
From the Evasion of Degradation to Ubiquitin-Dependent Protein Stabilization



Abstract
:1. Overview
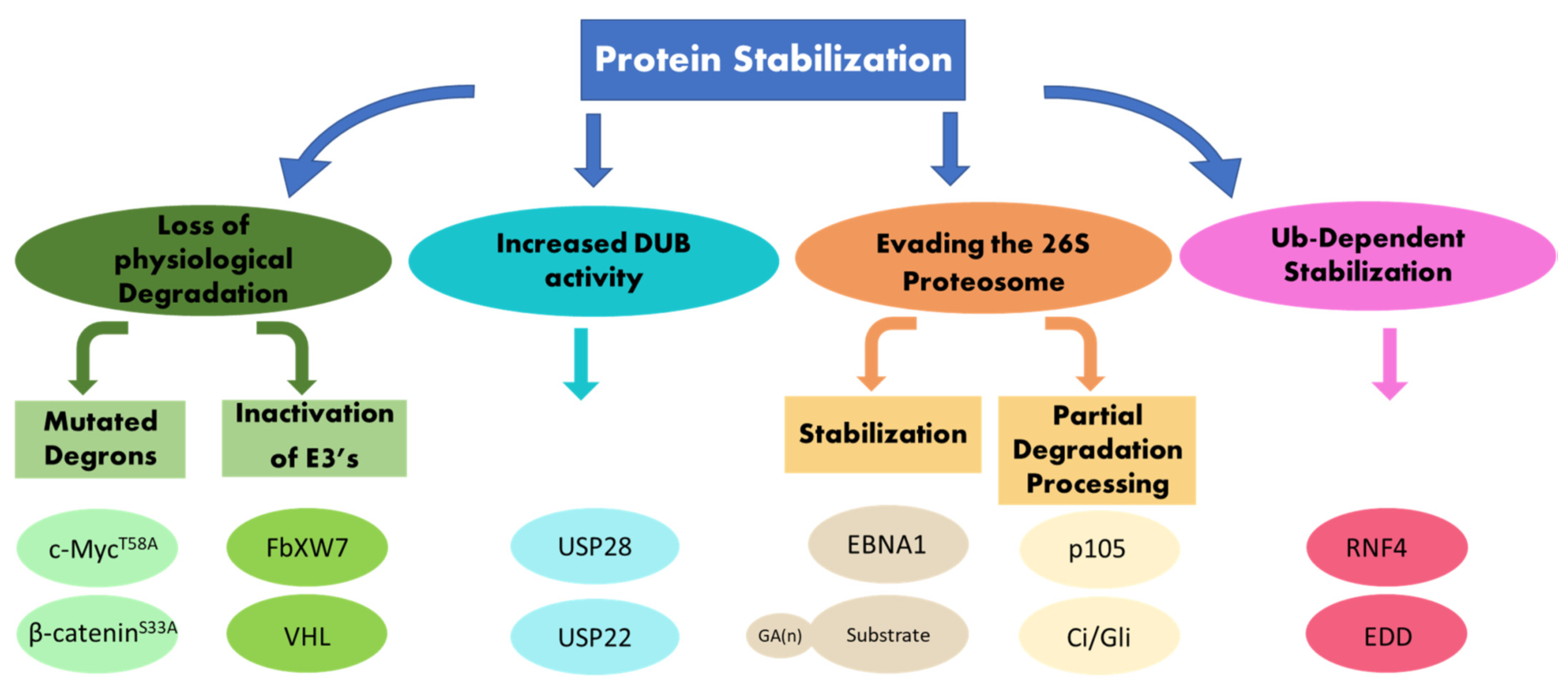
2. Evading Recognition I: Protein Stabilization Due to Lacking or Mutated Degradation Signals
3. Evading Recognition II: Protein Stabilization Due to Impaired E3 Ubiquitin Ligase Activity, and the Emergences of Degradation-Resistant Tumors
4. Evading the Proteasome: Stabilization and Limited Proteolysis Signals
5. Ubiquitin-Dependent Oncoprotein Stabilization
6. Heterotypic Ubiquitin Chains Mediate Protein Stabilization
7. Ubiquitin-Dependent Stabilization Is Evolutionarily Conserved
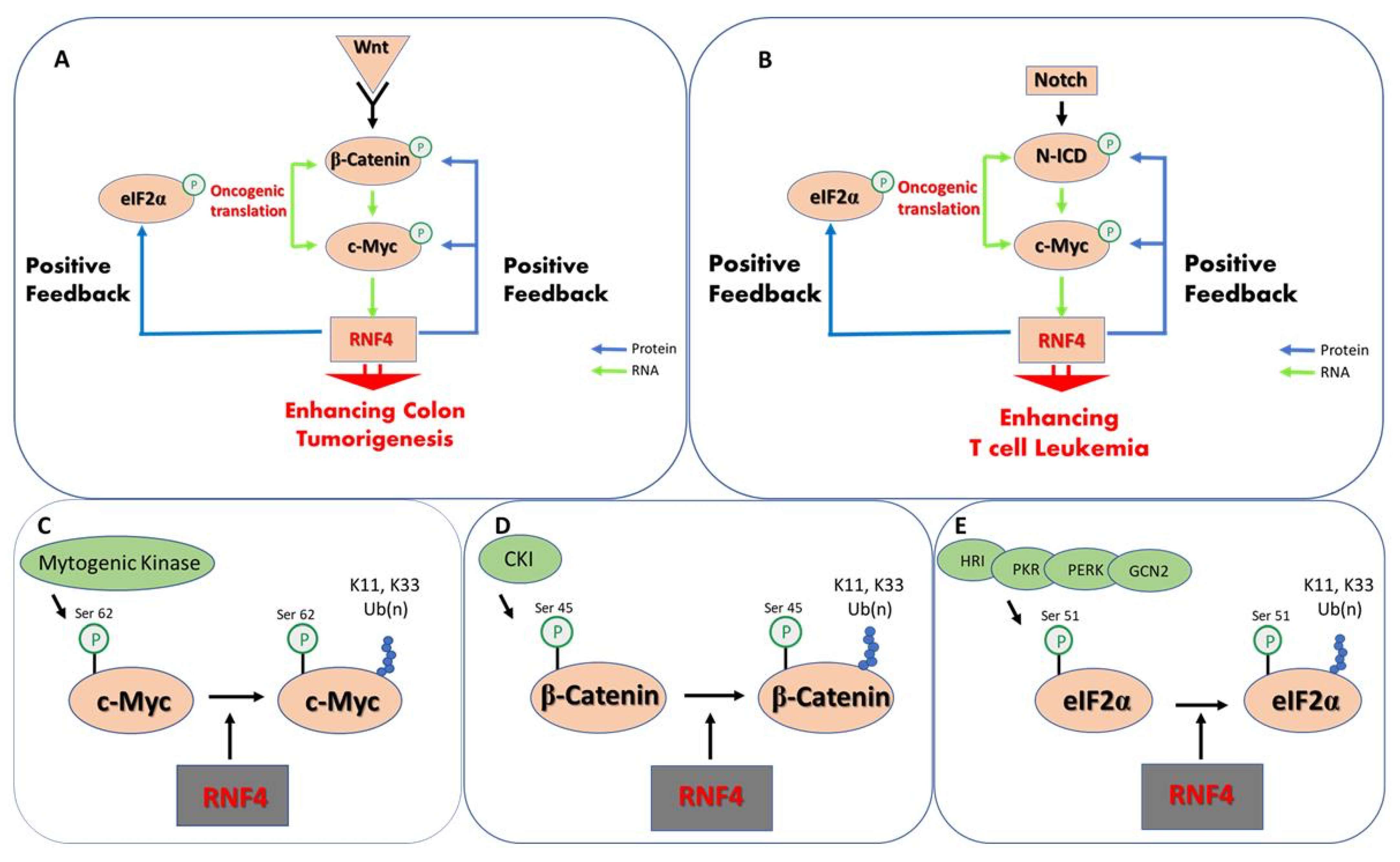
8. The Tumor-Promoting Activity of RNF4
9. RNF4 Is Associated with Resistance to Therapy and Poor Prognosis in Human Cancer
10. Implications of RNF4 and Ubiquitin-Dependent Protein Stabilization in Cancer

{kind=link}
{kind=link}
| SUMO-Dependent Substrates of RNF4 | |
| Protein | References |
| PML | [114,115,116,136] |
| JARID1B | [137] |
| SP100 * | [138] |
| IFI16 | [139] |
| CFTR | [140] |
| Pea3 | [141] |
| BLM | [109] |
| FANCI, FANCD2 (ID complex) | [142] |
| PIAS1, PIAS2, PIAS3 | [143] |
| ZNF451 | [143] |
| NSMCE2 | [143] |
| Ubc9 | [143] |
| BRCA1–BARD1 complex | [143] |
| MDC1 | [144] |
| PARIS/ZNF746 | [145] |
| KAP1 | [104] |
| Rta | [146] |
| NDRG2 | [147] |
| Tax | [148] |
| Ataxin-7 | [149] |
| FXR | [150] |
| SUMO-independent substrates of RNF4 | |
| Protein | Reference |
| c-Myc | [97] |
| c-Jun | [97] |
| β-catenin | [97] |
| NICD | [97] |
| PGC1 α | [97] |
| eIF2α | [98] |
| HIF1α | [98] |
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schoenheimer, R. The Dynamic State of Body Constituents. Cancer Res. 1942, 2, 810. [Google Scholar] [CrossRef]
- Cambridge, S.B.; Gnad, F.; Nguyen, C.; Bermejo, J.L.; Krüger, M.; Mann, M. Systems-wide proteomic analysis in mammalian cells reveals conserved, functional protein turnover. J. Proteome Res. 2011, 10, 5275–5284. [Google Scholar] [CrossRef] [PubMed]
- Toyama, B.H.; Savas, J.N.; Park, S.K.; Harris, M.S.; Ingolia, N.T.; Yates, J.R.; Hetzer, M.W. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 2013, 154, 971–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciechanover, A.; DiGiuseppe, J.A.; Bercovich, B.; Orian, A.; Richter, J.D.; Schwartz, A.L.; Brodeur, G.M. Degradation of nuclear oncoproteins by the ubiquitin system in vitro. Proc. Natl. Acad. Sci. USA 1991, 88, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Akopian, D.; Rape, M. Principles of Ubiquitin-Dependent Signaling. Annu. Rev. Cell Dev. Biol. 2018, 34, 137–162. [Google Scholar] [CrossRef]
- Mészáros, B.; Kumar, M.; Gibson, T.J.; Uyar, B.; Dosztányi, Z. Degrons in cancer. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Alkalay, I.; Yaron, A.; Hatzubai, A.; Orian, A.; Ciechanover, A.; Ben-Neriah, Y. Stimulation-dependent IκBα phosphorylation marks the NF-κb inhibitor for degradation via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1995, 92, 10599–10603. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-κB activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jiménez, F.; Muiños, F.; López-Arribillaga, E.; Lopez-Bigas, N.; Gonzalez-Perez, A. Systematic analysis of alterations in the ubiquitin proteolysis system reveals its contribution to driver mutations in cancer. Nat. Cancer 2020, 1, 122–135. [Google Scholar] [CrossRef] [Green Version]
- Diefenbacher, M.; Orian, A. Stabilization of nuclear oncoproteins by RNF4 and the ubiquitin system in cancer. Mol. Cell. Oncol. 2017, 4, e1260671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshavsky, A. N-degron and C-degron pathways of protein degradation. Proc. Natl. Acad. Sci. USA 2019, 116, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmair, A.; Finley, D.; Varshavsky, A. In vivo half-life of a protein is a function of its amino-terminal residue. Science 1986, 234, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cunningham, M.; Zhang, X.; Tokarz, S.; Laraway, B.; Troxell, M.; Sears, R.C. Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer Res. 2011, 71, 925–936. [Google Scholar] [CrossRef] [Green Version]
