
Intracellular Signaling Responses Induced by Radiation within an In Vitro Bone Metastasis Model after Pre-Treatment with an Estrone Analogue

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. General Cell Culture and Reagents
2.3. Compound and Experimental Protocols
2.4. Radiation
2.5. Controls
2.6. Cytotoxicity Studies: Dose–Response Curves of the Compound with and without Radiation on All Cell Lines
2.7. Cell Cycle Progression Assessment Using Flow Cytometry
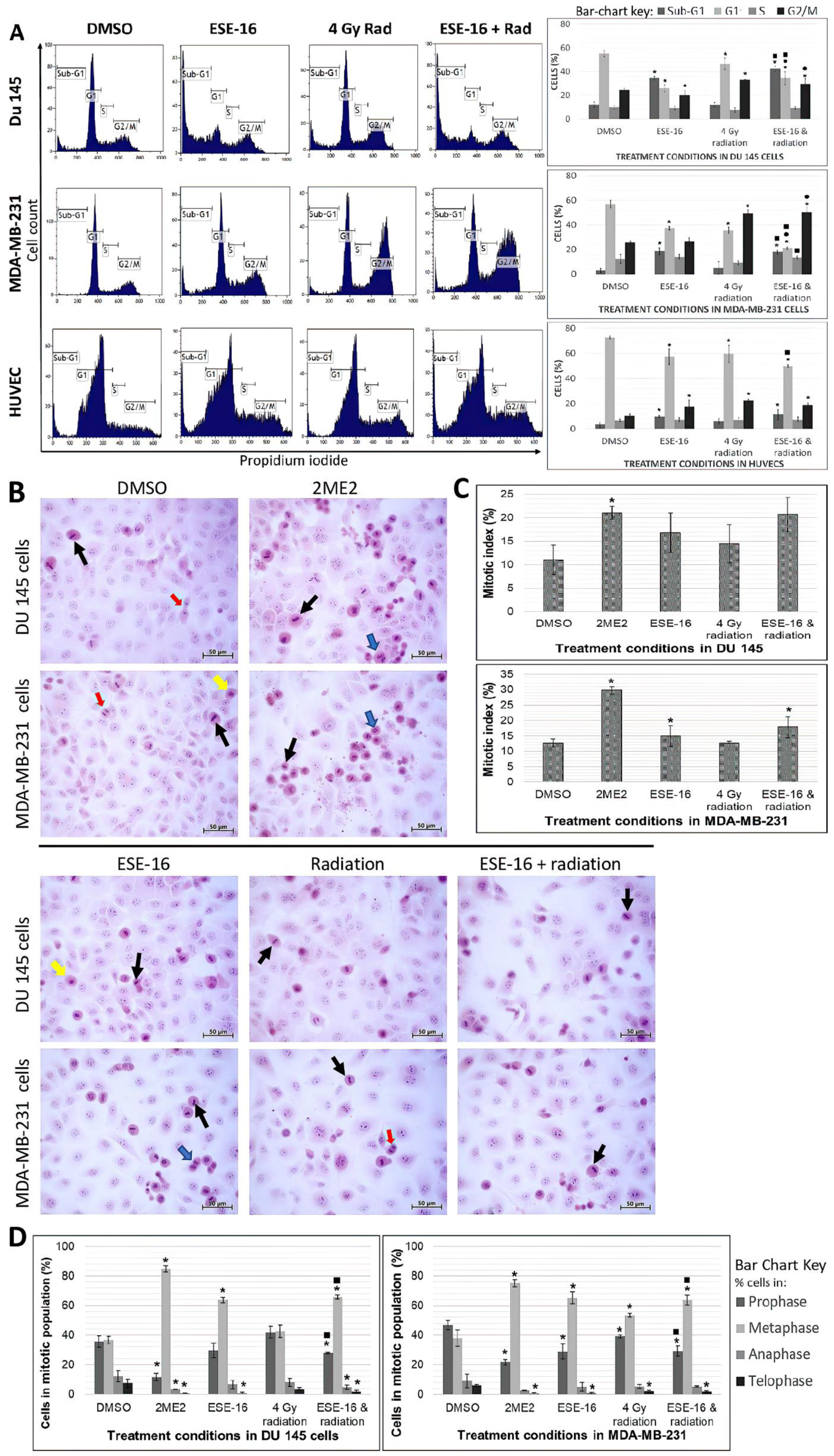
2.8. Cell Division: Mitotic Index Determination Using Light Microscopy
2.9. Apoptosis: Annexin V-FITC Detection by Flow Cytometry
2.10. Apoptotic Signaling: Colorimetric Determination of Caspase 3 Activity
2.11. Reactive Oxygen Species: Superoxide Detection by Flow Cytometry
2.12. DNA Damage: Micronuclei Quantification Using Light Microscopy
2.13. DNA Damage: Flow Cytometric Quantification of Histone H2A.X Phosphorylation
2.14. DNA Damage: γ-H2A.X Foci Using Fluorescence Microscopy
2.15. Metastatic Signaling: Western Blot Analyses of BMP-7 and MMP-9 Expression
2.16. Angiogenic Signaling: Western Blot Analysis of HIF-1α Expression
2.17. Effect of the Combination Treatment on the Bone Component of the Metastatic Lesion
2.18. Osteoclast Differentiation: TRAP Activity and Staining
2.19. Osteoclast Activity: Actin Ring Formation Using Fluorescence Microscopy
2.20. Statistical Analyses
3. Results
3.1. ESE-16 Is Cytotoxic to Cancer, Pre-Osteoclastic, and Endothelial Cells with Enhanced Sensitivity When Combined with Radiation Whilst Pre-Osteoblasts Are Spared
3.2. Subsequent Experimental Conditions
3.3. ESE-16-Pre-Sensitization Inhibits Cell Cycle Progression by Inducing G2/M Phase Arrest and Promoting Sub-G1 Phase Accumulation in Cancer and Endothelial Cells
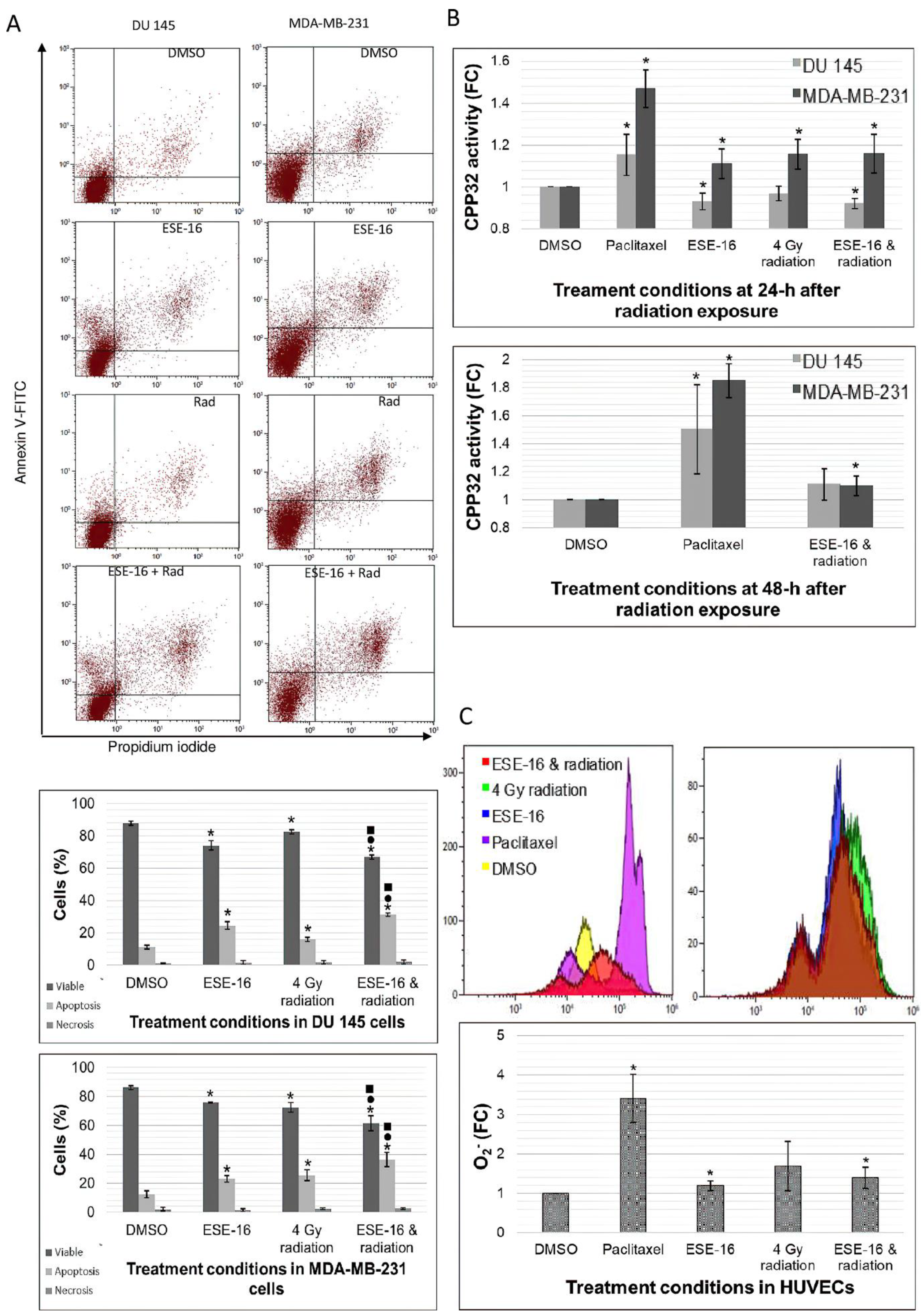
3.4. Apoptosis Was Induced in Combination-Treated Cancer Cells, with the Invovlement of CPP32 in MDA-MB-231 Cells
3.5. HUVECs Generate Superoxide Ions in Response to the Combination Treatment
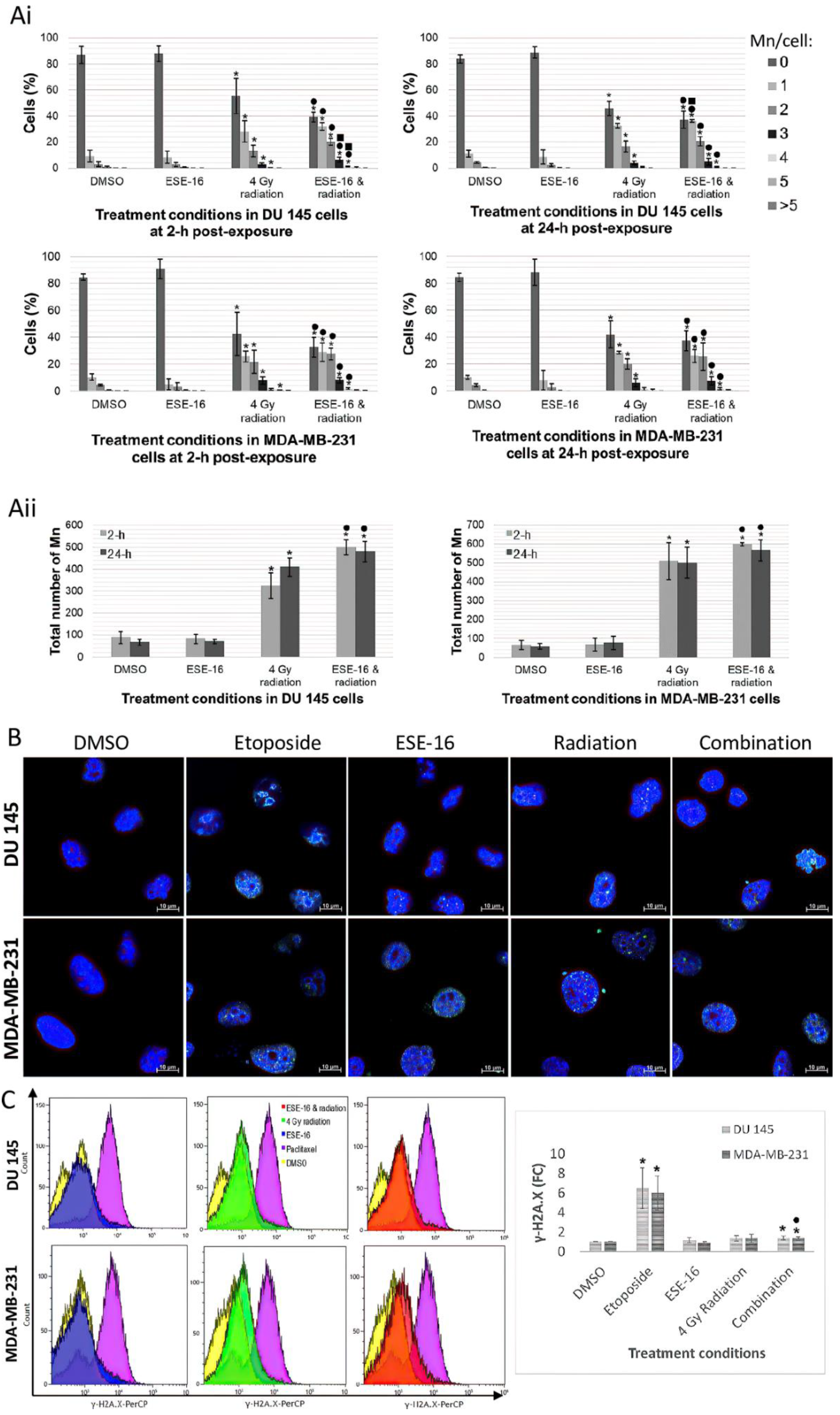
3.6. Drug Pretreated Cancer Cells Demonstrated Greater Radiation-Induced DNA Damage
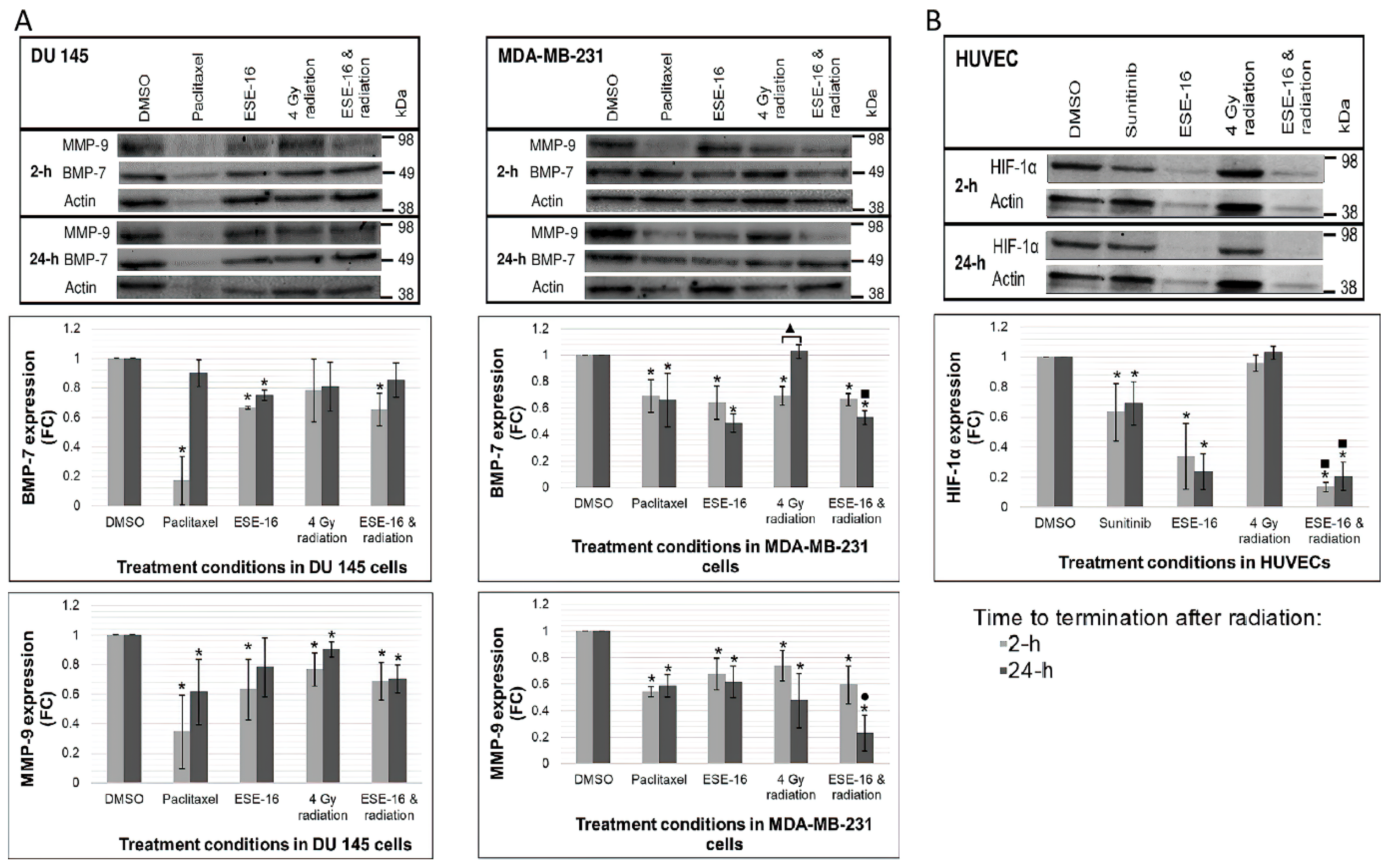
3.7. ESE-16/Radiation Inhibits Metastatic Signaling in Cancer Cells through the Downregulation of BMP-7 and MMP-9 Expression
3.8. ESE-16 Together with Radiation Inhibits HIF-1α Expression in HUVECs
3.9. Pre-Osteoclasts Retentained Theirr Differentiation Capacity after Treatment with the Comvination Therepy, and Actin Ring Formation Was Preserved in Mature Osteoclast
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2ME2 | 2-methoxyestradiol |
| ANOVA | analysis of variance |
| Apaf-1 | apoptosis protease activating factor-1 |
| ATCC | American Type Culture Collection |
| Bax | Bcl-2-associated X |
| BCA | bicinchoninic acid |
| Bcl-2 | B-cell lymphoma-2 |
| bFGF | basic fibroblast growth factor |
| BMP-7 | bone morphogenetic protein-7 |
| BSA | bovine serum albumin |
| CA | California |
| CANSA | Cancer Association of South Africa |
| CPP32 | cysteine protease P32 |
| DAPI | 4’,6-diamidino-2-phenylindole |
| ddH2O | double-distilled water |
