1. Introduction
Fibrosis is a major contributor to the chronic decline of organ function associated with end-stage organ failure. It is not unique to a single organ but manifests in any organ in the human body [
1]. In the United States, forty-five percent of all deaths can be attributed to fibrosis-related disease [
2,
3,
4]. Fibrosis is described as the remodeling of tissue architecture which results in loss of organ function due to the substitution of the functional parenchyma with mesenchymal tissue. While transient tissue fibrosis is a normal response to injury during wound healing, pathological fibrosis is characterized by relentless, non-resolving extracellular matrix (ECM) deposition and progressive tissue remodeling. Fibrosis progresses when a poorly characterized wound healing process is activated, leading to excessive remodeling and deposition of ECM by myofibroblasts, primarily fibronectin, type I and type III collagen [
5,
6]. Myofibroblasts are both ubiquitous and invasive as part of canonical wound healing; however, the origin of the myofibroblasts that fail to undergo programmed cell death or dedifferentiation following wound healing and result in progressive fibrosis remains ambiguous [
7,
8].
To date, there is no effective therapeutic to halt or reverse fibrotic progression and tractable therapeutic targets with pathological specificity are extremely limited [
5,
9,
10]. Only two therapeutics, Nintedanib and Pirfenidone, have been approved for fibrosis, and in only one organ system, the lungs. However, even with treatment idiopathic pulmonary fibrosis (IPF) continues to progress [
4]. Understandably, there have been several attempts at therapeutic intervention designed to target proteins differentially expressed by myofibroblasts, including several successful attempts at abolishing fibrosis in rodent models [
9,
11]. Notably, translating these models to the clinic has been challenging [
4]. The salient example of this is the significant number of compounds targeting TGF-β1 and its associated signaling. TGF-β1 is a critical growth factor with roles in normal wound healing and immunity, as well as cancer progression, myofibroblast activity and fibrogenesis [
9]. Neutralizing TGF-β1 signaling in rodent models abolishes fibrosis; however, molecules targeting TGF-β1 signaling in humans have failed to progress clinically [
11]. These failures to translate preclinical findings to the clinic represent an unmet need in the field to better identify therapeutic targets with specificity to fibrotic tissues.
Here we present data demonstrating that retention of the N-cadherin prodomain at the cell surface is a potential biomarker of pathological myofibroblasts and fibrosis associated tissues of the heart, lungs, and liver. Canonical N-cadherin protein processing occurs in the transgolgi network after the precursor N-cadherin is translated with an N-terminal precursor prodomain, termed pro-N-cadherin (PNC) [
10,
12,
13]. The precursor prodomain is then cleaved by proprotein convertases, generating the active mature N-cadherin protein which functions at the cell surface by forming homodimers at cellular adherens junctions [
10,
12]. The prodomain is non-adhesive and sterically restricts N-cadherin dimer formation by inhibiting N-cadherin tryptophan swapping [
12]. During certain developmental and pathological processes, PNC is localized to the cell surface with mature N-cadherin [
14,
15,
16]. To date, cell surface PNC expression has been observed in only two contexts: Perinatal synapse formation in vertebrates, and carcinogenesis [
14,
15,
16]. In this work, we identify a third clinically significant context in which PNC is expressed on the surface of myofibroblasts from pathological origins and aberrantly localized on tissues from failing heart, lung, and liver.
2. Materials and Methods
2.1. Cell Culture and Reagents
All cells were cultured at 37 °C, 5% CO2 in a humidified chamber. Cells were used between passages 3 and 10 and validated for alpha-smooth muscle actin (α-SMA) expression prior to use in experiments by immunoblot using α-SMA antibody (Cell Signaling 19245S, Danvers, MA, USA). The LL97A (ATCC CCL-191, Manassas, VA, USA), LL29 (ATCC CCL-134, Manassas, VA, USA) and CCD-16Lu (ATCC CCL-204, Manassas, VA, USA) cell lines were purchased from ATCC and grown by their specifications. Primary ventricular normal human cardiac fibroblasts (NHCF) from a healthy donor heart were purchased from Lonza (CC-2904, Morrisville, NC, USA) and grown in their recommended media. Primary ventricular cardiac fibroblasts (DCM-CF) from explant failed human hearts were isolated from tissue obtained from the Duke Human Heart Repository (IRB # Pro87831) and grown in DMEM;10% FBS; 1x Pen Strep. Primary normal human lung fibroblasts (NHLF) were purchased from the Duke Cell Culture Facility (Lonza CC-2512) and grown in MEM; 10% FBS; 1x Pen Strep.