- Myant, K.; Qiao, X.; Halonen, T.; Come, C.; Laine, A.; Janghorban, M.; Partanen, J.I.; Cassidy, J.; Ogg, E.L.; Cammareri, P.; et al. Serine 62-phosphorylated MYC Associates with nuclear lamins and its regulation by CIP2A is essential for regenerative proliferation. Cell Rep. 2015, 12, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Wong, C.C.; Gong, B.; Yu, J. Functional significance and therapeutic implication of ring-type E3 ligases in colorectal cancer. Oncogene 2018, 37, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treier, M.; Staszewski, L.M.; Bohmann, D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the δ domain. Cell 1994, 78, 787–798. [Google Scholar] [CrossRef]
- Musti, A.M.; Treier, M.; Peverali, F.A.; Bohmann, D. Differential regulation of c-Jun and JunD by ubiquitin-dependent protein degradation. Biol. Chem. 1996, 377, 619–624. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Yang, Y.; Ludwig, R.L.; Jensen, J.P.; Pierre, S.A.; Medaglia, M.V.; Davydov, I.V.; Safiran, Y.J.; Oberoi, P.; Kenten, J.H.; Phillips, A.C.; et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005, 7, 547–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, S.K.; Khatik, G.L.; Narang, R.; Monga, V.; Chopra, H.K. p53-Mdm2 Interaction Inhibitors as Novel Nongenotoxic Anticancer Agents. Curr. Cancer Drug Targets 2018, 18, 749–772. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Tamaskovic, R.; Yang, Z.; Brazil, D.P.; Merlo, A.; Hess, D.; Hemmings, B.A. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J. Biol. Chem. 2004, 279, 35510–35517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laine, A.; Ronai, Z. Regulation of p53 localization and transcription by the HECT domain E3 ligase WWP1. Oncogene 2007, 26, 1477–1483. [Google Scholar] [CrossRef]
- Xu, C.; Fan, C.D.; Wang, X. Regulation of Mdm2 protein stability and the p53 response by NEDD4-1 E3 ligase. Oncogene 2015, 34, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Yaron, A.; Hatzubai, A.; Davis, M.; Lavon, I.; Amit, S.; Manning, A.M.; Andersen, J.S.; Mann, M.; Mercurio, F.; Ben-Neriah, Y. Identification of the receptor component of the IκBα–ubiquitin ligase. Nature 1998, 396, 590–594. [Google Scholar] [CrossRef]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. β-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [Green Version]
- Hart, M.; Concordet, J.P.; Lassot, I.; Albert, I.; Del Los Santos, R.; Durand, H.; Perret, C.; Rubinfeld, B.; Margottin, F.; Benarous, R.; et al. The F-box protein β-TrCP associates with phosphorylated β-catenin and regulates its activity in the cell. Curr. Biol. 1999, 9, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Low, T.Y.; Peng, M.; Magliozzi, R.; Mohammed, S.; Guardavaccaro, D.; Heck, A.J.R. A systems-wide screen identifies substrates of the SCFβTrCP ubiquitin ligase. Sci. Signal. 2014, 7, rs8. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [Green Version]
- Rubinfeld, B.; Tice, D.A.; Polakis, P. Axin-dependent Phosphorylation of the Adenomatous Polyposis Coli Protein Mediated by Casein Kinase 1ε. J. Biol. Chem. 2001, 276, 39037–39045. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Kato, Y.; Zhang, Z.; Do, V.M.; Yankner, B.A.; He, X. β-Trcp couples β-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc. Natl. Acad. Sci. USA 1999, 96, 6273–6278. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.-H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of β-Catenin Phosphorylation/Degradation by a Dual-Kinase Mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Hay-Koren, A.; Caspi, M.; Zilberberg, A.; Rosin-Arbesfeld, R. The EDD E3 ubiquitin ligase ubiquitinates and up-regulates β-catenin. Mol. Biol. Cell 2011, 22, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Clurman, B.E. FBW7 ubiquitin ligase: A tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 2008, 8, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.H.; Kim, I.J.; Wu, D.; Climent, J.; Hio, C.K.; DelRosario, R.; Balmain, A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008, 321, 1499–1502. [Google Scholar] [CrossRef] [Green Version]