| DEVD | aspartate-glutamate-valine-aspartate |
| Dmax | photon beam electronic equilibrium depth |
| DMEM | Dulbecco’s modified Eagle medium |
| DMSO | dimethyl sulfoxide |
| DNA | deoxyribonucleic acid |
| DTT | dithiothreitol |
| DU 145 | metastatic human prostate carcinoma cells |
| EBM | endothelial basal medium |
| ECL | enhanced chemiluminescence |
| ECM | extracellular matrix |
| EDTA | ethylenediaminetetraacetic acid |
| EGF | epidermal growth factor |
| EGTA | ethylene glycol tetraacetic acid |
| EMT | epithelial-mesenchymal transition |
| ESE-15-ol | 2-ethyl-3-O-sulfamoyl-estra-1,3,5(10),15-tetraen-17-ol |
| ESE-16 | 2-ethyl-3-O-sulfamoyl-estra-1,3,5(10)16-tetraene |
| FADD | Fas-associated death domain |
| FBS | fetal bovine serum |
| FITC | fluorescein isothiocyanate |
| GI50 | half maximal growth inhibition concentration |
| HIF-1α | hypoxia-inducible factor 1-α |
| HRP | horse radish peroxidase |
| HUVECs | human umbilical vein endothelial cells |
| IGF | insulin growth factor |
| JHB | Johannesburg |
| LDS | lithium dodecyl sulfate |
| MA | Massachusetts |
| MC3T3-E1 | murine pre-osteoblasts |
| MD | Maryland |
| MDA-MB-231 | metastatic human breast adenocarcinoma cells |
| MI | mitotic index |
| MMP-9 | matrix metalloproteinase-9 |
| MMPs | matrix metalloproteinases |
| Mn | micronuclei |
| MO | Missouri |
| MOPS | 3-morpholinopropane-1-sulfonic acid |
| mRNA | messenger ribonucleic acid |
| MTAs | microtubule-targeting agents |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| MW | molecular weight |
| NRF | National Research Foundation |
| O2− | superoxide |
| PBS | phosphate buffer saline |
| PerCP | peridinin chlorophyll protein |
| PI | propidium iodide |
| pNA | p-nitroaniline |
| PS | phosphatidylserine |
| PTHrP | parathyroid hormone-related protein |
| PVDF | polyvinylidene fluoride |
| RANK | receptor activator of nuclear factor-κB |
| RANKL | receptor activator of nuclear factor κB ligand |
| RAW 264.7 | murine macrophages |
| RDP-UP | Research Development Programme of the University of Pretoria |
| RESCOM | School of Medicine Research Committee of the University of Pretoria |
| RIPA | radioimmunoprecipitation assay |
| RNase A | ribonuclease A |
| ROS | reactive oxygen species |
| RPMI | Roswell Park Memorial Institute |
| RT | room temperature |
| SAC | spindle assembly checkpoint |
| SD | standard deviation |
| SDS | sodium dodecyl sulfate |
| SSD | source-to-surface distance |
| TGF-β | transforming growth factor-β |
| TNF-α | tumor necrosis factor-α |
| TRAIL | tumor necrosis factor-related apoptosis inducing ligand |
| TRAP | tartrate-resistant acid phosphatase |
| USA | United States of America |
| VEGF-A | vascular endothelial growth factor-A |
| VT | Vermont |
References
- Maurizi, A.; Rucci, N. The osteoclast in bone metastasis: Player and target. Cancers 2018, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Weng, Y.; Ren, W.; Zhang, Z.; Wang, T.; Wang, J.; Jiang, Y.; Chen, Y.; Zhou, L.; He, T.; et al. Biological roles of human bone morphogenetic protein 9 in the bone microenvironment of human breast cancer MDA-MB-231 cells. Am. J. Transl. Res. 2015, 7, 1660–1674. [Google Scholar]
