
SNAP47 Interacts with ATG14 to Promote VP1 Conjugation and CVB3 Propagation



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Collection
2.2. Viral Infection and Titer Measurement
2.3. Chemical Treatment
2.4. Plasmids and siRNAs
2.5. In Vitro Cleavage Assay
2.6. Immunoprecipitation
2.7. Generation of SNAP47-Knockout and ATG14-Knockdown Cells Using CRISPR-Cas9
2.8. Cell Fractionation
2.9. Western Blot Analysis
2.10. Immunofluorescence and Confocal Microscopy
2.11. Statistical Analysis
3. Results
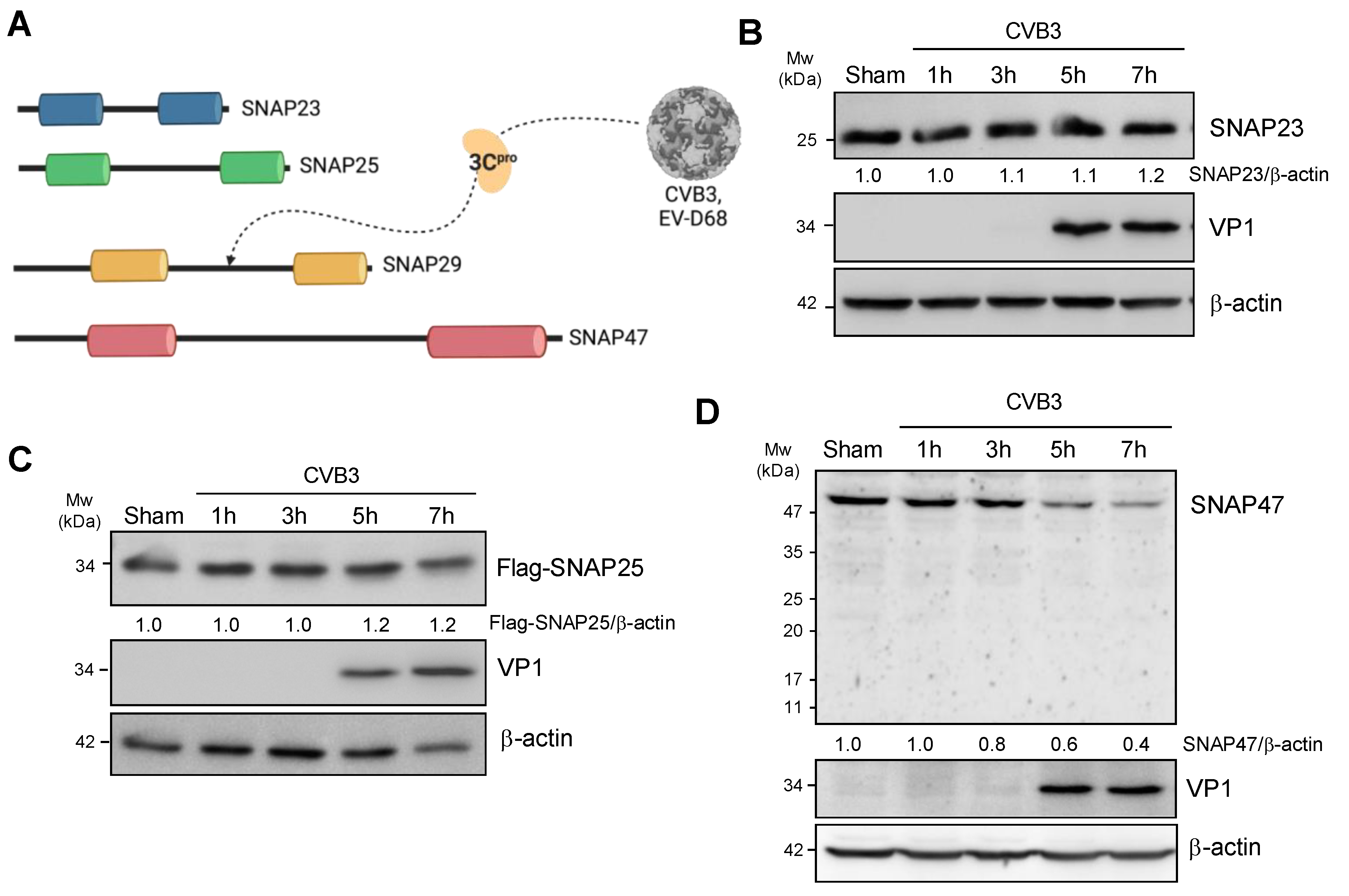
3.1. CVB3 Infection Perturbs SNAP47 but Not SNAP23 or SNAP25
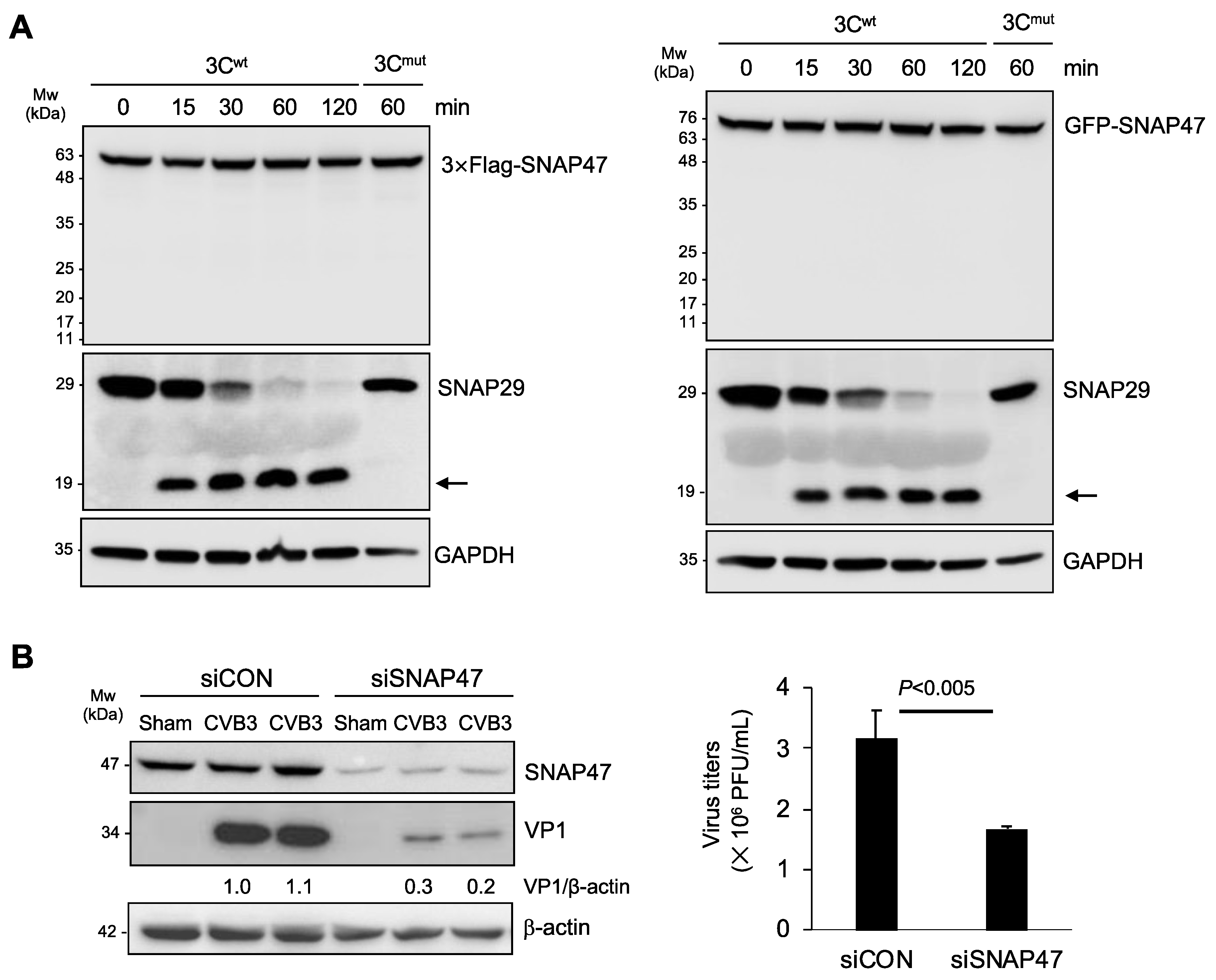
3.2. Gene-Silencing of SNAP47 Suppresses Viral Infection
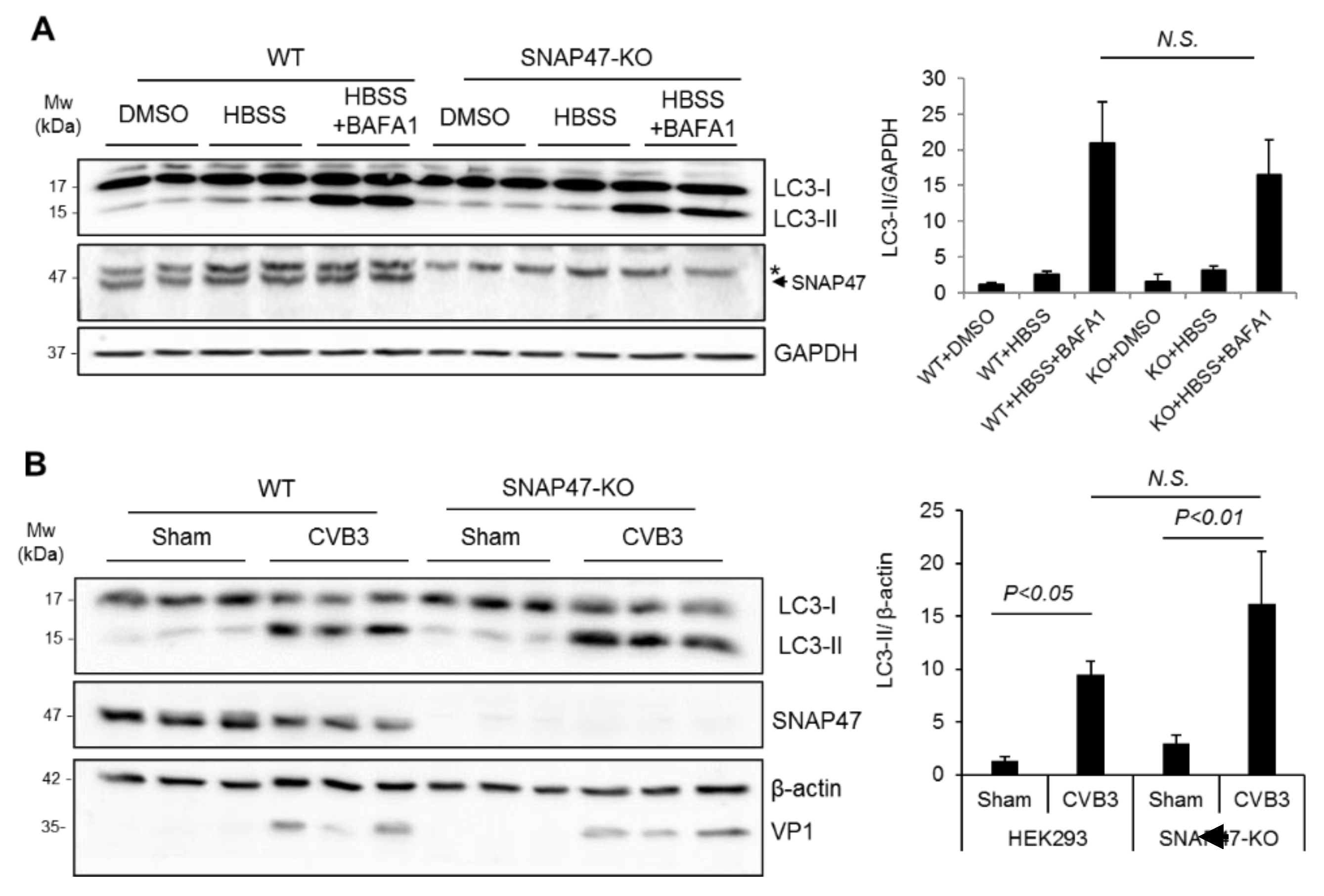
3.3. Ablation of SNAP47 Does Not Impair Starvation- or CVB3-Induced Autophagy
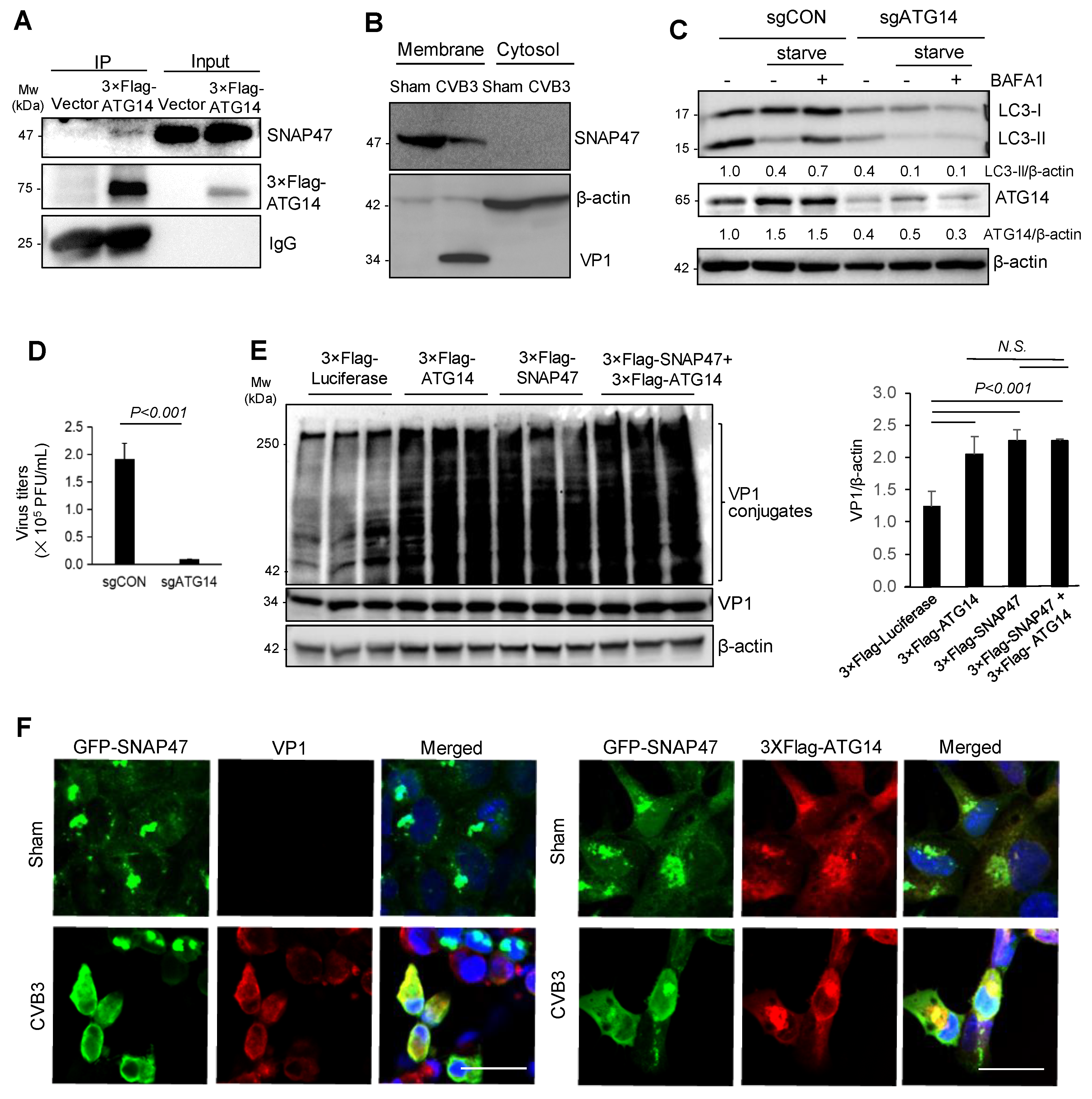
3.4. SNAP47 Interacts with ATG14 on the Cellular Membrane Fractions alongside VP1
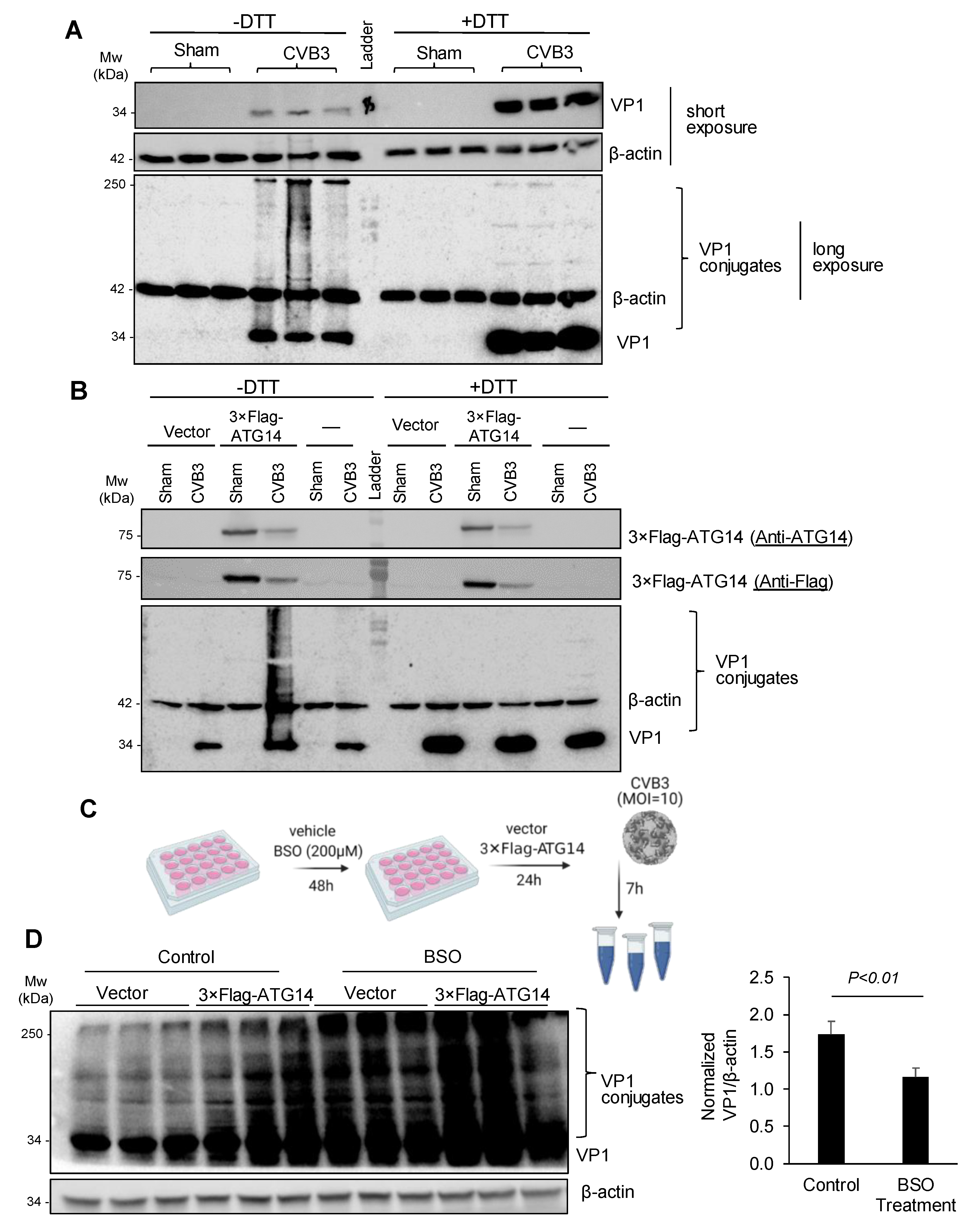
3.5. Atg14 Promotes Viral Capsid Maturation via Glutathione-Mediated Disulfide Bond Formation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garmaroudi, F.S.; Marchant, D.; Hendry, R.; Luo, H.; Yang, D.; Ye, X.; Shi, J.; McManus, B.M. Coxsackievirus B3 replication and pathogenesis. Future Microbiol. 2015, 10, 629–653. [Google Scholar] [CrossRef] [PubMed]