- Yumimoto, K.; Nakayama, K.I. Recent insight into the role of FBXW7 as a tumor suppressor. Semin. Cancer Biol. 2020, 67, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.A.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar] [CrossRef] [Green Version]
- Junttila, M.R.; Westermarck, J. Mechanisms of MYC stabilization in human malignancies. Cell Cycle 2008, 7, 592–596. [Google Scholar] [CrossRef]
- Farrell, A.S.; Sears, R.C. MYC degradation. Cold Spring Harb. Perspect. Med. 2014, 4, a014365. [Google Scholar] [CrossRef]
- Tokheim, C.; Wang, X.; Timms, R.T.; Zhang, B.; Mena, E.L.; Wang, B.; Chen, C.; Ge, J.; Chu, J.; Zhang, W.; et al. Systematic characterization of mutations altering protein degradation in human cancers. Mol. Cell 2021, 81, 1292–1308. [Google Scholar] [CrossRef]
- Acebron, S.P.; Karaulanov, E.; Berger, B.S.; Huang, Y.L.; Niehrs, C. Mitotic Wnt Signaling Promotes Protein Stabilization and Regulates Cell Size. Mol. Cell 2014, 54, 663–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Fagman, J.B.; Chen, C.; Alberti, S.; Liu, B. Protein phase separation and its role in tumorigenesis. eLife 2020, 9, e60264. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Hajdu-soltész, B.; Zeke, A.; Dosztányi, Z. Mutations of intrinsically disordered protein regions can drive cancer but lack therapeutic strategies. Biomolecules 2021, 11, 381. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Clurman, B.E. Oncoprotein ubiquitylation: Dimers, degrons, and degradation. Cell Cycle 2014, 13, 1829–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, S.; Pagano, M. Ubiquitin ligases in cancer: Functions and clinical potentials. Cell Chem. Biol. 2021, 28, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Kubbutat, M.H.G.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Hao, Q.; Chen, Y.; Zhou, X. The Janus Face of p53-Targeting Ubiquitin Ligases. Cells 2020, 9, 1656. [Google Scholar] [CrossRef]
- Levkowitz, G.; Waterman, H.; Ettenberg, S.A.; Katz, M.; Tsygankov, A.Y.; Alroy, I.; Lavi, S.; Iwai, K.; Reiss, Y.; Ciechanover, A.; et al. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell 1999, 4, 1029–1040. [Google Scholar] [CrossRef]
- Senft, D.; Qi, J.; Ronai, Z.A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Thien, C.B.F.; Langdon, W.Y. c-Cbl and Cbl-b ubiquitin ligases: Substrate diversity and the negative regulation of signalling responses. Biochem. J. 2005, 391, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Q. VHL and hypoxia signaling: Beyond HIF in cancer. Biomedicines 2018, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Levitskaya, J.; Coram, M.; Levitsky, V.; Imreh, S.; Steigerwald-Mullen, P.M.; Klein, G.; Kurilla, M.G.; Masucci, M.G. Inhibition of antigen processing by the internal repeat region of the epstein-barr virus nuclear antigen-1. Nature 1995, 375, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Goldberg, A.L. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu. Rev. Immunol. 1999, 17, 739–779. [Google Scholar] [CrossRef]
- Levitskaya, J.; Sharipo, A.; Leonchiks, A.; Ciechanover, A.; Masucci, M.G. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA 1997, 94, 12616–12621. [Google Scholar] [CrossRef] [Green Version]
- Heessen, S.; Dantuma, N.P.; Tessarz, P.; Jellne, M.; Masucci, M.G. Inhibition of ubiquitin/proteasome-dependent proteolysis in Saccharomyces cerevisiae by a Gly-Ala repeat. FEBS Lett. 2003, 555, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Heessen, S.; Leonchiks, A.; Issaeva, N.; Sharipo, A.; Selivanova, G.; Masucci, M.G.; Dantuma, N.P. Functional p53 chimeras containing the Epstein-Barr virus Gly-Ala repeat are protected from Mdm2- and HPV-E6-induced proteolysis. Proc. Natl. Acad. Sci. USA 2002, 99, 1532–1537. [Google Scholar] [CrossRef] [Green Version]