- Futakuchi, M.; Fukamachi, K.; Suzui, M. Heterogeneity of tumor cells in the bone microenvironment: Mechanisms and therapeutic targets for bone metastasis of prostate or breast cancer. Adv. Drug Deliv. Rev. 2016, 99, 206–211. [Google Scholar] [CrossRef]
- Fang, J.; Xu, Q. Differences of osteoblastic bone metastases and osteolytic bone metastases in clinical features and molecular characteristics. Clin. Transl. Oncol. 2015, 17, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Bonci, D.; Coppola, V.; Patrizii, M.; Addario, A.; Cannistraci, A.; Francescangeli, F.; Pecci, R.; Muto, G.; Collura, D.; Bedini, R.; et al. A microRNA code for prostate cancer metastasis. Oncogene 2016, 35, 1180–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 12, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Casimiro, S.; Mohammad, K.S.; Pires, R.; Tato-Costa, J.; Alho, I.; Teixeira, R.; Carvalho, A.; Ribeiro, S.; Lipton, A.; Guise, T.A.; et al. RANKL/RANK/MMP-1 molecular triad contributes to the metastatic phenotype of breast and prostate cancer cells In Vitro. PLoS ONE 2013, 8, e63153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abal, M.; Andreu, J.M.; Barasoain, I. Taxanes: Microtubule and centrosome targets, and cell cycle dependent mechanisms of action. Curr. Cancer Drug Targets 2003, 3, 193–203. [Google Scholar] [CrossRef]
- Göbel, A.; Dell’Endice, S.; Jaschke, N.; Pählig, S.; Shahid, A.; Hofbauer, L.C.; Rachner, T.D. The Role of inflammation in breast and prostate cancer metastasis to bone. Int. J. Mol. Sci. 2021, 22, 5078. [Google Scholar] [CrossRef]
- Kingsley, L.A.; Fournier, P.G.J.; Chirgwin, J.M.; Guise, T.A. Molecular biology of bone metastasis. Mol. Cancer Ther. 2007, 6, 2609. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhong, C.; Frenkel, B.; Reddi, A.H.; Roy-Burman, P. Diverse biological effect and Smad signaling of bone morphogenetic protein 7 in prostate tumor cells. Cancer Res. 2005, 65, 5769–5777. [Google Scholar] [CrossRef] [Green Version]
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.; Corey, E.; Padalecki, S.; Suva, L.; et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin. Cancer Res. 2006, 12, 6213s–6216s. [Google Scholar] [CrossRef] [Green Version]
- Sekita, A.; Matsugaki, A.; Nakano, T. Disruption of collagen/apatite alignment impairs bone mechanical function in osteoblastic metastasis induced by prostate cancer. Bone 2017, 97, 83–93. [Google Scholar] [CrossRef]
- Hensel, J.; Thalmann, G.N. Biology of bone metastases in prostate cancer. Urology 2016, 92, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F.; Gajate, C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis 2003, 8, 413–450. [Google Scholar] [CrossRef] [Green Version]
- Wickstead, B.; Gull, K. The evolution of the cytoskeleton. J. Cell Biol. 2011, 194, 513–525. [Google Scholar] [CrossRef]
- Parker, A.L.; Kavallaris, M.; McCarroll, J.A. Microtubules and their role in cellular stress in cancer. Front. Oncol. 2014, 4, 153. [Google Scholar] [CrossRef] [Green Version]