2.2. pro-N-Cadherin Antibody Purification
pro-N-cadherin monoclonal antibody-producing hybridoma clone 10A10 was a gift from the Wahl Laboratory at the University of Nebraska. Cells were acclimated to Hybridoma-SFM (Gibco, Grand Island, NE, USA) supplemented with 10% super low IgG HI-FBS (Hyclone, Logan, UT, USA) and grown to confluency. Hybridoma cells were passaged, and the media was replaced with serum free Hybridoma-SFM media and allowed to express for 5 days. Supernatant was collected by centrifugation, passed through a 0.22 µm filter and antibodies were purified by affinity chromatography using protein G sepharose prepacked column (GE Healthcare, Salt Lake City, UT, USA) following the manufacturer’s protocol using the Amersham Biosciences AKTA FPLC system. Antibodies were dialyzed into final buffer PBS pH 7.4 and stored at −20 °C for these studies.
2.3. Fibroblast Isolation from Explant Tissues
Tissues were minced into small pieces and enzymatically digested in appropriate volume of tissue dissociation solution comprised of PBS pH 7.4, 5 mg/mL Collagenase Type IV (Gibco, Grand Island, NE, USA), 1.3 mg/mL Dispase II (Gibco, Grand Island, NE, USA), 0.05% Trypsin, with agitation at 37° Celsius for 1 h. After incubation in dissociation solution, 25 mL of HBSS (Gibco, Grand Island, NE, USA) was added, followed by serial pipetting for manual dissociation and centrifugation at 1200 RPM, 4 °C, for 10 min. The supernatant was then aspirated and the pellet was resuspended in 5 mL HBSS then passed through a 100 µm filter followed by a 70 µm filter. The filtered cell suspension was then centrifuged at 1200 RPM, 4 °C, for 5 min, the supernatant was aspirated and the pellet resuspended into DMEM; 10% FBS; 1x Pen Strep. Cells were plated and passaged to remove plasma and myocyte contaminant.
2.4. Tissue Procurement and Processing
Fresh explant cardiac tissues were received from the Duke HHR (IRB Pro00087831) within 12 h of explant in PBS pH 7.4, 1x Pen Strep (Gibco, Grand Island, NE, USA), on ice and processed same day. Tissues were divided and a portion was immediately processed for fibroblast isolation (above). The remaining portion was fixed overnight at room temperature in 4% paraformaldehyde and PBS pH 7.4 followed by 70% EtOH and embedded in paraffin. Additional flash-frozen cardiac tissues from the Duke HHR, including a subset of previously characterized samples [
17] were embedded in OCT medium and sectioned on a Leica cryostat into 5 µm sections and adhered to charged glass slides for immunohistochemical analysis. Liver tissue specimens were provided by the DUHS Nonalcoholic Fatty Liver Disease Research Database and Specimen Repository under Pro00005368 and Abdominal Transplant Repository under Pro00107246. Formalin-fixed paraffin embedded heart and liver tissues were cut on a Leica microtome into 3–5 µm sections and adhered to charged glass slides for immunohistochemical analysis. Lung tissue sections were obtained from the Duke Bio Repository and Precision Pathology Center (BRPC) on charged glass slides under an IRB exemption. Additional heart, lung, and liver tissue microarrays were obtained commercially from USA Biomax, Inc. (Rockville, MD, USA) as formalin-fixed paraffin embedded 1.5 mm cores on charged glass slides.
