [CrossRef]
- Spittaels, K.; Van Den Haute, C.; Van Dorpe, J.; Geerts, H.; Mercken, M.; Bruynseels, K.; Lasrado, R.; Vandezande, K.; Laenen, I.; Boon, T.; et al. Glycogen Synthase Kinase-3β Phosphorylates Protein Tau and Rescues the Axonopathy in the Central Nervous System of Human Four-Repeat Tau Transgenic Mice. J. Biol. Chem. 2000, 275, 41340–41349. [Google Scholar] [CrossRef] [Green Version]
- Spittaels, K.; Van den Haute, C.; Van Dorpe, J.; Terwel, D.; Vandezande, K.; Lasrado, R.; Bruynseels, K.; Irizarry, M.; Verhoye, M.; Van Lint, J.; et al. Neonatal Neuronal Overexpression of Glycogen Synthase Kinase-3β Reduces Brain Size in Transgenic Mice. Neuroscience 2002, 113, 797–808. [Google Scholar] [CrossRef]
- Cui, H.; Meng, Y.; Bulleit, R.F. Inhibition of Glycogen Synthase Kinase 3β Activity Regulates Proliferation of Cultured Cerebellar Granule Cells. Dev. Brain Res. 1998, 111, 177–188. [Google Scholar] [CrossRef]
- Boku, S.; Nakagawa, S.; Masuda, T.; Nishikawa, H.; Kato, A.; Kitaichi, Y.; Inoue, T.; Koyama, T. Glucocorticoids and Lithium Reciprocally Regulate the Proliferation of Adult Dentate Gyrus-Derived Neural Precursor Cells through GSK-3β and β-Catenin/TCF Pathway. Neuropsychopharmacology 2009, 34, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Maurer, M.H.; Brömme, J.O.; Feldmann, R.E.; Järve, A.; Sabouri, F.; Bürgers, H.F.; Schelshorn, D.W.; Krüger, C.; Schneider, A.; Kuschinsky, W. Glycogen Synthase Kinase 3β (GSK3β) Regulates Differentiation and Proliferation in Neural Stem Cells from the Rat Subventricular Zone. J. Proteome Res. 2007, 6, 1198–1208. [Google Scholar] [CrossRef]
- Yeste-Velasco, M.; Folch, J.; Trullàs, R.; Abad, M.A.; Enguita, M.; Pallàs, M.; Camins, A. Glycogen Synthase Kinase-3 Is Involved in the Regulation of the Cell Cycle in Cerebellar Granule Cells. Neuropharmacology 2007, 53, 295–307. [Google Scholar] [CrossRef]
- Cheng, T.S.; Hsiao, Y.L.; Lin, C.C.; Yu, C.T.R.; Hsu, C.M.; Chang, M.S.; Lee, C.I.; Huang, C.Y.F.; Howng, S.L.; Hong, Y.R. Glycogen Synthase Kinase 3β Interacts with and Phosphorylates the Spindle-Associated Protein Astrin. J. Biol. Chem. 2008, 283, 2454–2464. [Google Scholar] [CrossRef] [Green Version]
- Mack, G.J.; Compton, D.A. Analysis of Mitotic Microtubule-Associated Proteins Using Mass Spectrometry Identifies Astrin, a Spindle-Associated Protein. Proc. Natl. Acad. Sci. USA 2001, 98, 14434–14439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tighe, A.; Ray-Sinha, A.; Staples, O.D.; Taylor, S.S. GSK-3 Inhibitors Induce Chromosome Instability. BMC Cell Biol. 2007, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.R.; Chen, C.H.; Chang, J.H.; Wang, S.K.; Sy, W.D.; Chou, C.K.; Howng, S.L. Cloning and Characterization of a Novel Human Ninein Protein That Interacts with the Glycogen Synthase Kinase 3β. Biochim. Biophys. Acta-Gene Struct. Expr. 2000, 1492, 513–516. [Google Scholar] [CrossRef]
- Howng, S.L.; Hsu, H.C.; Cheng, T.S.; Lee, Y.L.; Chang, L.K.; Lu, P.J.; Hong, Y.R. A Novel Ninein-Interaction Protein, CGI-99, Blocks Ninein Phosphorylation by GSK3β and Is Highly Expressed in Brain Tumors. FEBS Lett. 2004, 566, 162–168. [Google Scholar] [CrossRef]
- Ma, C.; Wang, J.; Gao, Y.; Gao, T.W.; Chen, G.; Bower, K.A.; Odetallah, M.; Ding, M.; Ke, Z.; Luo, J. The Role of Glycogen Synthase Kinase 3β in the Transformation of Epidermal Cells. Cancer Res. 2007, 67, 7756–7764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J. Glycogen Synthase Kinase 3β (GSK3β) in Tumorigenesis and Cancer Chemotherapy. Cancer Lett. 2009, 273, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Malliri, A.; Symons, M.; Hennigan, R.F.; Hurlstone, A.F.L.; Lamb, R.F.; Wheeler, T.; Ozanne, B.W. The Transcription Factor AP-1 Is Required for EGF-Induced Activation of Rho-like GTPases, Cytoskeletal Rearrangements, Motility, and in Vitro Invasion of A431 Cells. J. Cell Biol. 1998, 143, 1087–1099. [Google Scholar] [CrossRef] [Green Version]
- Spiering, D.; Hodgson, L. Dynamics of the Rho-Family Small GTPases in Actin Regulation and Motility. Cell Adhes. Migr. 2011, 5, 170–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govek, E.E.; Newey, S.E.; Van Aelst, L. The Role of the Rho GTPases in Neuronal Development. Genes Dev. 2005, 19, 1–49. [Google Scholar] [CrossRef] [Green Version]
- Kalpachidou, T.; Spiecker, L.; Kress, M.; Quarta, S. Rho GTPases in the Physiology and Pathophysiology of Peripheral Sensory Neurons. Cells 2019, 8, 591. [Google Scholar] [CrossRef] [Green Version]
- Sui, Z.; Sniderhan, L.F.; Fan, S.; Kazmierczak, K.; Reisinger, E.; Kovács, A.D.; Potash, M.J.; Dewhurst, S.; Gelbard, H.A.; Maggirwar, S.B. Human Immunodeficiency Virus-Encoded Tat Activates Glycogen Synthase Kinase-3β to Antagonize Nuclear Factor-ΚB Survival Pathway in Neurons. Eur. J. Neurosci. 2006, 23, 2623–2634. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.D.; Yang, S.I.; Kim, H.C.; Shin, C.Y.; Ko, K.H. Inhibition of GSK-3β Mediates Expression of MMP-9 through ERK1/2 Activation and Translocation of NF-ΚB in Rat Primary Astrocyte. Brain Res. 2007, 1186, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Kagawa, T.; Inoue, T.; Nonaka, A.; Takada, S.; Aburatani, H.; Taga, T. Stabilized β-Catenin Functions through TCF/LEF Proteins and the Notch/RBP-Jκ Complex to Promote Proliferation and Suppress Differentiation of Neural Precursor Cells. Mol. Cell. Biol. 2008, 28, 7427–7441. [Google Scholar] [CrossRef] [Green Version]
