
Metabolic Implications of Immune Checkpoint Proteins in Cancer


Abstract
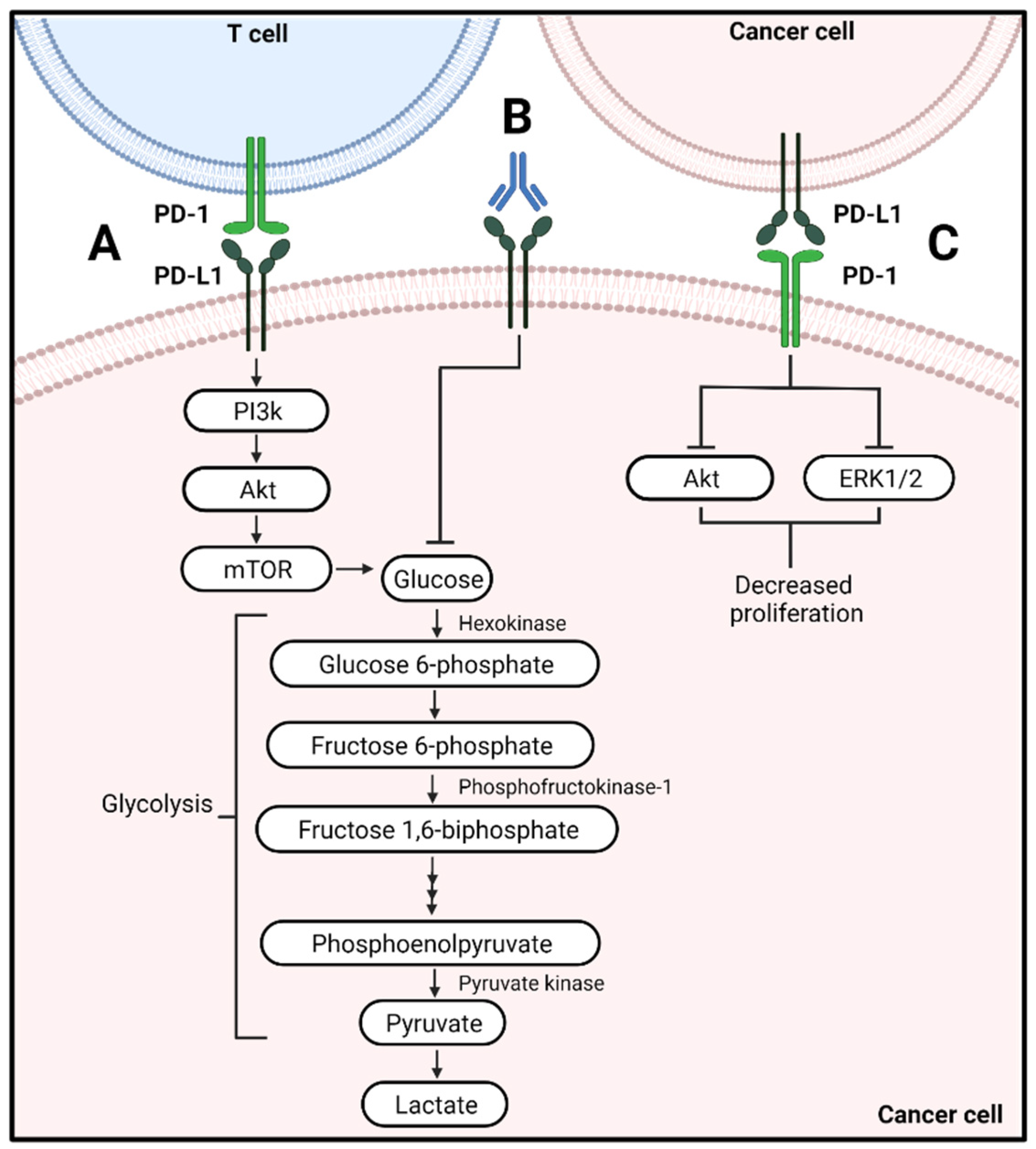
:1. Introduction
2. Metabolic Effects of Inhibitory Immune Checkpoint Protein Activation on Immune Cells
2.1. Effector T Cells
2.2. Exhausted T Cells
2.3. Memory T Cells
2.4. Macrophage and Dendritic Cells
3. Metabolic Effects of Stimulatory Immune Checkpoint Protein Activation on Immune Cells
3.1. CD28
3.2. Inducible Costimulator (ICOS)
3.3. Glucocorticoid-Induced TNFR-Related Protein (GITR)
3.4. 4-1BB
4. Metabolic Effects of Immune Checkpoint Protein Activation on Cancer Cells
4.1. Metabolic Alterations of Cancer Cells Associated with PD-L1 Signaling
4.2. Metabolic Alterations by PD-1 on Cancer Cells
4.3. Regulation of PD-L1 Expression by Metabolic Pathways
5. Impact of Diet and the Microbiome on Immune Checkpoint Blockade Response
5.1. Dietary Interventions
5.2. Obesity
5.3. Microbiome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | Study Size | Geographical Location | Treatment | Microbiota Associated with Favorable Response | Reference |
|---|---|---|---|---|---|
| Melanoma | N = 39 | Texas, USA | I, N, I+N, P (all) IN P | B. caccae F. prausnitzii, B. thetaiotamicron D. formicogenerans | Frankel et al., 2017 [126] |
| NSCLC and RCC | N = 100 | Paris, France | N | Akkermansia muciniphilia | Routy et al., 2018 [125] |
| Melanoma | N = 42 | Illinois, USA | I, N, or P | Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium | Matson et al., 2018 [127] |
| Melanoma | N = 53 | Texas, USA | N or P | Faecalibacterium prausnitzii | Gopalakrishnan et al., 2018 [124] |
| NSCLC | N = 37 | Shanghai, China | N | Alistipes putredinis, Bifidobacterium longum, and Prevotella copri | Jin et al., 2019 [130] |
| GI | N = 74 | Beijing, China | I, N, P, or A | Prevotella/Bacteroides ratio, Lactobacillus, Akkermansia muciniphilia | Peng et al., 2020 [128] |
6. Metabolism and Immune-Related Adverse Events (irAEs) Associated with Immune Checkpoint Blockade Therapy
7. Clinical Perspective of Immune Checkpoint Blockade Therapy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Roos, D.; Loos, J.A. Changes in the carbohydrate metabolism of mitogenically stimulated human peripheral lymphocytes. II. Relative importance of glycolysis and oxidative phosphorylation on phytohaemagglutinin stimulation. Exp. Cell Res. 1973, 77, 127–135. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 701–703. [Google Scholar] [CrossRef]