2.5. Immunohistochemistry
Immunohistochemical detection of PNC was performed on formalin fixed paraffin embedded tissue samples sectioned at 3–5 µm. Sections were deparaffinated with xylene, rehydrated, and treated with 3% H2O2 to quench endogenous peroxidase. Heat-mediated antigen retrieval was performed in a citrate buffer (pH 6) and blocked with 5% horse serum. PNC was detected using purified m-α-PNC mAb clone 10A10 at 5–10 µg/mL at 4 °C overnight. An avidin-biotin amplification step and chromogenic detection (DAB) of α-mouse HRP-conjugated secondary antibody was used to visualize pro-N-cadherin localization and expression. Tissues were counter-stained with Mayer’s hematoxylin and mounted with Cytoseal 60 (Thermo Fisher, Grand Island, NE, USA) mounting media for imaging.
2.6. Plasma Procurement
Healthy human donor plasma was purchased commercially from Innovative Research, Inc, Novi, MI, USA. Plasma from heart failure patients was provided by the Duke HHR IRB Pro00087831. Plasma from NAFLD-cirrhosis patients was provided by the Duke Nonalcoholic Fatty Liver Disease Research Database and Specimen Repository under Pro00005368 and Abdominal Transplant Repository under Pro00107246. Plasma from IPF patients was purchased commercially from Innovative Research, Inc.
2.7. Immunoprecipitations
LL29 conditioned media was incubated at 4 °C with 10 µg of murine 10A10 antibody for 1 h. Protein G Sepharose (GE Healthcare, Salt Lake City, UT, USA) was blocked with 1% BSA in PBS then 5 µL of resin was added to the conditioned media. After incubating at 4 °C for 30 min, the resin was washed three times with PBS then eluted using 1x LDS sample buffer (Thermo Fisher, Grand Island, NE, USA) with B-mercaptoethanol. Elutes were boiled, immunoblotted, and developed using humanized 10A10 chimeric antibody.
For plasma samples, 10A10 and mouse IgG1 isotype control (Thermo Fisher, Grand Island, NE, USA) antibodies were first crosslinked to protein G sepharose (GE Healthcare, Salt Lake City, UT, USA) using Pierce crosslink IP kit (Thermo Fisher, Grand Island, NE, USA). Equivalent volumes of healthy donor and diseased donor plasma samples were pooled then diluted 1:10 in PBS, centrifuged at 16,000 G and filtered using a 0.22 µm filter. Samples were then rotated overnight with antibody crosslinked protein G sepharose, washed and eluted following the manufacturer’s protocol. Elutes were immunoblotted and developed using murine 10A10 antibody.
2.8. SDS-PAGE and Immunoblotting
Total cell lysates were prepared using RIPA buffer adjusted to 1% w/v SDS (Sigma, Saint Louis, USA) supplemented with Halt Protease & Phosphatase Inhibitor Cocktail (Thermo Fisher, Grand Island, NE, USA) and Benzonase (Millipore Sigma, Saint Louis, MO, USA). Cell surface proteins were isolated using the Pierce cell surface biotinylation and isolation kit (Thermo Fisher, Grand Island, NE, USA) following the manufacturer’s protocol. Plasma membrane loading control Na,k-ATPase α-1 (cell signaling 23565T, Massachusetts, USA) was used for cell surface protein isolates. Protein concentration of each lysate was measured using Pierce BCA protein assay (Thermo Fisher, Grand Island, NE, USA). For total lysates, precleared lysates were boiled in sample buffer (Thermo Fisher, Grand Island, NE, USA) and 40 µg of protein was loaded. All samples were run on 10% NuPage gels containing 0.1% SDS under reducing conditions. A discontinuous Laemmli buffer system was used. The proteins were transferred from the gels to nitrocellulose membranes. The molecular weights were assessed using Precision Plus Prestained Marker (Bio-Rad, Hercules, CA, USA). The membranes were thoroughly washed with tris-buffered saline (TBS) and then blocked with infrared blocking buffer (Rockland, Pottstown, PA, USA) for 1 h at room temperature. Membranes were incubated with antibodies overnight at 4 °C in 5% BSA, 1x TBS, 0.1% Tween-20. RPL13A (Cell Signaling 2765S, Danvers, MA USA) was used for total cell lysate loading control, along with mature N-cadherin (Sigma, GC-4 clone, C3865, Saint Louis, MO, USA) and α-PNC mAb. After incubation with the primary antibody, the membranes were washed three times for 5 min each with 1x TBS containing 0.1% Tween 20 (TBST). The membranes were then incubated with the manufacturer’s recommended dilution of appropriate Alexa Fluor conjugated secondary (Thermo Fisher, Grand Island, NE, USA). The membranes were washed twice for 5 min each with TBST and once with TBS for 5 min. The probed membranes were scanned on a Li-Cor Odyssey System (Li-Cor Biosciences, Lincoln, NE, USA).