- Minde, D.P.; Anvarian, Z.; Rüdiger, S.G.D.; Maurice, M.M. Messing up Disorder: How Do Missense Mutations in the Tumor Suppressor Protein APC Lead to Cancer? Mol. Cancer 2011, 10, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minde, D.P.; Radli, M.; Forneris, F.; Maurice, M.M.; Rüdiger, S.G.D. Large Extent of Disorder in Adenomatous Polyposis Coli Offers a Strategy to Guard Wnt Signalling against Point Mutations. PLoS ONE 2013, 8, e77257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Rao, T.P.; Kühl, M. An Updated Overview on Wnt Signaling Pathways: A Prelude for More. Circ. Res. 2010, 106, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Krylova, O.; Messenger, M.J.; Salinas, P.C. Dishevelled-1 Regulates Microtubule Stability: A New Function Mediated by Glycogen Synthase Kinase-3β. J. Cell Biol. 2000, 151, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Staal, F.J.T.; Clevers, H. Tcf/Lef Transcription Factors during T-Cell Development: Unique and Overlapping Functions. Hematol. J. 2000, 1, 3–6. [Google Scholar] [CrossRef]
- Bhat, R.V.; Shanley, J.; Correll, M.P.; Fieles, W.E.; Keith, R.A.; Scott, C.W.; Lee, C.M. Regulation and Localization of Tyrosine216 Phosphorylation of Glycogen Synthase Kinase-3β in Cellular and Animal Models of Neuronal Degeneration. Proc. Natl. Acad. Sci. USA 2000, 97, 11074–11079. [Google Scholar] [CrossRef] [Green Version]
- Ciani, L.; Salinas, P.C. C-Jun N-Terminal Kinase (JNK) Cooperates with GSK3β to Regulate Dishevelled-Mediated Microtubule Stability. BMC Cell Biol. 2007, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, A.I.M.; Caro-Gonzalez, H.Y.; Nelson, W.J. Role of Adenomatous Polyposis Coli (APC) and Microtubules in Directional Cell Migration and Neuronal Polarization. Semin. Cell Dev. Biol. 2008, 19, 245–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meixner, A.; Haverkamp, S.; Wässle, H.; Führer, S.; Thalhammer, J.; Kropf, N.; Bittner, R.E.; Lassmann, H.; Wiche, G.; Propst, F. MAP1B Is Required for Axon Guidance and Is Involved in the Development of the Central and Peripheral Nervous System. J. Cell Biol. 2000, 151, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- González-Billault, C.; Del Río, J.A.; Ureña, J.M.; Jiménez-Mateos, E.M.; Barallobre, M.J.; Pascual, M.; Pujadas, L.; Simó, S.; La Torre, A.; Gavin, R.; et al. A Role of MAP1B in Reelin-Dependent Neuronal Migration. Cereb. Cortex 2005, 15, 1134–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, T.Y.; Hong, C.J.; Hsueh, Y.P. Bcl11A/CTIP1 Regulates Expression of DCC and MAP1b in Control of Axon Branching and Dendrite Outgrowth. Mol. Cell. Neurosci. 2009, 42, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Bouquet, C.; Soares, S.; Von Boxberg, Y.; Ravaille-Veron, M.; Propst, F.; Nothias, F. Microtubule-Associate Protein 1B Controls Directionality of Growth Cone Migration and Axonal Branching in Regeneration of Adult Dorsal Root Ganglia Neurons. J. Neurosci. 2004, 24, 7204–7213. [Google Scholar] [CrossRef] [Green Version]
- Fabry, B.; Klemm, A.H.; Kienle, S.; Schäffer, T.E.; Goldmann, W.H. Focal Adhesion Kinase Stabilizes the Cytoskeleton. Biophys. J. 2011, 101, 2131–2138. [Google Scholar] [CrossRef] [Green Version]
- Burridge, K.; Turner, C.E.; Romer, L.H. Tyrosine Phosphorylation of Paxillin and Pp125FAK Accompanies Cell Adhesion to Extracellular Matrix: A Role in Cytoskeletal Assembly. J. Cell Biol. 1992, 119, 893–903. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Cdc42 Regulates GSK-3β and Adenomatous Polyposis Coli to Control Cell Polarity. Nature 2003, 421, 753–756. [Google Scholar] [CrossRef]
- Brozzi, F.; Arcuri, C.; Giambanco, I.; Donato, R. S100B Protein Regulates Astrocyte Shape and Migration via Interaction with Src Kinase: Implications for Astrocyte Development, Activation, and Tumor Growth. J. Biol. Chem. 2009, 284, 8797–8811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.H.; Cheng, T.; Jan, L.Y.; Jan, Y.N. APC and GSK-3β Are Involved in MPar3 Targeting to the Nascent Axon and Establishment of Neuronal Polarity. Curr. Biol. 2004, 14, 2025–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelly, M.; Cancedda, L.; Heilshorn, S.; Sumbre, G.; Poo, M. ming LKB1/STRAD Promotes Axon Initiation During Neuronal Polarization. Cell 2007, 129, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Di Nardo, A.; Kramvis, I.; Meikle, L.; Kwiatkowski, D.J.; Sahin, M.; He, X. Tuberous Sclerosis Complex Proteins Control Axon Formation. Genes Dev. 2008, 22, 2485–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.H.; Werner, H.; Püschel, A.W. Rheb and MTOR Regulate Neuronal Polarity through Rap1B. J. Biol. Chem. 2008, 283, 33784–33792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Li, C.; Qin, C.; Xie, L.; Wang, X.; Gao, Z.; Qiangbacuozhen; Wang, T.; Yu, L.; Liu, H. Role of the PI3K/AKT Pathway in Modulating Cytoskeleton Rearrangements and Phenotype Switching in Rat Pulmonary Arterial Vascular Smooth Muscle Cells. DNA Cell Biol. 2014, 33, 12–19. [Google Scholar] [CrossRef]