- Di Cesare, E.; Verrico, A.; Miele, A.; Giubettini, M.; Rovella, P.; Coluccia, A.; Famiglini, V.; La Regina, G.; Cundari, E.; Silvestri, R.; et al. Mitotic cell death induction by targeting the mitotic spindle with tubulin-inhibitory indole derivative molecules. Oncotarget 2017, 8, 19738–19759. [Google Scholar] [CrossRef] [Green Version]
- Raviraj, J.; Bokkasam, V.; Kumar, V.; Reddy, U.; Suman, V. Radiosensitizers, radioprotectors, and radiation mitigators. Indian J. Dent. Res. 2014, 25, 83–90. [Google Scholar] [CrossRef]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [Google Scholar] [CrossRef]
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. Drugs that target dynamic microtubules: A new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. [Google Scholar] [CrossRef] [Green Version]
- Tangutur, A.; Kumar, D.; Krishna, K.; Kantevari, S. Microtubule targeting agents as cancer chemotherapeutics: An overview of molecular hybrids as stabilizing and destabilizing agents. Curr. Top. Med. Chem 2017, 17, 2523–2537. [Google Scholar] [CrossRef] [PubMed]
- Farhat, M.; Poissonnier, A.; Hamze, A.; Ouk-Martin, C.; Brion, J.; Alami, M.; Feuillard, J.; Jayat-Vignoles, C. Reversion of apoptotic resistance of TP53-mutated Burkitt lymphoma B-cells to spindle poisons by exogenous activation of JNK and p38 MAP kinases. Cell Death Dis. 2014, 5, e1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, R.; Katrukha, E.A.; Doodhi, H.; Smal, I.; Meijering, E.; Kapitein, L.C.; Steinmetz, M.O.; Akhmanova, A. End-binding proteins sensitize microtubules to the action of microtubule-targeting agents. Proc. Natl. Acad. Sci. USA 2013, 110, 8900–8905. [Google Scholar] [CrossRef] [Green Version]
- Mooberry, S.L. Mechanism of action of 2-methoxyestradiol: New developments. Drug Resist. Updates 2004, 6, 355–361. [Google Scholar] [CrossRef]
- Li, L.; Bu, S.; Bäckström, T.; Ländström, M.; Ulmsten, U.; Fu, X. Induction of apoptosis and G2/M arrest by 2-methoxyestradiol in human cervical cancer HeLa cells. Anticancer Res. 2004, 24, 873–880. [Google Scholar]
- Casarez, E.V.; Dunlap-Brown, M.E.; Conaway, M.R.; Amorino, G.P. Radiosensitisation and modulation of p44/42 mitogen-activated protein kinase by 2-Methoxyestradiol in prostate cancer models. Cancer Res. 2007, 67, 8316–8324. [Google Scholar] [CrossRef] [Green Version]
- Amorino, G.P.; Freeman, M.L.; Choy, H. Enhancement of radiation effects in vitro by the estrogen metabolite 2-methoxyestradiol. Radiat. Res. 2000, 153, 384–391. [Google Scholar] [CrossRef]
- Zhao, H.; Jiang, H.; Li, Z.; Zhuang, Y.; Liu, Y.; Zhou, S.; Zhou, Y. 2-Methoxyestradiol enhances radiosensitivity in radioresistant melanoma MDA-MB-435R cells by regulating glycolysis via HIF-1α/PDK1 axis. Int. J. Oncol. 2017, 50, 1531–1540. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, A.L.; Wilhelmson, A.S.; Fagman, J.B.; Ryberg, H.; Koskela, A.; Tuukkanen, J.; Tivesten, Å.; Ohlsson, C. The bone sparing effects of 2-methoxyestradiol are mediated via estrogen receptor-α in male mice. Endocrinology 2016, 157, 4200–4205. [Google Scholar] [CrossRef] [Green Version]