2.9. Flow Cytometry
Cells were plated in complete media in 10 cm dishes and allowed to anchor overnight. The following day, cells were washed with PBS pH 7.4, then detached using PBS pH 7.4; 0.5 mM EDTA; 10% Glycerol at 37 °C for approximately 5–10 min or until cell rounding followed by scraping. Cells were kept on ice for the duration of the staining procedure. Cells were pelleted at 1200 RPM, 4 °C, for 5 min followed by supernatant aspiration, PBS pH 7.4 wash, and resuspension in PBS pH 7.4; 1% BSA; 0.09% sodium azide. Cells were incubated with either α-N-cadherin antibody (Sigma, GC-4 clone, C3865), α-PNC antibody (5 µg/mL), or 5 µg/mL mouse IgG1 isotype control (Thermo Fisher, Grand Island, NE, USA) for 30 min. Cells were washed with PBS pH 7.4; 1% BSA; 0.09% sodium azide followed by 5 µg/mL Alexa Fluor 488 secondary antibody (Thermo Fisher, Grand Island, NE, USA) incubation for 30 min. Cells were then washed with PBS pH 7.4; 1% BSA; 0.9% sodium azide and stained with 7AAD (BioLegend, San Diego, CA, USA) following the manufacturer’s protocol. Cells were analyzed using the Guava EasyCyte (Luminex, Austin, TX, USA) flow cytometer and the latest version of Flowjo software, gating and excluding 7AAD positive cells. At least 20,000 events were collected for each experiment. To calculate background, the same sample was ran using the isotype control antibody four times independently and histograms were stacked to determine baseline chi-square and SE dymax % positive.
2.10. Immunofluorescent Microscopy
For immunofluorescent microscopy, cells were plated into multi-well chamber slides coated with 5 µg/mL human Collagen type I (Sigma, Saint Louis, MO, USA) in PBS. After cells were allowed to adhere overnight, each well was aspirated, washed with PBS pH 7.4 and fixed in 1% formaldehyde for 30 min at room temperature. For perinuclear PNC staining, cells were permeabilized using 0.05% Triton in PBS. Cells were then blocked with 5% goat serum (Abcam, Cambridge, UK); PBS pH 7.4 for 1 h at room temperature. Primary antibody against pro-N-cadherin was incubated at 2 µg/mL on cells overnight at 4 °C followed by Qdot 655 conjugated secondary (Thermo Fisher, Grand Island, NE, USA) for 1 h at room temperature. For colocalization, FN1 antibody (Cell Signaling 26836S, Danvers, MA, USA) was used following the manufacturer’s protocol followed by AlexaFluor 488 conjugated secondary (Thermo Fisher, Grand Island, NE, USA). DAPI was used following the manufacturers protocol (Sigma, Saint Louis, MO, USA) to visualize DNA. Coverslips were applied using 50% glycerol in PBS and sealed with nail polish. Images were taken using the Leica DMI400 B.