- Jiang, H.; Guo, W.; Liang, X.; Rao, Y. Both the Establishment and the Maintenance of Neuronal Polarity Require Active Mechanisms: Critical Roles of GSK-3β and Its Upstream Regulators. Cell 2005, 120, 123–135. [Google Scholar]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; Kawabata, S.; Kikuchi, A.; Kaibuchi, K. GSK-3β Regulates Phosphorylation of CRMP-2 and Neuronal Polarity. Cell 2005, 120, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.R.; Knebel, A.; Morrice, N.A.; Robertson, L.A.; Irving, A.J.; Connolly, C.N.; Sutherland, C. GSK-3 Phosphorylation of the Alzheimer Epitope within Collapsin Response Mediator Proteins Regulates Axon Elongation in Primary Neurons. J. Biol. Chem. 2004, 279, 50176–50180. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.Y.; Zhou, F.Q.; Zhou, J.; Yokota, Y.; Wang, Y.M.; Yoshimura, T.; Kaibuchi, K.; Woodgett, J.R.R.; Anton, E.S.; Snider, W.D.D. Essential Roles for GSK-3s and GSK-3-Primed Substrates in Neurotrophin-Induced and Hippocampal Axon Growth. Neuron 2006, 52, 981–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumbrunn, J.; Kinoshita, K.; Hyman, A.A.; Näthke, I.S. Binding of the Adenomatous Polyposis Coli Protein to Microtubules Increases Microtubule Stability and Is Regulated by GSK3β Phosphorylation. Curr. Biol. 2001, 11, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.Q.; Zhou, J.; Dedhar, S.; Wu, Y.H.; Snider, W.D. NGF-Induced Axon Growth Is Mediated by Localized Inactivation of GSK-3β and Functions of the Microtubule plus End Binding Protein APC. Neuron 2004, 42, 897–912. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Wang, Z.; Gu, Y.; Feil, R.; Hofmann, F.; Ma, L. Regulate Axon Branching by the Cyclic GMP Pathway via Inhibition of Glycogen Synthase Kinase 3 in Dorsal Root Ganglion Sensory Neurons. J. Neurosci. 2009, 29, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Ma, L. Regulation of Axonal Development by Natriuretic Peptide Hormones. Proc. Natl. Acad. Sci. USA 2009, 106, 18016–18021. [Google Scholar] [CrossRef] [Green Version]
- Krylova, O.; Herreros, J.; Cleverley, K.E.; Ehler, E.; Henriquez, J.P.; Hughes, S.M.; Salinas, P.C. WNT-3, Expressed by Motoneurons, Regulates Terminal Arborization of Neurotrophin-3-Responsive Spinal Sensory Neurons. Neuron 2002, 35, 1043–1056. [Google Scholar] [CrossRef] [Green Version]
- Bilimoria, P.M.; De La Torre-Ubieta, L.; Ikeuchi, Y.; Becker, E.B.E.; Reiner, O.; Bonni, A. A JIP3-Regulated GSK3β/DCX Signaling Pathway Restricts Axon Branching. J. Neurosci. 2010, 30, 16766–16776. [Google Scholar] [CrossRef]
- Kwon, C.H.; Luikart, B.W.; Powell, C.M.; Zhou, J.; Matheny, S.A.; Zhang, W.; Li, Y.; Baker, S.J.; Parada, L.F. Pten Regulates Neuronal Arborization and Social Interaction in Mice. Neuron 2006, 50, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Drinjakovic, J.; Jung, H.; Campbell, D.S.; Strochlic, L.; Dwivedy, A.; Holt, C.E. E3 Ligase Nedd4 Promotes Axon Branching by Downregulating PTEN. Neuron 2010, 65, 341–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dent, E.W.; Kalil, K. Axon Branching Requires Interactions between Dynamic Microtubules and Actin Filaments. J. Neurosci. 2001, 21, 9757–9769. [Google Scholar] [CrossRef]
- Gerrow, K.; Triller, A. Synaptic Stability and Plasticity in a Floating World. Curr. Opin. Neurobiol. 2010, 20, 631–639. [Google Scholar] [CrossRef]
- Gaiarsa, J.L.; Caillard, O.; Ben-Ari, Y. Long-Term Plasticity at GABAergic and Glycinergic Synapses: Mechanisms and Functional Significance. Trends Neurosci. 2002, 25, 564–570. [Google Scholar] [CrossRef]
- Sorra, K.E.; Harris, K.M. Overview on the Structure, Composition, Function, Development, and Plasticity of Hippocampal Dendritic Spines. Hippocampus 2000, 10, 501–511. [Google Scholar] [CrossRef]
- Lei, W.; Omotade, O.F.; Myers, K.R.; Zheng, J.Q. Actin Cytoskeleton in Dendritic Spine Development and Plasticity. Curr. Opin. Neurobiol. 2016, 39, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Hlushchenko, I.; Koskinen, M.; Hotulainen, P. Dendritic Spine Actin Dynamics in Neuronal Maturation and Synaptic Plasticity. Cytoskeleton 2016, 73, 435–441. [Google Scholar] [CrossRef]
- Koivisto, L.; Häkkinen, L.; Matsumoto, K.; McCulloch, C.A.; Yamada, K.M.; Larjava, H. Glycogen Synthase Kinase-3 Regulates Cytoskeleton and Translocation of Rac1 in Long Cellular Extensions of Human Keratinocytes. Exp. Cell Res. 2004, 293, 68–80. [Google Scholar] [CrossRef]
- Salinas, P.C. Retrograde Signalling at the Synapse: A Role for Wnt Proteins. In Proceedings of the Biochemical Society Transactions. Biochem. Soc. Trans. 2005, 33, 1295–1298. [Google Scholar] [CrossRef] [Green Version]
- Lucas, F.R.; Goold, R.G.; Gordon-Weeks, P.R.; Salinas, P.C. Inhibition of GSK-3β Leading to the Loss of Phosphorylated MAP-1B Is an Early Event in Axonal Remodelling Induced by WNT-7a or Lithium. J. Cell Sci. 1998, 111, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.C.; Lucas, F.R.; Salinas, P.C. Axonal Remodeling and Synaptic Differentiation in the Cerebellum Is Regulated by WNT-7a Signaling. Cell 2000, 100, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Chin, L.S.; Li, L.; Ferreira, A.; Kosik, K.S.; Greengard, P. Impairment of Axonal Development and of Synaptogenesis in Hippocampal Neurons of Synapsin I-Deficient Mice. Proc. Natl. Acad. Sci. USA 1995, 92, 9230–9234. [Google Scholar] [CrossRef] [Green Version]