- Fung, G.; Luo, H.; Qiu, Y.; Yang, D.; McManus, B. Myocarditis. Circ. Res. 2016, 118, 496–514. [Google Scholar] [CrossRef]
- Wong, A.H.; Lau, C.; Cheng, P.K.; Ng, A.Y.; Lim, W.W. Coxsackievirus B3-associated aseptic meningitis: An emerging infection in Hong Kong. J. Med. Virol. 2011, 83, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Song, Y.; Li, Y.; Liu, Y.; Jiang, P.; Lin, X.; Liu, G.; Song, L.; Wang, H.; Xu, A. Coxsackievirus B3, Shandong province, china, 1990–2010. Emerg. Infect. Dis. 2012, 18, 1865. [Google Scholar] [CrossRef]
- Mohamud, Y.; Luo, H. The intertwined life cycles of enterovirus and autophagy. Virulence 2019, 10, 470–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Obara, K.; Ohsumi, Y. Atg14: A key player in orchestrating autophagy. Int. J. Cell Biol. 2011, 2011, 713435. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Nassiri, A.; Zhong, Q. Autophagosome targeting and membrane curvature sensing by Barkor/Atg14 (L). Proc. Natl. Acad. Sci. USA 2011, 108, 7769–7774. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Thapa, N.; Liao, Y.; Choi, S.; Anderson, R.A. PtdIns (4, 5) P2 signaling regulates ATG14 and autophagy. Proc. Natl. Acad. Sci. USA 2016, 113, 10896–10901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Pluhackova, K.; Bockmann, R.A. The multifaceted role of SNARE proteins in membrane fusion. Front. Physiol. 2017, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Sudhof, T.C.; Rothman, J.E. Membrane fusion: Grappling with SNARE and SM proteins. Science 2009, 323, 474–477. [Google Scholar] [CrossRef] [Green Version]
- Nair, U.; Jotwani, A.; Geng, J.; Gammoh, N.; Richerson, D.; Yen, W.L.; Griffith, J.; Nag, S.; Wang, K.; Moss, T.; et al. SNARE proteins are required for macroautophagy. Cell 2011, 146, 290–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corona Velazquez, A.; Corona, A.K.; Klein, K.A.; Jackson, W.T. Poliovirus induces autophagic signaling independent of the ULK1 complex. Autophagy 2018, 14, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Mohamud, Y.; Shi, J.; Tang, H.; Xiang, P.; Xue, Y.C.; Liu, H.; Ng, C.S.; Luo, H. Coxsackievirus infection induces a non-canonical autophagy independent of the ULK and PI3K complexes. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Corona, A.K.; Saulsbery, H.M.; Velazquez, A.F.C.; Jackson, W.T. Enteroviruses remodel autophagic trafficking through regulation of host SNARE proteins to promote virus replication and cell exit. Cell Rep. 2018, 22, 3304–3314. [Google Scholar] [CrossRef] [Green Version]