- Dantuma, N.P.; Heessen, S.; Lindsten, K.; Jellne, M.; Masucci, M.G. Inhibition of proteasomal degradation by the Gly-Ala repeat of Epstein-Barr virus is influenced by the length of the repeat and the strength of the degradation signal. Proc. Natl. Acad. Sci. USA 2000, 97, 8381–8385. [Google Scholar] [CrossRef] [Green Version]
- Hoyt, M.A.; Zich, J.; Takeuchi, J.; Zhang, M.; Govaerts, C.; Coffino, P. Glycine-alanine repeats impair proper substrate unfolding by the proteasome. EMBO J. 2006, 25, 1720–1729. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Ghosh, S. A glycine-rich region in NF-kappaB p105 functions as a processing signal for the generation of the p50 subunit. Mol. Cell. Biol. 1996, 16, 2248–2254. [Google Scholar] [CrossRef] [Green Version]
- Orian, A.; Schwartz, A.L.; Israël, A.; Whiteside, S.; Kahana, C.; Ciechanover, A. Structural Motifs Involved in Ubiquitin-Mediated Processing of the NF-κB Precursor p105: Roles of the Glycine-Rich Region and a Downstream Ubiquitination Domain. Mol. Cell. Biol. 1999, 19, 3664–3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Dries, D.R.; Mayer, P.; Good, S.K.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 Is Directed to Stress Granules by Sorbitol, a Novel Physiological Osmotic and Oxidative Stressor. Mol. Cell. Biol. 2011, 31, 1098–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimonaka, S.; Nonaka, T.; Suzuki, G.; Hisanaga, S.I.; Hasegawa, M. Templated aggregation of TAR DNA-binding protein of 43 kDa (TDP-43) by seeding with TDP-43 peptide fibrils. J. Biol. Chem. 2016, 291, 8896–8907. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Struhl, G. Regulation of the Hedgehog and Wingless signalling pathways by the F- box/WD40-repeat protein Slimb. Nature 1998, 391, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Price, M.A. A Unique Protection Signal in Cubitus interruptus Prevents Its Complete Proteasomal Degradation. Mol. Cell. Biol. 2008, 28, 5555–5568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J. Regulation of Hh/Gli signaling by dual ubiquitin pathways. Cell Cycle 2006, 5, 2457–2463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, L.; Wang, B.; Ou, C.Y.; Chien, C.T.; Jiang, J. A Hedgehog-Induced BTB Protein Modulates Hedgehog Signaling by Degrading Ci/Gli Transcription Factor. Dev. Cell 2006, 10, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravtsova-Ivantsiv, Y.; Shomer, I.; Cohen-Kaplan, V.; Snijder, B.; Superti-Furga, G.; Gonen, H.; Sommer, T.; Ziv, T.; Admon, A.; Naroditsky, I.; et al. KPC1-mediated ubiquitination and proteasomal processing of nf-κb1 p105 to p50 restricts tumor growth. Cell 2015, 161, 333–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belich, M.P.; Salmerón, A.; Johnston, L.H.; Ley, S.C. TPL-2 kinase regulates the proteolysis of the NF-κB-inhibitory protein NF-κB1 p105. Nature 1999, 397, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Orian, A.; Gonen, H.; Bercovich, B.; Fajerman, I.; Eytan, E.; Israël, A.; Mercurio, F.; Iwai, K.; Schwartz, A.L.; Ciechanover, A. SCF(beta)(-TrCP) ubiquitin ligase-mediated processing of NF-kappaB p105 requires phosphorylation of its C-terminus by IkappaB kinase. EMBO J. 2000, 19, 2580–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, Z.B.; Kofahl, B.; Beaudette, P.; Baum, K.; Ipenberg, I.; Weih, F.; Wolf, J.; Dittmar, G.; Scheidereit, C. Quantitative Dissection and Modeling of the NF-κB p100-p105 Module Reveals Interdependent Precursor Proteolysis. Cell Rep. 2014, 9, 1756–1769. [Google Scholar] [CrossRef] [Green Version]
- Heessen, S.; Masucci, M.G.; Dantuma, N.P. The UBA2 domain functions as an intrinsic stabilization signal that protects rad23 from proteasomal degradation. Mol. Cell 2005, 18, 225–235. [Google Scholar] [CrossRef]
- Heinen, C.; Ács, K.; Hoogstraten, D.; Dantuma, N.P. C-terminal UBA domains protect ubiquitin receptors by preventing initiation of protein degradation. Nat. Commun. 2011, 2, 191. [Google Scholar] [CrossRef] [Green Version]