- Rosahl, T.W.; Spillane, D.; Missler, M.; Herz, J.; Selig, D.K.; Wolff, J.R.; Hammer, R.E.; Malenka, R.C.; Südhof, T.C. Essential Functions of Synapsins I and II in Synaptic Vesicle Regulation. Nature 1995, 375, 488–493. [Google Scholar] [CrossRef]
- Lucas, F.R.; Salinas, P.C. WNT-7a Induces Axonal Remodeling and Increases Synapsin I Levels in Cerebellar Neurons. Dev. Biol. 1997, 192, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.Q.; Wang, S.H.; Liu, D.; Yin, Y.Y.; Tian, Q.; Wang, X.C.; Wang, Q.; Chen, J.G.; Wang, J.Z. Activation of Glycogen Synthase Kinase-3 Inhibits Long-Term Potentiation with Synapse-Associated Impairments. J. Neurosci. 2007, 27, 12211–12220. [Google Scholar] [CrossRef]
- Hooper, C.; Markevich, V.; Plattner, F.; Killick, R.; Schofield, E.; Engel, T.; Hernandez, F.; Anderton, B.; Rosenblum, K.; Bliss, T.; et al. Glycogen Synthase Kinase-3 Inhibition Is Integral to Long-Term Potentiation. Eur. J. Neurosci. 2007, 25, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Greengard, P.; Valtorta, F.; Czernik, A.J.; Benfenati, F. Synaptic Vesicle Phosphoproteins and Regulation of Synaptic Function. Science 1993, 259, 780–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Chang, S.P.; Tang, S.J. Activity-Dependent Synaptic Wnt Release Regulates Hippocampal Long Term Potentiation. J. Biol. Chem. 2006, 281, 11910–11916. [Google Scholar] [CrossRef] [Green Version]
- Opazo, P.; Watabe, A.M.; Grant, S.G.N.; O’Dell, T.J. Phosphatidylinositol 3-Kinase Regulates the Induction of Long-Term Potentiation through Extracellular Signal-Related Kinase-Independent Mechanisms. J. Neurosci. 2003, 23, 3679–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, E.L.; Sue, N.; Smillie, K.J.; O’Leary, T.; Bache, N.; Cheung, G.; Cole, A.R.; Wyllie, D.J.; Sutherland, C.; Robinson, P.J.; et al. Dynamin i Phosphorylation by GSK3 Controls Activity-Dependent Bulk Endocytosis of Synaptic Vesicles. Nat. Neurosci. 2010, 13, 845–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La, T.M.; Tachibana, H.; Li, S.A.; Abe, T.; Seiriki, S.; Nagaoka, H.; Takashima, E.; Takeda, T.; Ogawa, D.; Makino, S.; et al. Dynamin 1 Is Important for Microtubule Organization and Stabilization in Glomerular Podocytes. FASEB J. 2020, 34, 16449–16463. [Google Scholar] [CrossRef]
- Šamaj, J.; Baluška, F.; Voigt, B.; Schlicht, M.; Volkmann, D.; Menzel, D. Endocytosis, Actin Cytoskeleton, and Signaling. Plant. Physiol. 2004, 135, 1150–1161. [Google Scholar] [CrossRef] [Green Version]
- Lossi, L.; Merighi, A. In Vivo Cellular and Molecular Mechanisms of Neuronal Apoptosis in the Mammalian CNS. Prog. Neurobiol. 2003, 69, 287–312. [Google Scholar] [CrossRef]
- Gibson, R.M. Does Apoptosis Have a Role in Neurodegeneration? Br. Med. J. 2001, 322, 1539–1540. [Google Scholar] [CrossRef] [Green Version]
- D’Mello, S.R.; Anelli, R.; Calissano, P. Lithium Induces Apoptosis in Immature Cerebellar Granule Cells but Promotes Survival of Mature Neurons. Exp. Cell Res. 1994, 211, 332–338. [Google Scholar] [CrossRef]
- Hetman, M.; Cavanaugh, J.E.; Kimelman, D.; Zhengui, X. Role of Glycogen Synthase Kinase-3β in Neuronal Apoptosis Induced by Trophic Withdrawal. J. Neurosci. 2000, 20, 2567–2574. [Google Scholar] [CrossRef] [Green Version]
- Brewster, J.L.; Linseman, D.A.; Bouchard, R.J.; Loucks, F.A.; Precht, T.A.; Esch, E.A.; Heidenreich, K.A. Endoplasmic Reticulum Stress and Trophic Factor Withdrawal Activate Distinct Signaling Cascades That Induce Glycogen Synthase Kinase-3β and a Caspase-9-Dependent Apoptosis in Cerebellar Granule Neurons. Mol. Cell. Neurosci. 2006, 3, 242–253. [Google Scholar] [CrossRef]
- Lee, K.Y.; Koh, S.H.; Noh, M.Y.; Park, K.W.; Lee, Y.J.; Kim, S.H. Glycogen Synthase Kinase-3β Activity Plays Very Important Roles in Determining the Fate of Oxidative Stress-Inflicted Neuronal Cells. Brain Res. 2007, 1129, 89–99. [Google Scholar] [CrossRef]
- Tsukane, M.; Yoshizaki, C.; Yamauchi, T. Development and Specific Induction of Apoptosis of Cultured Cell Models Overexpressing Human Tau during Neural Differentiation: Implication in Alzheimer’s Disease. Anal. Biochem. 2007, 360, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen Synthase Kinase-3 (GSK3): Inflammation, Diseases, and Therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuskaitis, C.J.; Jope, R.S. Glycogen Synthase Kinase-3 Regulates Microglial Migration, Inflammation, and Inflammation-Induced Neurotoxicity. Cell. Signal. 2009, 21, 264–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, M.R.; St-Pierre, M.K.; Wendeln, A.C.; Makoni, N.J.; Gouwens, L.K.; Garrad, E.C.; Sohrabi, M.; Neher, J.J.; Tremblay, M.E.; Combs, C.K. Inflammatory Mechanisms in Neurodegeneration. J. Neurochem. 2019, 149, 562–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.Y.; Wang, W.L.; Wang, S.M.; Chu, Y.Y.; Chang, W.C.; Wang, J.M. Glycogen Synthase Kinase-3β-Mediated CCAAT/Enhancer-Binding Protein Delta Phosphorylation in Astrocytes Promot Es Migration and Activation of Microglia/Macrophages. Neurobiol. Aging. 2014, 35, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Paul, C.D.; Mistriotis, P.; Konstantopoulos, K. Cancer cell motility: Lessons from migration in confined spaces. Nat. Rev. Cancer. 2017, 17, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Farooqui, R.; Zhu, S.; Fenteany, G. Glycogen synthase kinase-3 acts upstream of ADP-ribosylation factor 6 and Rac1 to regulate epithelial cell migration. Exp. Cell Res. 2006, 312, 1514–1525. [Google Scholar] [CrossRef] [PubMed]