- Mohamud, Y.; Shi, J.; Qu, J.; Poon, T.; Xue, Y.C.; Deng, H.; Zhang, J.; Luo, H. Enteroviral infection inhibits autophagic flux via disruption of the SNARE complex to enhance viral replication. Cell Rep. 2018, 22, 3292–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemball, C.C.; Alirezaei, M.; Flynn, C.T.; Wood, M.R.; Harkins, S.; Kiosses, W.B.; Whitton, J.L. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 2010, 84, 12110. [Google Scholar] [CrossRef] [Green Version]
- Tabor-Godwin, J.M.; Tsueng, G.; Sayen, M.R.; Gottlieb, R.A.; Feuer, R. The role of autophagy during coxsackievirus infection of neural progenitor and stem cells. Autophagy 2012, 8, 938–953. [Google Scholar] [CrossRef] [Green Version]
- Holt, M.; Varoqueaux, F.; Wiederhold, K.; Takamori, S.; Urlaub, H.; Fasshauer, D.; Jahn, R. Identification of SNAP-47, a novel Qbc-SNARE with ubiquitous expression. J. Biol. Chem. 2006, 281, 17076–17083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamud, Y.; Xue, Y.C.; Liu, H.; Ng, C.S.; Bahreyni, A.; Luo, H. Autophagy receptor protein Tax1-binding protein 1/TRAF6-binding protein is a cellular substrate of enteroviral proteinase. Front. Microbiol. 