- Theron, A.; Prudent, R.; Nolte, E.; van den Bout, I.; Punchoo, R.; Marais, S.; du Toit, P.; Hlophe, Y.; van Papendorp, D.; Lafanechère, L.; et al. Novel in silico-designed estradiol analogues are cytotoxic to a multidrug-resistant cell line at nanomolar concentrations. Cancer Chemother. Pharmacol. 2015, 75, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Amin, M.L. P-glycoprotein inhibition for optimal drug delivery. Drug Target. Insights 2013, 7, 27–34. [Google Scholar] [CrossRef]
- Newman, S.P.; Ireson, C.R.; Tutill, H.J.; Day, J.M.; Parsons, M.F.C.; Leese, M.P.; Potter, B.V.L.; Reed, M.J.; Purohit, A. The role of 17β-hydroxysteroid dehydrogenases in modulating the activity of 2-methoxyestradiol in breast cancer cells. Cancer Res. 2006, 66, 324–330. [Google Scholar] [CrossRef] [Green Version]
- Stander, A.; Joubert, F.; Joubert, A. Docking, synthesis, and in vitro evaluation of antimitotic estrone analogs. Chem. Biol. Drug Des. 2011, 77, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stander, X.X.; Stander, B.A.; Joubert, A.M. In vitro effects of an in silico-modelled 17β-estradiol derivative in combination with dichloroacetic acid on MCF-7 and MCF-12A cells. Cell Prolif. 2011, 44, 567–581. [Google Scholar] [CrossRef]
- Stander, B.A.; Joubert, F.; Tu, C.; Sippel, K.H.; McKenna, R.; Joubert, A.M. Signaling pathways of ESE-16, an antimitotic and anticarbonic anhydrase estradiol analog, in breast cancer cells. PLoS ONE 2013, 8, e53853. [Google Scholar] [CrossRef]
- Theron, A.E.; Nolte, E.M.; Lafanechere, L.; Joubert, A.M. Molecular crosstalk between apoptosis and autophagy induced by a novel 2-methoxyestradiol analogue in cervical adenocarcinoma cells. Cancer Cell Int. 2013, 13, 87. [Google Scholar] [CrossRef]
- Mothibeli, K.; Mercier, A.; Cromarty, A.; Rheeder, M.; Naidoo, V.; Olorunju, S.; Joubert, A. Confirming oral bioavailability of novel oestradiol analogues by liquid chromatography-tandem mass spectrometry in a murine model. Biomed. Res. 2018, 29, 3267–3275. [Google Scholar]
- Mercier, A.E.; Prudent, R.; Pepper, M.S.; De Koning, L.; Nolte, E.; Peronne, L.; Nel, M.; Lafanechère, L.; Joubert, A.M. Characterization of signalling pathways that link apoptosis and autophagy to cell death induced by estrone Analogues which reversibly depolymerize microtubules. Molecules 2021, 26, 706. [Google Scholar] [CrossRef] [PubMed]
- Wolmarans, E.; Sippel, K.; McKenna, R.; Joubert, A. Induction of the intrinsic apoptotic pathway via a new antimitotic agent in an esophageal carcinoma cell line. Cell Biosci. 2014, 4, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repsold, L.; Pretorius, E.; Joubert, A.M. An estrogen analogue and promising anticancer agent refrains from inducing morphological damage and reactive oxygen species generation in erythrocytes, fibrin and platelets: A pilot study. Cancer Cell Int. 2014, 14, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poruchynsky, M.S.; Komlodi-Pasztor, E.; Trostel, S.; Wilkerson, J.; Regairaz, M.; Pommier, Y.; Zhang, X.; Kumar Maity, T.; Robey, R.; Burotto, M.; et al. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 1571–1576. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, D.; Ha, G.; Ruggieri, R.; Symons, M. Microtubule-targeting agents can sensitize cancer cells to ionizing radiation by an interphase-based mechanism. OncoTargets Ther. 2017, 10, 5633. [Google Scholar] [CrossRef] [Green Version]