2.11. Sandwich Enzyme-Linked Immunosorbent Assay
Recombinant prodomain (rPro) of N-cadherin, amino acids 26–159 (Accession # AAB22854) was generated and supplied by GenScript (Piscataway, NJ, USA) and used to optimize a PNC sandwich enzyme-linked immunosorbent assay (ELISA) and later used as an antagonist to cell surface PNC in migration assays. High binding ELISA plates (Costar, Kennebunk, ME, USA) were used to bind 1 µg/well of α-PNC antibody 10A10 as the capture antibody. Washes were performed using PBS pH 7.4 0.1% Tween 20. Three washes (300 µL/well) were performed between each of the following steps using Biotek (Winooski, VT, USA) ELx405 Select CW automated plate washer. All steps were performed at room temperature with room temperature equilibrated buffers. Capture antibody was bound overnight at room temperature in PBS pH 7.4 followed by blocking with 300 µL per well blocking buffer 5% non-fat dry milk (Bio-Rad, Hercules, CA, USA) in 1x PBS (Gibco, Grand Island, NE, USA) with 0.1% Tween 20 for 1 h. Plasma samples and rPro analyte standard were applied 100 µL per well in 1% BSA, PBS pH 7.4, 5 mM EDTA, 0.1% Tween 20 for 1 h followed by 100 µL/well 1:800 dilution of biotinylated polyclonal sheep α-PNC detection antibody (R&D BAF1388, Minneapolis, MN, USA) in 1% BSA, PBS pH 7.4, 0.1% Tween 20 for 1 h. Streptavidin horseradish peroxidase conjugate (Thermo Fisher, Grand Island, NE, USA ) was applied at 100 µL per well, 1:800 in 2% BSA, PBS pH 7.4 for 20 min followed by 150 µL of ABTS (Thermo Fisher, Grand Island, NE, USA ) for 15 min and read using the Biotek (Winooski, VT, USA) Cytation 3 Imager Reader at absorbance (Abs) 410 nm.
2.12. Solid Phase Enzyme Immunoassays
To find rPro binding partners, a solid phase enzyme immunoassay was used. Medium binding ELISA plates (Costar, Kennebunk, ME, USA) were coated with either 10 µg/mL human plasma fibronectin (Sigma, Saint Louis, MO, USA), human type I collagen (Sigma, Saint Louis, MO, USA) or human type III collagen (Sigma, Saint Louis, MO, USA) overnight at room temperature in PBS. All washes were done with PBS 0.1% Tween-20, 300 µL/well, three times using the Biotek ELx405 Select CW automated plate washer. After immobilizing fibronectin, collagen type I, or BSA, wells were washed then blocked using 300 µL/well 1% BSA PBS in wash buffer for 1 h at room temperature. Wells were washed then a serial dilution starting at 10 µg/mL of His tagged rPro peptide diluted in blocking buffer was incubated over the immobilized proteins for one hour at room temperature. Wells were washed and incubated with 1:2000 mouse α-His biotinylated antibody (Invitrogen MA1-21315-BTIN, Grand Island, NE, USA) in block for 1 h at room temperature followed by 1:2000 streptavidin HRP (Thermo Fisher, Grand Island, NE, USA) in 2% BSA PBS at room temperature for 15 min. Immunoassay was developed using TMB Ultra (R&D, Minneapolis, MN, USA) following the manufacturers recommended protocol. Absorbance 450 nm was read using the Biotek Cytation 3 indicating bound his tagged rPro peptide.
For antibody displacement assays, medium binding ELISA plates (Costar, Kennebunk, ME, USA) were coated with human plasma fibronectin (Sigma, Saint Louis, MO, USA) at 10 µg/mL in PBS overnight at room temperature. All washes were done with PBS 0.1% Tween-20, 300 µL/well, three times using the Biotek ELx405 Select CW automated plate washer. After immobilizing fibronectin, wells were washed then blocked using 300 µL/well 5% human plasma in wash buffer for 1 h at room temperature. While blocking, antibodies were added at the respective concentration to blocking buffer containing 1.5 µg/mL recombinant his-tagged prodomain. Wells were washed and samples were applied for 1 h at room temperature. Wells were washed and incubated with 1:2000 mouse α-His biotinylated antibody (Invitrogen MA1-21315-BTIN, Grand Island, NE, USA) for 1 h at room temperature followed by 1:2000 streptavidin HRP (Thermo Fisher, Grand Island, NE, USA) at room temperature for 15 min. Immunoassay was developed using TMB Ultra (R&D, Minneapolis, MN, USA) following the manufacturers recommended protocol. Absorbance 450 nm was read using the Biotek Cytation 3 indicating bound his tagged rPro peptide.