- Chikano, Y.; Domoto, T.; Furuta, T.; Sabit, H.; Kitano-Tamura, A.; Pyko, I.V.; Takino, T.; Sai, Y.; Hayashi, Y.; Sato, H.; et al. Glycogen synthase kinase 3β sustains invasion of glioblastoma via the focal adhesion kinase, Rac1, and c-Jun N-terminal kinase-mediated pathway. Mol. Cancer Ther. 2015, 14, 564–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitano, A.; Shimasaki, S.; Chikano, Y.; Nakada, M.; Hirose, M.; Higashi, T.; Ishigaki, Y.; Endo, Y.; Takino, T.; Sato, H.; et al. Aberrant glycogen synthase kinase 3β is involved in pancreatic cancer cell invasion and resistance to therapy. PLoS ONE 2013, 8, e55289. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.L.; Hong, T.M.; Chang, Y.L.; Wu, C.T.; Pan, S.H.; Yang, P.C. Phosphorylation of LCRMP-1 by GSK3β promotes filopoda formation, migration and invasion abilities in lung cancer cells. PLoS ONE 2012, 7, e31689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Grand, M.; Rovini, A.; Bourgarel-Rey, V.; Honore, S.; Bastonero, S.; Braguer, D.; Carre, M. ROS-mediated EB1 phosphorylation through Akt/GSK3β pathway: Implication in cancer cell response to microtubule-targeting agents. Oncotarget 2014, 5, 3408–3423. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.G.; Kim, M.J.; Choi, W.J.; Moon, M.Y.; Kim, H.J.; Lee, J.Y.; Kim, J.; Kim, S.C.; Kang, S.G.; Seo, G.Y.; et al. Wnt3A Induces GSK-3β Phosphorylation and β-Catenin Accumulation Through RhoA/ROCK. J. Cell Physiol. 2017, 232, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lee, Y.K.; Lee, W.S.; Park, O.J.; Kim, Y.M. The involvement of AMPK/GSK3-beta signals in the control of metastasis and proliferation in hepato-carcinoma cells treated with anthocyanins extracted from Korea wild berry Meoru. BMC Complement. Altern. Med. 2014, 14, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 2010, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Schaks, M.; Giannone, G.; Rottner, K. Actin dynamics in cell migration. Essays Biochem. 2019, 63, 483–495. [Google Scholar]
- Yamazaki, D.; Kurisu, S.; Takenawa, T. Involvement of Rac and Rho signaling in cancer cell motility in 3D substrates. Oncogene. 2009, 28, 1570–1583. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Wang, C.H.; Yan, H.; Zhang, R.; Zhao, J.B.; Qian, C.F.; Xiao, H.; Liu, H.Y. Inhibition of the Rac1-WAVE2-Arp2/3 signaling pathway promotes radiosensitivity via downregulation of cofilin-1 in U251 human glioma cells. Mol. Med. Rep. 2016, 13, 4414–4420. [Google Scholar] [CrossRef] [Green Version]
- To, C.; Roy, A.; Chan, E.; Prado, M.A.M.; Di Guglielmo, G.M. Synthetic triterpenoids inhibit GSK3β activity and localization and affect focal adhesions and cell migration. Biochim Biophys Acta Mol. Cell Res. 2017, 1864, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- To, C.; Shilton, B.H.; Di Guglielmo, G.M. Synthetic triterpenoids target the Arp2/3 complex and inhibit branched actin polymerization. J. Biol Chem. 2010, 285, 27944–27957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rom, S.; Fan, S.; Reichenbach, N.; Dykstra, H.; Ramirez, S.H.; Persidsky, Y. Glycogen synthase kinase 3β inhibition prevents monocyte migration across brain endothelial cells via Rac1-GTPase suppression and down-regulation of active integrin conformation. Am. J. Pathol. 2012, 181, 1414–1425. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, Y.; Suzuki, M.; Takahashi, H.; Ishioka, C. Inhibition of invasion by glycogen synthase kinase-3 beta inhibitors through dysregulation of actin re-organisation via down-regulation of WAVE2. Biochem Biophys Res. Commun. 2015, 464, 275–280. [Google Scholar] [CrossRef]
- Seiz, J.R.; Klinke, J.; Scharlibbe, L.; Lohfink, D.; Heipel, M.; Ungefroren, H.; Giehl, K.; Menke, A. Different signaling and functionality of Rac1 and Rac1b in the progression of lung adenocarcinoma. Biol Chem. 2020, 401, 517–531. [Google Scholar] [CrossRef]
- Suraneni, P.; Fogelson, B.; Rubinstein, B.; Noguera, P.; Volkmann, N.; Hanein, D.; Mogilner, A.; Li, R. A mechanism of leading-edge protrusion in the absence of Arp2/3 complex. Mol. Biol Cell. 2015, 26, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Jacquemet, G.; Hamidi, H.; Ivaska, J. Filopodia in cell adhesion, 3D migration and cancer cell invasion. Curr Opin Cell Biol. 2015, 36, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, S.H.; Chao, Y.C.; Chen, H.Y.; Hung, P.F.; Lin, P.Y.; Lin, C.W.; Chang, Y.L.; Wu, C.T.; Lee, Y.C.; Yang, S.C.; et al. Long form collapsin response mediator protein-1 (LCRMP-1) expression is associated with clinical outcome and lymph node metastasis in non-small cell lung cancer patients. Lung Cancer. 2010, 67, 93–100. [Google Scholar] [CrossRef]
- Krugmann, S.; Jordens, I.; Gevaert, K.; Driessens, M.; Vandekerckhove, J.; Hall, A. Cdc42 induces filopodia by promoting the formation of an IRSp53:Mena complex. Curr. Biol. 2001, 11, 1645–1655. [Google Scholar] [CrossRef] [Green Version]