- Boeyens, J.; Deepak, V.; Chua, W.-H.; Kruger, M.; Joubert, A.; Coetzee, M. Effects of ω3- and ω6-Polyunsaturated Fatty Acids on RANKL-Induced Osteoclast Differentiation of RAW264.7 Cells: A Comparative in Vitro Study. Nutrients 2014, 6, 2584. [Google Scholar] [CrossRef] [Green Version]
- Avwioro, G. Histochemical uses of haematoxylin—A review. J. Pharm. Clin. Sci. 2011, 1, 24–34. [Google Scholar]
- Rieger, A.M.; Nelson, K.L.; Konowalchuk, J.D.; Barreda, D.R. Modified annexin V/propidium iodide apoptosis assay for accurate assessment of cell death. J. Vis. Exp. 2011, 2597. [Google Scholar] [CrossRef]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Kalsbeek, D.; Golsteyn, R.M. G2/M-phase checkpoint adaptation and micronuclei formation as mechanisms that contribute to genomic instability in human cells. Int. J. Mol. Sci. 2017, 18, 2344. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Halicka, D.; Traganos, F.; Darzynkiewicz, Z. Cytometric analysis of DNA damage: Phosphorylation of histone H2AX as a marker of DNA double-strand breaks (DSBs). In Chromatin Protocols, 2nd ed.; Chellappan, S.P., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 161–168. [Google Scholar]
- Guppy, M. The hypoxic core: A possible answer to the cancer paradox. Biochem. Biophys. Res. Commun. 2002, 299, 676–680. [Google Scholar] [CrossRef]
- Wu, X.-H.; Qian, C.; Yuan, K. Correlations of hypoxia-inducible factor-1α/hypoxia-inducible factor -2α expression with angiogenesis factors expression and prognosis in non-small cell lung cancer. Chin. Med. J. 2011, 124, 11–18. [Google Scholar]
- Marino, S.; Logan, J.G.; Mellis, D.; Capulli, M. Generation and culture of osteoclasts. BoneKEy Rep. 2014, 3, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolte, E.; Joubert, A.; Lakier, R.; van Rensburg, A.; Mercier, A. Exposure of breast and lung cancer cells to a novel estrone analog prior to radiation enhances Bcl-2-mediated cell death. Int. J. Mol. Sci. 2018, 19, 2887. [Google Scholar] [CrossRef] [Green Version]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H.E. Molecular mechanisms of radiation-induced cancer cell death: A primer. Front. Cell Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef]
- Sergeeva, T.F.; Shirmanova, M.V.; Zlobovskaya, O.A.; Gavrina, A.I.; Dudenkova, V.V.; Lukina, M.M.; Lukyanov, K.A.; Zagaynova, E.V. Relationship between intracellular pH, metabolic co-factors and caspase-3 activation in cancer cells during apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 604–611. [Google Scholar] [CrossRef]
- Flanagan, L.; Meyer, M.; Fay, J.; Curry, S.; Bacon, O.; Duessmann, H.; John, K.; Boland, K.C.; McNamara, D.A.; Kay, E.W.; et al. Low levels of Caspase-3 predict favourable response to 5FU-based chemotherapy in advanced colorectal cancer: Caspase-3 inhibition as a therapeutic approach. Cell Death Dis. 2016, 7, e2087. [Google Scholar] [CrossRef] [Green Version]
- Boyd, L.S.; Gozuacik, D.; Joubert, A.M. The in vitro effects of a novel estradiol analog on cell proliferation and morphology in human epithelial cervical carcinoma. Cell. Mol. Biol. Lett. 2018, 23, 10. [Google Scholar] [CrossRef] [Green Version]