2.13. Humanization of Murine pro-N-Cadherin Antibody 10A10
Fusion Antibodies, PLC (Belfast, Northern Ireland) was contracted to humanize the murine α-PNC mAb 10A10. CDR regions of the murine α-PNC mAb were cloned onto human IgG
4 heavy and light chain constant domains. The variable regions of the murine α-PNC mAb were modified in-silico to reduce T-cell epitope antigenicity and increase binding affinity to the prodomain of N-cadherin. These 22 humanized antibody designs were expressed, purified, and ranked for binding affinity using bio-layer interferometry (octet) technology by Fusion Antibodies to the rPro peptide (
Supplementary Figure S3A).
2.14. Migration Assays
Transwell permeable supports with a 6.5 mm polycarbonate membrane and 8 µm pores were used to separate the upper and lower chambers of a 24-well plate. Both sides of the membrane were coated with 1 µg/mL human plasma fibronectin (Sigma, Saint Louis, MO, USA). Bovine fibronectin was depleted from the complete media using gelatin Sepharose (GE Healthcare, Salt Lake City, UT, USA) following the manufacturers protocol. Complete media containing 10 ng/mL TGF-β1 was then added to the lower chamber at 600 µL/well. Cells were trypsinized, pelleted, resuspended in low serum media containing 0.5% FBS and added into the upper chamber at 1.0 × 104 cells/well in 100μL media per well. After allowing attachment, antibody treatment, rPro or PBS blank was added to the lower chamber and the plate was incubated for 5 h at 37 °C, 5% CO2. Media was aspirated, and cells were fixed using 4% Formaldehyde for 15 min at room temperature. Cells were removed from the top of the upper chamber using a sterile cotton swab and wells were washed three times with PBS. Adherent cells on the apical side of wells were then stained by applying DAPI solution (Sigma, Saint Louis, MO, USA) following the manufacturers protocol. Nuclei were visualized by fluorescent microscopy and counted using the Biotek Cytation 3 and latest Gen5 v3.11 software (Winooski, VT, USA). Samples were imaged using the 2.5× objective and total cell numbers were counted in each frame.
2.15. Statistical Analysis
Statistical analysis was performed using T-tests and one-way ANOVA with Post hoc multiple comparisons analyses where appropriate using the latest version of GraphPad Prism 9 (San Diego, CA, USA) software (* p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001). Post hoc analyses are indicated in corresponding figure legends. For flow cytometry, chi-squared test was performed using Flowjo v10.7.2 (Vancouver, BC, Canada) analysis software. Chi-squared ≥ 4 is statistically significant.
4. Discussion
Our study suggests the need to further investigate N-cadherin biology regarding tissue fibrosis, as reagents developed to study and/or target N-cadherin in this context may or may not cross-react with the pro-N-cadherin precursor protein. This study and others suggest PNC has a unique and divergent role from what is typical of mature N-cadherin [
13,
15,
16,
24]. In its mature form, N-cadherin functions as a cell–cell adhesion molecule [
10,
26]. In its precursor form, the data suggest that it may serve a role in cell-ECM focal adhesions. This is particularly interesting when considering the cardiac structure of the intercalated disc. The major cell adhesion molecule within the fascia adherens of the cardiac intercalated discs is N-cadherin [
18,
20]. Mature N-cadherin is essential in maintaining the structure and function of the intercalated discs, as well as myofibrillar organization and myocyte shape [
19]. Conversely, loss of N-cadherin results in disassembly of intercalated disc structure in the mammalian heart, dilated cardiomyopathy, impaired cardiac function and death [
18,
27]. The biomechanical consequences of PNC localization to intercalated discs could play a role in the progression of heart failure.