- van Leeuwen, F.W.; de Kleijn, D.P.V.; van den Hurk, H.H.; Neubauer, A.; Sonnemans, M.A.F.; Sluijs, J.A.; Köycü, S.; Ramdjielal, R.D.J.; Salehi, A.; Martens, G.J.M.; et al. Frameshift Mutants of β Amyloid Precursor Protein and Ubiquitin-B in Alzheimer’s and Down Patients. Science 1998, 279, 242–247. [Google Scholar] [CrossRef]
- Lindsten, K.; de Vrij, F.M.S.; Verhoef, L.G.G.C.; Fischer, D.F.; van Leeuwen, F.W.; Hol, E.M.; Masucci, M.G.; Dantuma, N.P. Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J. Cell Biol. 2002, 157, 417–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Leeuwen, F.W.; Hol, E.M.; Fischer, D.F. Frameshift proteins in Alzheimer’s disease and in other conformational disorders: Time for the ubiquitin-proteasome system. J. Alzheimers Dis. 2006, 9, 319–325. [Google Scholar] [CrossRef]
- Tank, E.M.H.; True, H.L. Disease-Associated Mutant Ubiquitin Causes Proteasomal Impairment and Enhances the Toxicity of Protein Aggregates. PLoS Genet. 2009, 5, e1000382. [Google Scholar] [CrossRef] [Green Version]
- Krutauz, D.; Reis, N.; Nakasone, M.A.; Siman, P.; Zhang, D.; Kirkpatrick, D.S.; Gygi, S.P.; Brik, A.; Fushman, D.; Glickman, M.H. Extended ubiquitin species are protein-based DUB inhibitors. Nat. Chem. Biol. 2014, 10, 664–670. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ramot, Y.; Torrelo, A.; Paller, A.S.; Si, N.; Babay, S.; Kim, P.W.; Sheikh, A.; Lee, C.-C.R.; Chen, Y.; et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012, 64, 895–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, A.; Jacks, J.; Kessler, M.; Emanuel, P.D.; Gao, L. Proteasome-associated autoinflammatory syndromes: Advances in pathogeneses, clinical presentations, diagnosis, and management. Int. J. Dermatol. 2015, 54, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef] [PubMed]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr. Genom. 2014, 15, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boland, B.; Haung Yu, W.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Publ. Gr. 2018, 17, 660–688. [Google Scholar] [CrossRef]
- Maltzman, W.; Czyzyk, L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol. Cell. Biol. 1984, 4, 1689–1694. [Google Scholar] [CrossRef] [Green Version]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 Protein in the Cellular Response to DNA Damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef] [Green Version]
- Banin, S.; Moyal, L.; Shieh, S.Y.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef]
- Buschmann, T.; Potapova, O.; Bar-Shira, A.; Ivanov, V.N.; Fuchs, S.Y.; Henderson, S.; Fried, V.A.; Minamoto, T.; Alarcon-Vargas, D.; Pincus, M.R.; et al. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol. Cell. Biol. 2001, 21, 2743–2754. [Google Scholar] [CrossRef] [Green Version]
- Lavin, M.F.; Gueven, N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006, 13, 941–950. [Google Scholar] [CrossRef]
- Amit, S.; Hatzubai, A.; Birman, Y.; Andersen, J.S.; Ben-Shushan, E.; Mann, M.; Ben-Neriah, Y.; Alkalay, I. Axin-mediated CKI phosphorylation of β-catenin at Ser 45: A molecular switch for the Wnt pathway. Genes Dev. 2002, 16, 1066–1076. [Google Scholar] [CrossRef] [Green Version]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [Green Version]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 2004, 6, 308–318. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.J.; Abed, M.; Heuberger, J.; Novak, R.; Zohar, Y.; Beltran Lopez, A.P.; Trausch-Azar, J.S.; Ilagan, M.X.G.; Benhamou, D.; Dittmar, G.; et al. RNF4-Dependent Oncogene Activation by Protein Stabilization. Cell Rep. 2016, 16, 3388–3400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avitan-Hersh, E.; Feng, Y.; Oknin Vaisman, A.; Abu Ahmad, Y.; Zohar, Y.; Zhang, T.; Lee, J.S.; Lazar, I.; Sheikh Khalil, S.; Feiler, Y.; et al. Regulation of eIF2α by RNF4 Promotes Melanoma Tumorigenesis and Therapy Resistance. J. Investig. Dermatol. 2020, 140, 2466–2477. [Google Scholar] [CrossRef] [PubMed]