- Visagie, M.H.; Birkholtz, L.-M.; Joubert, A.M. A 2-methoxyestradiol bis-sulphamoylated derivative induces apoptosis in breast cell lines. Cell Biosci. 2015, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Kang, M.; Cho, Y. Low production of reactive oxygen species and high DNA repair: Mechanism of radioresistance of prostate cancer stem cells. Anticancer Res. 2013, 33, 4469–4474. [Google Scholar]
- Ramadhani, D.W.I.; Purnami, S. Automated detection of binucleated cell and micronuclei using CellProfiler 2.0 software. HAYATI J. Biosci. 2013, 20, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Herd, O.; Francies, F.; Kotzen, J.; Smith, T.; Nxumalo, Z.; Muller, X.; Slabbert, J.; Vral, A.; Baeyens, A. Chromosomal radiosensitivity of human immunodeficiency virus positive/negative cervical cancer patients in South Africa. Mol. Med. Rep. 2016, 13, 130–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, W.S.; Banerjee, S.; Crawford, D.F. G2E3 is a nucleo-cytoplasmic shuttling protein with DNA damage responsive localization. Exp. Cell Res. 2007, 313, 665–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Huang, X.; Halicka, H.D.; Zhao, H.; Traganos, F.; Albino, A.P.; Dai, W.; Darzynkiewicz, Z. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytom. A 2007, 71, 648–661. [Google Scholar] [CrossRef]
- Redon, C.E.; Nakamura, A.J.; Zhang, Y.-W.; Ji, J.; Bonner, W.M.; Kinders, R.J.; Parchment, R.E.; Doroshow, J.H.; Pommier, Y. Histone γH2AX and Poly(ADP-Ribose) as clinical pharmacodynamic biomarkers. Clin. Cancer Res. 2010, 16, 4532–4542. [Google Scholar] [CrossRef] [Green Version]
- Solier, S.; Pommier, Y. The nuclear γ-H2AX apoptotic ring: Implications for cancers and autoimmune diseases. Cell. Mol. Life Sci. 2014, 71, 2289–2297. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Morris, R.J. The Yin and Yang of bone morphogenetic proteins in cancer. Cytokine Growth Factor Rev. 2010, 21, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Pivetta, E.; Scapolan, M.; Pecolo, M.; Wassermann, B.; Abu-Rumeileh, I.; Balestreri, L.; Borsatti, E.; Tripodo, C.; Colombatti, A.; Spessotto, P. MMP-13 stimulates osteoclast differentiation and activation in tumour breast bone metastases. Breast Cancer Res. 2011, 13, R105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P. The role of hypoxia-induced factors in tumor progression. Oncologist 2004, 9, 10–17. [Google Scholar] [CrossRef]
- Zhang, L.; Ye, S.-B.; Li, Z.-L.; Ma, G.; Chen, S.-P.; He, J.; Liu, W.-L.; Xie, D.; Zeng, Y.-X.; Li, J. Increased HIF-1alpha expression in tumor cells and lymphocytes of tumor microenvironments predicts unfavorable survival in esophageal squamous cell carcinoma patients. Int. J. Clin. Exp. Pathol. 2014, 7, 3887–3897. [Google Scholar]
- Blumer, M.J.F.; Hausott, B.; Schwarzer, C.; Hayman, A.R.; Stempel, J.; Fritsch, H. Role of tartrate-resistant acid phosphatase (TRAP) in long bone development. Mech. Dev. 2012, 129, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Maran, A.; Gorny, G.; Oursler, M.J.; Zhang, M.; Shogren, K.L.; Yaszemski, M.J.; Turner, R.T. 2-Methoxyestradiol inhibits differentiation and is cytotoxic to osteoclasts. J. Cell. Biochem. 2006, 99, 425–434. [Google Scholar] [CrossRef]