- Schoumacher, M.; Goldman, R.D.; Louvard, D.; Vignjevic, D.M. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J. Cell Biol. 2010, 189, 541–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Yamamoto, N.; Domoto, T.; Bolidong, D.; Hayashi, K.; Takeuchi, A.; Miwa, S.; Igarashi, K.; Inatani, H.; Aoki, Y.; et al. Glycogen synthase kinase 3β as a potential therapeutic target in synovial sarcoma and fibrosarcoma. Cancer Sci. 2020, 111, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Jing, L.; Ma, S.; Li, Q.; Luo, X.; Pan, Z.; Feng, Y.; Feng, P. GSK3β mediates pancreatic cancer cell invasion in vitro via the CXCR4/MMP-2 Pathway. Cancer Cell Int. 2015, 15, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, W.; Kong, L.; Yu, H.; Bao, J.; Song, C.; Qu, G. Glycogen synthase kinase 3β promotes osteosarcoma invasion and migration via regulating PTEN and phosphorylation of focal adhesion kinase. Biosci Rep. 2021, BSR20193514. [Google Scholar] [CrossRef]
- Eddy, R.J.; Weidmann, M.D.; Sharma, V.P.; Condeelis, J.S. Tumor Cell Invadopodia: Invasive Protrusions that Orchestrate Metastasis. Trends Cell Biol. 2017, 27, 595–607. [Google Scholar] [CrossRef]
- Paluch, E.K.; Raz, E. The role and regulation of blebs in cell migration. Curr. Opin. Cell Biol. 2013, 25, 582–590. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, Y.; Xu, R.; Du, J.; Hu, Z.; Yang, L.; Chen, Y.; Zhu, Y.; Gu, L. PI3K/Akt-dependent phosphorylation of GSK3β and activation of RhoA regulate Wnt5a-induced gastric cancer cell migration. Cell. Signal. 2013, 25, 447–456. [Google Scholar] [CrossRef]
- Kremenevskaja, N.; von Wasielewski, R.; Rao, A.S.; Schöfl, C.; Andersson, T.; Brabant, G. Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene 2005, 24, 2144–2154. [Google Scholar] [CrossRef] [Green Version]
- Islam, R.; Kim, J.G.; Park, Y.; Cho, J.Y.; Cap, K.C.; Kho, A.R. Chung WS, Suh SW, Park JB. Insulin induces phosphorylation of pyruvate dehydrogenase through RhoA activation pathway in HepG2 cells. FASEB J. 2019, 33, 2072–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Betson, M.; Mulloy, R.; Foster, R.; Lévay, M.; Ligeti, E.; Settleman, J. p190A RhoGAP is a glycogen synthase kinase-3-beta substrate required for polarized cell migration. J. Biol. Chem. 2008, 283, 20978–20988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etienne-Manneville, S. Microtubules in cell migration. Annu. Rev. Cell Dev. Biol. 2013, 29, 471–499. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S. APC in cell migration. Adv. Exp. Med. Biol. 2009, 656, 30–40. [Google Scholar]
- Zaoui, K.; Honoré, S.; Isnardon, D.; Braguer, D.; Badache, A. Memo-RhoA-mDia1 signaling controls microtubules, the actin network, and adhesion site formation in migrating cells. J. Cell Biol. 2008, 183, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Eng, C.H.; Schmoranzer, J.; Cabrera-Poch, N.; Morris, E.J.; Chen, M.; Wallar, B.J.; Alberts, A.S.; Gundersen, G.G. EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat. Cell Biol. 2004, 6, 820–830. [Google Scholar] [CrossRef]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109, djw332. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.T.; Liao, C.K.; Shen, C.C.; Tang, T.K.; Jow, G.M.; Wang, H.S.; Wu, J.C. HYS-32-Induced Microtubule Catastrophes in Rat Astrocytes Involves the PI3K-GSK3beta Signaling Pathway. PLoS ONE 2015, 10, e0126217. [Google Scholar] [CrossRef]
- Nehlig, A.; Molina, A.; Rodrigues-Ferreira, S.; Honoré, S.; Nahmias, C. Regulation of end-binding protein EB1 in the control of microtubule dynamics. Cell Mol. Life Sci. 2017, 74, 2381–2393. [Google Scholar] [CrossRef] [Green Version]
- Dong, B.; Zhang, S.S.; Gao, W.; Su, H.; Chen, J.; Jin, F.; Bhargava, A.; Chen, X.; Jorgensen, L.; Alberts, A.S.; et al. Mammalian diaphanous-related formin 1 regulates GSK3β-dependent microtubule dynamics required for T cell migratory polarization. PLoS ONE 2013, 8, e80500. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, T.; Waterman-Storer, C.M. Spatial regulation of CLASP affinity for microtubules by Rac1 and GSK3beta in migrating epithelial cells. J. Cell Biol. 2005, 169, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Zaoui, K.; Benseddik, K.; Daou, P.; Salaün, D.; Badache, A. ErbB2 receptor controls microtubule capture by recruiting ACF7 to the plasma membrane of migrating cells. Proc. Natl. Acad. Sci. USA 2010, 107, 18517–18522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Lyle, K.S.; Gierke, S.; Matov, A.; Danuser, G.; Wittmann, T. GSK3beta phosphorylation modulates CLASP-microtubule association and lamella microtubule attachment. J. Cell Biol. 2009, 184, 895–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Shen, Q.T.; Oristian, D.S.; Lu, C.P.; Zheng, Q.; Wang, H.W.; Fuchs, E. Skin stem cells orchestrate directional migration by regulating microtubule-ACF7 connections through GSK3β. Cell 2011, 144, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Kanchanawong, P.; Shtengel, G.; Pasapera, A.M.; Ramko, E.B.; Davidson, M.W.; Hess, H.F.; Waterman, C.M. Nanoscale architecture of integrin-based cell adhesions. Nature 2010, 468, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, D.D.; Mitra, S.K. Multiple connections link FAK to cell motility and invasion. Curr. Opin. Genet. Dev. 2004, 14, 92–101. [Google Scholar] [CrossRef]