- Sriramachandran, A.M.; Dohmen, R.J. SUMO-targeted ubiquitin ligases. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1843, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Abed, M.; Bitman-Lotan, E.; Orian, A. The biology of SUMO-targeted ubiquitin ligases in Drosophila development, immunity, and cancer. J. Dev. Biol. 2018, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Sabapathy, K. RNF4—A Paradigm for SUMOylation-Mediated Ubiquitination. Proteomics 2019, 19, 1900185. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Barry, K.C.; Kenyagin, D.; Koltun, B.; Phippen, T.M.; Delrow, J.J.; Parkhurst, S.M.; Orian, A. Degringolade, a SUMO-targeted ubiquitin ligase, inhibits Hairy/Groucho-mediated repression. EMBO J. 2011, 30, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, K.C.; Abed, M.; Kenyagin, D.; Werwie, T.R.; Boico, O.; Orian, A.; Parkhurst, S.M. The Drosophila STUbL protein Degringolade limits HES functions during embryogenesis. Development 2011, 138, 1759–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.Y.; Li, X.; Kong, X.Q.; Luo, C.; Chang, C.C.; Chung, Y.; Shih, H.M.; Li, K.K.; Ann, D.K. An arginine-rich motif of ring finger protein 4 (RNF4) oversees the recruitment and degradation of the phosphorylated and SUMOylated Krüppel-associated box domain-associated protein 1 (KAP1)/TRIM28 protein during genotoxic stress. J. Biol. Chem. 2014, 289, 20757–20772. [Google Scholar] [CrossRef] [Green Version]
- Moilanen, A.-M.; Poukka, H.; Karvonen, U.; Häkli, M.; Jänne, O.A.; Palvimo, J.J. Identification of a Novel RING Finger Protein as a Coregulator in Steroid Receptor-Mediated Gene Transcription. Mol. Cell. Biol. 1998, 18, 5128–5139. [Google Scholar] [CrossRef] [Green Version]
- Saville, B.; Poukka, H.; Wormke, M.; Jänne, O.; Palvimo, J.; Stoner, M.; Samudio, I.; Safe, S. Cooperative Coactivation of Estrogen Receptor α in ZR-75 Human Breast Cancer Cells by SNURF and TATA-binding Protein. J. Biol. Chem. 2002, 277, 2485–2497. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.V.; Rodrigues, T.M.A.; Tao, H.; Baker, R.K.; Miraglia, L.; Orth, A.P.; Lyons, G.E.; Schultz, P.G.; Wu, X. Identification of RING finger protein 4 (RNF4) as a modulator of DNA demethylation through a functional genomics screen. Proc. Natl. Acad. Sci. USA 2010, 107, 15087–15092. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y. RING finger protein 4 (RNF4) derepresses gene expression from DNA methylation. J. Biol. Chem. 2014, 289, 33808–33813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Jackson, S.P. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 2012, 26, 1179–1195. [Google Scholar] [CrossRef] [Green Version]
- Guzzo, C.M.; Berndsen, C.E.; Zhu, J.; Gupta, V.; Datta, A.; Greenberg, R.A.; Wolberger, C.; Matunis, M.J. RNF4-Dependent Hybrid SUMO-Ubiquitin Chains Are Signals for RAP80 and Thereby Mediate the Recruitment of BRCA1 to Sites of DNA Damage. Sci. Signal. 2012, 5, ra88. [Google Scholar] [CrossRef] [Green Version]
- Keiten-Schmitz, J.; Wagner, K.; Piller, T.; Kaulich, M.; Alberti, S.; Müller, S. The Nuclear SUMO-Targeted Ubiquitin Quality Control Network Regulates the Dynamics of Cytoplasmic Stress Granules. Mol. Cell 2020, 79, 54–67. [Google Scholar] [CrossRef]
- Nagai, S.; Davoodi, N.; Gasser, S.M. Nuclear organization in genome stability: SUMO connections. Cell Res. 2011, 21, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C.; Oram, M.K.; Bielinsky, A.K. Sumo-targeted ubiquitin ligases and their functions in maintaining genome stability. Int. J. Mol. Sci. 2021, 22, 5391. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Zhu, J.; Raught, B.; de Thé, H. Arsenic degrades PML or PML–RARα through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 2008, 10, 547–555. [Google Scholar] [CrossRef]
- Tatham, M.H.; Geoffroy, M.C.; Shen, L.; Plechanovova, A.; Hattersley, N.; Jaffray, E.G.; Palvimo, J.J.; Hay, R.T. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 2008, 10, 538–546. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Zhu, J.; Chen, Z.; De Thé, H. Curing APL through PML/RARA degradation by As2O3. Trends Mol. Med. 2012, 18, 36–42. [Google Scholar] [CrossRef]