Tissue fibrosis is observed anatomically as the accumulation of excessive extracellular matrix which stresses cell–cell contacts through increased tensile force and elicits maladaptive remodeling of tissues [
22,
28]. Cells can respond to increased mechanical load at the cell–cell junctions by forming focal adhesions to offload intercellular mechanical force and maintain tensional homeostasis [
28,
29,
30]. Conversely, it is also demonstrated that focal adhesions and cell–cell junctions can function inversely [
31]. In the heart, this is observed in developmental and pathological processes. Mimicking fibrosis in cardiac cells resulted in increased focal adhesion formation adjacent to the cell–cell interface [
28]. This was interpreted as the need for mechanical off-loading from the intercalated discs. In the pathological processes of cardiac fibrosis and remodeling, increased focal adhesion is correlated with lower working efficiency of the cardiac tissue [
28]. The observation of FN1 as a potential PNC binding partner leads us to postulate that post translational changes to N-cadherin processing, through retention of the prodomain could serve as a method for poised cellular response to changes in mechanical load. Future studies are required to understand the role of PNC at the cell surface in settings of fibrosis.
It is possible the role of PNC is cell dependent. In this study, PNC expression is observed in multiple different diseased tissues associated with fibrosis and tissue remodeling. Targeting PNC with a monoclonal antibody specific for an epitope on the prodomain results in reduction of migration by PNC expressing myofibroblasts suggesting a role for PNC in myofibroblast migration. This is consistent with the role of cell surface PNC in carcinogenesis and a recent report that cell surface PNC regulates apico-basal polarity in neural stem cells [
13,
15,
16,
24]. Cell surface PNC expression in vivo, but not mature N-cadherin, caused loss of apico-basal polarity, breach of basal membrane and invasion of neural precursors into the ventricle and surrounding mesenchyme of the neural tube [
13]. The observation that rPro antagonizes cell migration suggests cell surface PNC is a mediator of pathological myofibroblast migration, as opposed to the sPNC. In progressive fibrosis, myofibroblasts continually migrate into and within the affected organ without resolving, leading to disease progression. This is readily apparent in cardiac fibrosis and IPF disease progression [
22,
32]. In the case of cardiac fibrosis, myofibroblasts can be found widespread within the failing heart [
22]. It is thought that IPF is initiated at the periphery of the lungs and slowly migrates to encompass the entirety of the lungs [
33]. It has been demonstrated that myofibroblasts isolated from advanced IPF display a higher capacity to migrate than those isolated from less advanced disease stages [
34]. Targeting myofibroblasts migration is a reasonable therapeutic strategy for mitigating fibrotic conditions.
It has been postulated that cell surface prodomain cleavage from N-cadherin is a means of spatially and temporally regulating adhesion during development and synapse formation [
14]. Increased expression and proteolysis of the prodomain at the cell surface could explain elevated levels of sPNC found in the plasma of patients with pathological fibrosis. It could also be argued that increased cell death due to disease could contribute to the elevation of the 17 kDa prodomain in the plasma. Almost one third of the plasma proteome is made up of intercellular proteins that have escaped due to cellular turnover [
35]. With the origins of the 60 kDa protein unknown, future studies are necessary to understand the processing of PNC in tissue fibrosis.
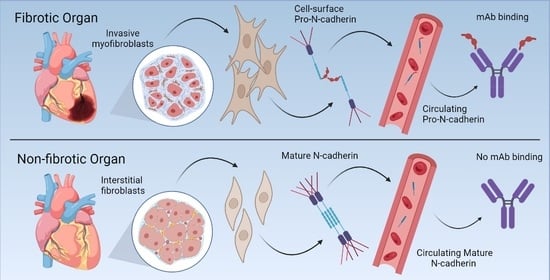
Our study is a preliminary report with a relatively limited and mostly retrospective sample set, highlighting the need for further studies to thoroughly evaluate PNC as both a biomarker and a therapeutic target in fibrotic disease. Additionally, our data (
Figure 1) indicate that epithelial cells in each respective tissue and not only the myofibroblasts may have an important role in PNC-driven fibrotic disease. This is important considering existing work that identifies PNC as a driver of migration and invasion in carcinogenesis [
15,
16].


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