- Hu, Y.L.; Lu, S.; Szeto, K.W.; Sun, J.; Wang, Y.; Lasheras, J.C.; Chien, S. FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Sci. Rep. 2014, 4, 6024. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Hino, S.; Oue, N.; Asahara, T.; Zollo, M.; Yasui, W.; Kikuchi, A. Glycogen synthase kinase 3 and h-prune regulate cell migration by modulating focal adhesions. Mol. Cell Biol. 2006, 26, 898–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, J.K.; Paraiso, K.H.; Rebecca, V.W.; Cantini, L.P.; Abel, E.V.; Pagano, N.; Meggers, E.; Mathew, R.; Krepler, C.; Izumi, V.; et al. GSK3β inhibition blocks melanoma cell/host interactions by downregulating N-cadherin expression and decreasing FAK phosphorylation. J. Investig. Dermatol. 2012, 132, 2818–28127. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Li, M.; Vrana, J.; Schaller, M.D. Glycogen synthase kinase 3- and extracellular signal-regulated kinase-dependent phosphorylation of paxillin regulates cytoskeletal rearrangement. Mol. Cell Biol. 2006, 26, 2857–2868. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Ge, Y.; Liu, Z.; Gong, R. Glycogen synthase kinase 3β dictates podocyte motility and focal adhesion turnover by modulating paxillin activity: Implications for the protective effect of low-dose lithium in podocytopathy. Am. J. Pathol. 2014, 184, 2742–2756. [Google Scholar] [CrossRef] [Green Version]
- Nayal, A.; Webb, D.J.; Brown, C.M.; Schaefer, E.M.; Vicente-Manzanares, M.; Horwitz, A.R. Paxillin phosphorylation at Ser273 localizes a GIT1-PIX-PAK complex and regulates adhesion and protrusion dynamics. J. Cell Biol. 2006, 173, 587–589. [Google Scholar] [CrossRef]
- Juanes, M.A.; Isnardon, D.; Badache, A.; Brasselet, S.; Mavrakis, M.; Goode, B.L. The role of APC-mediated actin assembly in microtubule capture and focal adhesion turnover. J. Cell Biol. 2019, 218, 3415–3435. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Kodama, A.; Fuchs, E. ACF7 regulates cytoskeletal-focal adhesion dynamics and migration and has ATPase activity. Cell 2008, 135, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Yue, J.; Zhang, Y.; Liang, W.G.; Gou, X.; Lee, P.; Liu, H.; Lyu, W.; Tang, W.J.; Chen, S.Y.; Yang, F.; et al. In vivo epidermal migration requires focal adhesion targeting of ACF7. Nat. Commun. 2016, 7, 11692. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Schaefer, A.; Wang, Y.; Hodge, R.G.; Blake, D.R.; Diehl, J.N.; Papageorge, A.G.; Stachler, M.D.; Liao, J.; Zhou, J.; et al. Gain-of-Function RHOA Mutations Promote Focal Adhesion Kinase Activation and Dependency in Diffuse Gastric Cancer. Cancer Discov. 2020, 10, 288–305. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Oliveros, A.; Jang, M.H. Dysfunctional mitochondrial bioenergetics and synaptic degeneration in Alzheimer disease. Int. Neurourol. J. 2019, 23, S5–S10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnery, P.F. Mitochondrial Disorders Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2014. [Google Scholar]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L.F. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovas, J.R.; Wang, X. The meaning of mitochondrial movement to a neuron’s life. Biochim. Biophys. Acta-Mol. Cell Res. 2013, 1833, 184–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederick, R.L.; Shaw, J.M. Moving mitochondria: Establishing distribution of an essential organelle. Traffic 2007, 8, 1668–1675. [Google Scholar] [CrossRef] [Green Version]
- Deheshi, S.; Pasqualotto, B.A.; Rintoul, G.L. Mitochondrial trafficking in neuropsychiatric diseases. Neurobiol. Dis. 2013, 51, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Mitochondria on the move: Emerging paradigms of organelle trafficking in tumour plasticity and metastasis. Br. J. Cancer 2017, 117, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Shanmughapriya, S.; Langford, D.; Natarajaseenivasan, K. Inter and Intracellular mitochondrial trafficking in health and disease. Ageing Res. Rev. 2020, 62, 101128. [Google Scholar] [CrossRef]
- Wang, Q.; Tian, J.; Chen, H.; Du, H.; Guo, L. Amyloid beta-mediated KIF5A deficiency disrupts anterograde axonal mitochondrial movement. Neurobiol. Dis. 2019, 127, 410–418. [Google Scholar] [CrossRef]
- Baas, P.W.; Qiang, L. Neuronal microtubules: When the MAP is the roadblock. Trends Cell Biol. 2005, 15, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am. J. Pathol. 2011, 179, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Morel, M.; Authelet, M.; Dedecker, R.; Brion, J.P. Glycogen synthase kinase-3β and the p25 activator of cyclin dependent kinase 5 increase pausing of mitochondria in neurons. Neuroscience 2010, 167, 1044–1056. [Google Scholar] [CrossRef] [PubMed]
- Griebel, G.; Stemmelin, J.; Lopez-Grancha, M.; Boulay, D.; Boquet, G.; Slowinski, F.; Pichat, P.; Beeské, S.; Tanaka, S.; Mori, A.; et al. The selective GSK3 inhibitor, SAR502250, displays neuroprotective activity and attenuates behavioral impairments in models of neuropsychiatric symptoms of Alzheimer’s disease in rodents. Sci. Rep. 2019, 9, 18045. [Google Scholar] [CrossRef] [Green Version]