- Behrends, C.; Harper, J.W. Constructing and decoding unconventional ubiquitin chains. Nat. Struct. Mol. Biol. 2011, 18, 520–528. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, M.E.; Koehler, C.F.; Hunter, T. Emerging functions of branched ubiquitin chains. Cell Discov. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Karin, M. Emerging roles of Lys63-linked polyubiquitylation in immune responses. Immunol. Rev. 2015, 266, 161–174. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Haakonsen, D.L.; Rape, M. Branching Out: Improved Signaling by Heterotypic Ubiquitin Chains. Trends Cell Biol. 2019, 29, 704–716. [Google Scholar] [CrossRef]
- Rodrigues, L.; Popov, N.; Kaye, K.M.; Simas, J.P. Stabilization of Myc through Heterotypic Poly-Ubiquitination by mLANA Is Critical for γ-Herpesvirus Lymphoproliferation. PLoS Pathog. 2013, 9, e1003554. [Google Scholar] [CrossRef] [PubMed]
- Ben-Saadon, R.; Zaaroor, D.; Ziv, T.; Ciechanover, A. The Polycomb Protein Ring1B Generates Self Atypical Mixed Ubiquitin Chains Required for Its In Vitro Histone H2A Ligase Activity. Mol. Cell 2006, 24, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Zucchelli, S.; Codrich, M.; Marcuzzi, F.; Pinto, M.; Vilotti, S.; Biagioli, M.; Ferrer, I.; Gustincich, S. TRAF6 promotes atypical ubiquitination of mutant DJ-1 and alpha-synuclein and is localized to Lewy bodies in sporadic Parkinson’s disease brains. Hum. Mol. Genet. 2010, 19, 3759–3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitcomb, E.A.; Tsai, Y.C.; Basappa, J.; Liu, K.; Le Feuvre, A.K.; Weissman, A.M.; Taylor, A. Stabilization of p27Kip1/CDKN1B by UBCH7/UBE2L3 catalyzed ubiquitinylation: A new paradigm in cell-cycle control. FASEB J. 2019, 33, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Koltun, B.; Shackelford, E.; Bonnay, F.; Matt, N.; Reichhart, J.M.; Orian, A. The SUMO-targeted ubiquitin ligase, Dgrn, is essential for Drosophila innate immunity. Int. J. Dev. Biol. 2017, 61, 319–327. [Google Scholar] [CrossRef]
- Sendoel, A.; Dunn, J.G.; Rodriguez, E.H.; Naik, S.; Gomez, N.C.; Hurwitz, B.; Levorse, J.; Dill, B.D.; Schramek, D.; Molina, H.; et al. Translation from unconventional 5′ start sites drives tumour initiation. Nature 2017, 541, 494–499. [Google Scholar] [CrossRef]
- Holcik, M. Could the eIF2α-independent translation be the achilles heel of cancer? Front. Oncol. 2015, 5, 264. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.C.L.; Kanellos, G.; Vlahov, N.; Alexandrou, C.; Willis, A.E.; Knight, J.R.P.; Sansom, O.J. Translation initiation in cancer at a glance. J. Cell Sci. 2021, 134, jcs248476. [Google Scholar] [CrossRef]
- Koromilas, A.E. Roles of the translation initiation factor eIF2α serine 51 phosphorylation in cancer formation and treatment. Biochim. Biophys. Acta 2015, 1849, 871–880. [Google Scholar] [CrossRef]
- Győrffy, B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef]
- Palomero, T.; Wei, K.L.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [Green Version]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; Da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Reymann, S.; Borlak, J. Transcription profiling of lung adenocarcinomas of c-myc-transgenic mice: Identification of the c-myc regulatory gene network. BMC Syst. Biol. 2008, 2, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koidl, S.; Eisenhardt, N.; Fatouros, C.; Droescher, M.; Chaugule, V.K.; Pichler, A. The SUMO2/3 specific E3 ligase ZNF451-1 regulates PML stability. Int. J. Biochem. Cell Biol. 2016, 79, 478–487. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Treffers, L.W.; Verlaan-de Vries, M.; Olsen, J.V.; Vertegaal, A.C.O. SUMO-2 Orchestrates Chromatin Modifiers in Response to DNA Damage. Cell Rep. 2015, 10, 1778–1791. [Google Scholar] [CrossRef] [Green Version]