- Desai, S.; Kumar, A.; Laskar, S.; Pandey, B.N. Cytokine profile of conditioned medium from human tumor cell lines after acute and fractionated doses of gamma radiation and its effect on survival of bystander tumor cells. Cytokine 2013, 61, 54–62. [Google Scholar] [CrossRef]
- Kwak, H.B.; Jin, H.-M.; Ha, H.; Kang, M.-J.; Lee, S.B.; Kim, H.-H.; Lee, Z.H. Tumor necrosis factor-alpha induces differentiation of human peripheral blood mononuclear cells into osteoclasts through the induction of p21(WAF1/Cip1). Biochem. Biophys. Res. Commun. 2005, 330, 1080–1086. [Google Scholar] [CrossRef]
- Zwerina, J.; Hayer, S.; Redlich, K.; Bobacz, K.; Kollias, G.; Smolen, J.S.; Schett, G. Activation of p38 MAPK is a key step in tumor necrosis factor–mediated inflammatory bone destruction. Arthritis Rheum. 2006, 54, 463–472. [Google Scholar] [CrossRef]
- Eck, S.M.; Côté, A.L.; Winkelman, W.D.; Brinckerhoff, C.E. CXCR4 and matrix metalloproteinase-1 are elevated in breast carcinoma-associated fibroblasts and in normal mammary fibroblasts exposed to factors secreted by breast cancer cells. Mol. Cancer Res. 2009, 7, 1033–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Nakayama, J.; Hayashi, Y.; Jeong, S.; Futakuchi, M.; Ito, E.; Watanabe, S.; Semba, K. Establishment and characterization of highly osteolytic luminal breast cancer cell lines by intracaudal arterial injection. Genes Cells 2020, 25, 111–123. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell lines | GI50 (µM) ± SD | |
|---|---|---|
| Cancer cells | ||
| ESE-16-only dose curve | ||
| DU 145 | 0.625 ± 0.039 | |
| MDA-MB-231 | 0.469 ± 0.078 | |
| Bone- and endothelial cells | ||
| ESE-16 | ESE-16 & 4 Gy radiation | |
| MC3T3-E1 | >10 | >10 |
| RAW 264.7 | 0.625 ± 0.156 | <0.039 |
| HUVEC | 0.156 ± 0.02 | <0.039 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helena, J.; Joubert, A.; Mabeta, P.; Coetzee, M.; Lakier, R.; Mercier, A. Intracellular Signaling Responses Induced by Radiation within an In Vitro Bone Metastasis Model after Pre-Treatment with an Estrone Analogue. Cells 2021, 10, 2105. https://doi.org/10.3390/cells10082105
Helena J, Joubert A, Mabeta P, Coetzee M, Lakier R, Mercier A. Intracellular Signaling Responses Induced by Radiation within an In Vitro Bone Metastasis Model after Pre-Treatment with an Estrone Analogue. Cells. 2021; 10(8):2105. https://doi.org/10.3390/cells10082105
Chicago/Turabian StyleHelena, Jolene, Anna Joubert, Peace Mabeta, Magdalena Coetzee, Roy Lakier, and Anne Mercier. 2021. "Intracellular Signaling Responses Induced by Radiation within an In Vitro Bone Metastasis Model after Pre-Treatment with an Estrone Analogue" Cells 10, no. 8: 2105. https://doi.org/10.3390/cells10082105
APA StyleHelena, J., Joubert, A., Mabeta, P., Coetzee, M., Lakier, R., & Mercier, A. (2021). Intracellular Signaling Responses Induced by Radiation within an In Vitro Bone Metastasis Model after Pre-Treatment with an Estrone Analogue. Cells, 10(8), 2105. https://doi.org/10.3390/cells10082105