2021, 12, 647410. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Dawson, H. Glutathione is required for efficient production of infectious picornavirus virions. Virology 2006, 353, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Jagdeo, J.M.; Dufour, A.; Klein, T.; Solis, N.; Kleifeld, O.; Kizhakkedathu, J.; Luo, H.; Overall, C.M.; Jan, E. N-terminomics TAILS identifies host cell substrates of poliovirus and coxsackievirus B3 3C proteinases that modulate virus infection. J. Virol. 2018, 92, e02211–e02217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Yoder, J.D.; Cifuente, J.O.; Pan, J.; Bergelson, J.M.; Hafenstein, S. The crystal structure of a coxsackievirus B3-RD variant and a refined 9-angstrom cryo-electron microscopy reconstruction of the virus complexed with decay-accelerating factor (DAF) provide a new footprint of DAF on the virus surface. J. Virol. 2012, 86, 12571–12581. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Wolff, G.; Melia, C.E.; Snijder, E.J.; Barcena, M. Double-membrane vesicles as platforms for viral replication. Trends Microbiol. 2020, 28, 1022–1033. [Google Scholar] [CrossRef]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome Supports Coxsackievirus B3 Replication in Host Cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, E.; Roingeard, P. Virus-induced double-membrane vesicles. Cell Microbiol. 2015, 17, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthi, S.; Jessop, C.E.; Bulleid, N.J. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006, 7, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Tsunoda, S.; Avezov, E.; Zyryanova, A.; Konno, T.; Mendes-Silva, L.; Melo, E.P.; Harding, H.P.; Ron, D. Intact protein folding in the glutathione-depleted endoplasmic reticulum implicates alternative protein thiol reductants. Elife 2014, 3, e03421. [Google Scholar] [CrossRef]
- Chakravarthi, S.; Bulleid, N.J. Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J. Biol. Chem. 2004, 279, 39872–39879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dörsam, B.; Fahrer, J. The disulfide compound α-lipoic acid and its derivatives: A novel class of anticancer agents targeting mitochondria. Cancer Lett. 2016, 371, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Abernathy, E.; Mateo, R.; Majzoub, K.; van Buuren, N.; Bird, S.W.; Carette, J.E.; Kirkegaard, K. Differential and convergent utilization of autophagy components by positive-strand RNA viruses. PLoS Biol. 2019, 17, e2006926. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemicals, Peptides, and Recombinant Proteins | ||