- Maarifi, G.; Maroui, M.A.; Dutrieux, J.; Dianoux, L.; Nisole, S.; Chelbi-Alix, M.K. Small Ubiquitin-like Modifier Alters IFN Response. J. Immunol. 2015, 195, 2312–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Asmi, F.; McManus, F.P.; Brantis-de-Carvalho, C.E.; Valle-Casuso, J.C.; Thibault, P.; Chelbi-Alix, M.K. Cross-talk between SUMOylation and ISGylation in response to interferon. Cytokine 2020, 129, 155025. [Google Scholar] [CrossRef]
- Ahner, A.; Gong, X.; Schmidt, B.Z.; Peters, K.W.; Rabeh, W.M.; Thibodeau, P.H.; Lukacs, G.L.; Frizzell, R.A. Small heat shock proteins target mutant cystic fibrosis transmembrane conductance regulator for degradation via a small ubiquitin-like modifier-dependent pathway. Mol. Biol. Cell 2013, 24, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Sharrocks, A.D. Extracellular signal-regulated kinase mitogen-activated protein kinase signaling initiates a dynamic interplay between sumoylation and ubiquitination to regulate the activity of the transcriptional activator PEA3. Mol. Cell. Biol. 2009, 29, 3204–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs-Seymour, I.; Oka, Y.; Rajendra, E.; Weinert, B.T.; Passmore, L.A.; Patel, K.J.; Olsen, J.V.; Choudhary, C.; Bekker-Jensen, S.; Mailand, N. Ubiquitin-SUMO circuitry controls activated fanconi anemia ID complex dosage in response to DNA damage. Mol. Cell 2015, 57, 150–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; González-Prieto, R.; Xiao, Z.; Verlaan-De Vries, M.; Vertegaal, A.C.O. The STUbL RNF4 regulates protein group SUMOylation by targeting the SUMO conjugation machinery. Nat. Commun. 2017, 8, 1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, K.; Zhang, H.; Wang, L.; Yuan, J.; Lou, Z. Sumoylation of MDC1 is important for proper DNA damage response. EMBO J. 2012, 31, 3008–3019. [Google Scholar] [CrossRef] [Green Version]
- Nishida, T.; Yamada, Y. RNF4-mediated SUMO-targeted ubiquitination relieves PARIS/ZNF746-mediated transcriptional repression. Biochem. Biophys. Res. Commun. 2020, 526, 110–116. [Google Scholar] [CrossRef]
- Yang, Y.-C.; Yoshikai, Y.; Hsu, S.-W.; Saitoh, H.; Chang, L.-K. Role of RNF4 in the ubiquitination of Rta of Epstein-Barr virus. J. Biol. Chem. 2013, 288, 12866–12879. [Google Scholar] [CrossRef] [Green Version]
- Tantai, J.; Pan, X.; Hu, D. RNF4-mediated SUMOylation is essential for NDRG2 suppression of lung adenocarcinoma. Oncotarget 2016, 7, 26837–26843. [Google Scholar] [CrossRef] [Green Version]
- Fryrear, K.A.; Guo, X.; Kerscher, O.; Semmes, O.J. The Sumo-targeted ubiquitin ligase RNF4 regulates the localization and function of the HTLV-1 oncoprotein Tax. Blood 2012, 119, 1173–1181. [Google Scholar] [CrossRef]
- Marinello, M.; Werner, A.; Giannone, M.; Tahiri, K.; Alves, S.; Tesson, C.; den Dunnen, W.; Seeler, J.-S.; Brice, A.; Sittler, A. SUMOylation by SUMO2 is implicated in the degradation of misfolded ataxin-7 via RNF4 in SCA7 models. Dis. Model. Mech. 2019, 12, dmm036145. [Google Scholar] [CrossRef] [Green Version]
- Bilodeau, S.; Caron, V.; Gagnon, J.; Kuftedjian, A.; Tremblay, A. A CK2-RNF4 interplay coordinates non-canonical SUMOylation and degradation of nuclear receptor FXR. J. Mol. Cell Biol. 2017, 9, 195–208. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abu Ahmad, Y.; Oknin-Vaisman, A.; Bitman-Lotan, E.; Orian, A. From the Evasion of Degradation to Ubiquitin-Dependent Protein Stabilization. Cells 2021, 10, 2374. https://doi.org/10.3390/cells10092374
Abu Ahmad Y, Oknin-Vaisman A, Bitman-Lotan E, Orian A. From the Evasion of Degradation to Ubiquitin-Dependent Protein Stabilization. Cells. 2021; 10(9):2374. https://doi.org/10.3390/cells10092374
Chicago/Turabian StyleAbu Ahmad, Yamen, Avital Oknin-Vaisman, Eliya Bitman-Lotan, and Amir Orian. 2021. "From the Evasion of Degradation to Ubiquitin-Dependent Protein Stabilization" Cells 10, no. 9: 2374. https://doi.org/10.3390/cells10092374
APA StyleAbu Ahmad, Y., Oknin-Vaisman, A., Bitman-Lotan, E., & Orian, A. (2021). From the Evasion of Degradation to Ubiquitin-Dependent Protein Stabilization. Cells, 10(9), 2374. https://doi.org/10.3390/cells10092374