- Cavendish, J.Z.; Sarkar, S.N.; Colantonio, M.A.; Quintana, D.D.; Ahmed, N.; White, B.A.; Engler-Chiurazzi, E.B.; Simpkins, J.W. Mitochondrial Movement and Number Deficits in Embryonic Cortical Neurons from 3xTg-AD Mice. J. Alzheimer’s Dis. 2019, 70, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Martín, M.; López-Doménech, G.; Soriano, E.; Avila, J. GSK3β is involved in the relief of mitochondria pausing in a Tau-dependent manner. PLoS ONE 2011, 6, e27686. [Google Scholar] [CrossRef] [Green Version]
- Emamian, E.S.; Hall, D.; Birnbaum, M.J.; Karayiorgou, M.; Gogos, J.A. Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat. Genet. 2004, 36, 131–137. [Google Scholar] [CrossRef]
- Chan, M.H.; Chiu, P.H.; Lin, C.Y.; Chen, H.H. Inhibition of glycogen synthase kinase-3 attenuates psychotomimetic effects of ketamine. Schizophr. Res. 2012, 136, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, F.; Murphy, L.C.; Malavasi, E.L.V.; O’Sullivan, S.T.; Torrance, H.S.; Porteous, D.J.; Millar, J.K. NDE1 and GSK3β Associate with TRAK1 and Regulate Axonal Mitochondrial Motility: Identification of Cyclic AMP as a Novel Modulator of Axonal Mitochondrial Trafficking. ACS Chem. Neurosci. 2016, 7, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Emamian, E.S. AKT/GSK3 signaling pathway and schizophrenia. Front. Mol. Neurosci. 2012, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Cheong, J.H. Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells. Cells 2020, 9, 1691. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, B.; McKenzie, A.J.; Heintz, N.H.; Howe, A.K. AMPK activity regulates trafficking of Mitochondria to the leading edge during cell migration and matrix invasion. Mol. Biol. Cell 2016, 27, 2662–2674. [Google Scholar] [CrossRef]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef]
- Caino, M.C.; Ghosh, J.C.; Chae, Y.C.; Vaira, V.; Rivadeneira, D.B.; Faversani, A.; Rampini, P.; Kossenkov, A.V.; Aird, K.M.; Zhang, R.; et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc. Natl. Acad. Sci. USA 2015, 112, 8638–8643. [Google Scholar] [CrossRef] [Green Version]
- Yi, M.; Weaver, D.; Hajnóczky, G. Control of mitochondrial motility and distribution by the calcium signal: A homeostatic circuit. J. Cell Biol. 2004, 167, 661–672. [Google Scholar] [CrossRef]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetić, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell. Dev. Biol. 2021, 8, 624216. [Google Scholar] [CrossRef]
- Viola, H.M.; Davies, S.M.; Filipovska, A.; Hool, L.C. L-type Ca(2+) channel contributes to alterations in mitochondrial calcium handling in the mdx ventricular myocyte. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H767–H775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Sanz, C.; Ruiz-Meana, M.; Miro-Casas, E.; Nuñez, E.; Castellano, J.; Loureiro, M.; Barba, I.; Poncelas, M.; Rodriguez-Sinovas, A.; Vázquez, J.; et al. Defective sarcoplasmic reticulum-mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 2014, 5, e1573. [Google Scholar] [CrossRef] [Green Version]
- Saotome, M.; Safiulina, D.; Szabadkai, G.; Das, S.; Fransson, A.; Aspenstrom, P.; Rizzuto, R.; Hajnóczky, G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 20728–20733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, K.M.; Brocardo, M.G.; Henderson, B.R. APC binds the Miro/Milton motor complex to stimulate transport of mitochondria to the plasma membrane. Mol. Biol. Cell 2016, 27, 466–482. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Structure | Formula | Molecular Weight | IC50 | Soluble in | Selectivity |
|---|---|---|---|---|---|---|
| Lithium (LiCl) | Li–Cl | LiCl | 42.39 | 1.0 mM | DMSO, ethanol, water, and other solvents | Also inhibits protein kinase A, casein kinase II, MEK/ERK, ERK1, SAPK, Raf, and Trk |
| SB216763 |  | C19H12N2O2Cl2 | 371.22 | 34.3 nM | DMSO | high |
| SB415286 |  | C16H10ClN3O5 | 359.72 | 78 nM | DMSO ethanol | high |
| I3′M |  | C16H11N3O2 | 277.28 | 22nM | DMSO | Also inhibits CDK5/p25, CDK1/cyclin B and 5-Lipoxygenase |
| Kenpaullone |  | C16H11BrN2O | 327.18 | 23 nM | DMSO | Also inhibits CDK2/cyclin A, CDK2/cyclin E, and CDK5/p25 |
| 1-Azakenpaullone |  | C15H10BrN3O | 328.16 | 18 nM | DMSO | Also inhibits CDK1/cyclin B and CDK5/p25 |
| GSK 3 Inhibitor IX |  | C16H10BrN3O2 | 356.17 | 5 nM | DMSO | Also inhibits CDK1 and CDK5 |
| AR-A0114418 |  | C12H12N4O4S | 308.31 | 104 nM | DMSO | high |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajka, D.; Budziak, B.; Pietras, Ł.; Duda, P.; McCubrey, J.A.; Gizak, A. GSK3 as a Regulator of Cytoskeleton Architecture: Consequences for Health and Disease. Cells 2021, 10, 2092. https://doi.org/10.3390/cells10082092
Hajka D, Budziak B, Pietras Ł, Duda P, McCubrey JA, Gizak A. GSK3 as a Regulator of Cytoskeleton Architecture: Consequences for Health and Disease. Cells. 2021; 10(8):2092. https://doi.org/10.3390/cells10082092
Chicago/Turabian StyleHajka, Daria, Bartosz Budziak, Łukasz Pietras, Przemysław Duda, James A. McCubrey, and Agnieszka Gizak. 2021. "GSK3 as a Regulator of Cytoskeleton Architecture: Consequences for Health and Disease" Cells 10, no. 8: 2092. https://doi.org/10.3390/cells10082092
APA StyleHajka, D., Budziak, B., Pietras, Ł., Duda, P., McCubrey, J. A., & Gizak, A. (2021). GSK3 as a Regulator of Cytoskeleton Architecture: Consequences for Health and Disease. Cells, 10(8), 2092. https://doi.org/10.3390/cells10082092