|---|---|---|
| Bafilomycin A1 | Sigma-Aldrich | B1793 |
| Buthionine Sulfoximine | Cayman Chemicals | 14484 |
| CVB3 recombinant 3wt, 3Cmut | Dr. Eric Jan (University of British Columbia, Vancouver, Canada | Jagdeo et al., 2015 |
| Experimental model: Cell lines | ||
| HeLa | ATCC | CCL-2 |
| HEK-293T | ATCC | CRL-3216 |
| Recombinant DNA | ||
| 3×Flag-SNAP29 | Dr. Qing Zhong (University of Texas Southwestern Medical Center, TX, USA) | (Diao et al., 2015) |
| GFP-SNAP47 | Current study | |
| 3XFLAG-ATG14 | Dr. Qing Zhong (University of Texas Southwestern Medical Center, TX, USA) | (Diao et al., 2015) |
| Software and Algorithms | ||
| ImageJ | NIH | Version 1.46r |
| Antibodies | ||
| LC3-I/II (rabbit) | Novus Biologicals, Cat. No. NB100-2220 | |
| GAPDH (rabbit) | Abcam, Cat. No. ab8245 | |
| SNAP 23 (mouse) | Santa Cruz, Cat. No. sc-374215 | |
| SNAP 29 (rabbit) | Abcam, Cat. No. ab181151 | |
| SANAP47 (rabbit) | Abcam, Cat. No. ab172609 | |
| β-Actin (mouse) | Sigma-Aldrich, Cat. No. A5316 | |
| VP1 (mouse) | Dako Cat.No. M706401-1 | |
| Flag (mouse) | Sigma-Aldrich Cat.No. F1804 | |
| DAPI | Sigma-Aldrich, F6057 | |
| Goat-anti-mouse IgG, HRP-linked antibody | Santa Cruz, Cat. No. SC-2005 | |
| Goat-anti-rabbit IgG, HRP-linked antibody | Cell Signaling Technology, Cat. No. 7074 | |
| Oligonucleotides | ||
| SNAP47 | Forward 5′AAATTTAGATCTATGCGCGCGGCTCGC 3′ Reverse 5′AAATTTGGTACCCTAGGTCAGCCTCTTCAT CCGCCTGTT 3′ | Current study |
| SNAP47-KO | Forward 5′CACCGAGGCTCACCGTCCTTGTGTC 3′ Reverse 5′AAACGACACAAGGACGGTGAGCCT C 3′ | Current study |
| sgATG14 | Forward 5′ CACCGGACTCCGTGGACGATGCGG 3′ | Current study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, P.; Mohamud, Y.; Luo, H. SNAP47 Interacts with ATG14 to Promote VP1 Conjugation and CVB3 Propagation. Cells 2021, 10, 2141. https://doi.org/10.3390/cells10082141
Xiang P, Mohamud Y, Luo H. SNAP47 Interacts with ATG14 to Promote VP1 Conjugation and CVB3 Propagation. Cells. 2021; 10(8):2141. https://doi.org/10.3390/cells10082141
Chicago/Turabian StyleXiang, Pinhao, Yasir Mohamud, and Honglin Luo. 2021. "SNAP47 Interacts with ATG14 to Promote VP1 Conjugation and CVB3 Propagation" Cells 10, no. 8: 2141. https://doi.org/10.3390/cells10082141
APA StyleXiang, P., Mohamud, Y., & Luo, H. (2021). SNAP47 Interacts with ATG14 to Promote VP1 Conjugation and CVB3 Propagation. Cells, 10(8), 2141. https://doi.org/10.3390/cells10082141