- Van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily--CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, L.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [Green Version]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.V.; Brodie, D.W.; Gilbert, R.J.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; van der Merwe, P.A.; Davis, S.J. The interaction properties of costimulatory molecules revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Kane, L.P.; Andres, P.G.; Howland, K.C.; Abbas, A.K.; Weiss, A. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-gamma but not TH2 cytokines. Nat. Immunol. 2001, 2, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Reif, K.; Smith, G.; Sansom, D.M.; Hemmings, B.A.; Ward, S.G. Ligation of the T cell co-stimulatory receptor CD28 activates the serine-threonine protein kinase protein kinase B. Eur. J. Immunol. 1997, 27, 2495–2501. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Bullock, K.; Gurjao, C.; Braun, D.; Shukla, S.A.; Bosse, D.; Lalani, A.A.; Gopal, S.; Jin, C.; Horak, C.; et al. Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat. Commun. 2019, 10, 4346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amobi-McCloud, A.; Muthuswamy, R.; Battaglia, S.; Yu, H.; Liu, T.; Wang, J.; Putluri, V.; Singh, P.K.; Qian, F.; Huang, R.Y.; et al. IDO1 Expression in Ovarian Cancer Induces PD-1 in T Cells via Aryl Hydrocarbon Receptor Activation. Front. Immunol. 2021, 12, 678999. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, U.; Orabona, C.; Fallarino, F.; Vacca, C.; Calcinaro, F.; Falorni, A.; Candeloro, P.; Belladonna, M.L.; Bianchi, R.; Fioretti, M.C.; et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 2002, 3, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Crisosto, C.; Bravo-Sagua, R.; Rodriguez-Pena, M.; Mera, C.; Castro, P.F.; Quest, A.F.; Rothermel, B.A.; Cifuentes, M.; Lavandero, S. ER-to-mitochondria miscommunication and metabolic diseases. Biochim. Biophys. Acta. 2015, 1852, 2096–2105. [Google Scholar] [CrossRef] [Green Version]
- Rieusset, J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death Dis. 2018, 9, 388. [Google Scholar] [CrossRef] [Green Version]
- Freitas, C.M.T.; Johnson, D.K.; Weber, K.S. T Cell Calcium Signaling Regulation by the Co-Receptor CD5. Int. J. Mol. Sci. 2018, 19, 1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurst, K.E.; Lawrence, K.A.; Essman, M.T.; Walton, Z.J.; Leddy, L.R.; Thaxton, J.E. Endoplasmic Reticulum Stress Contributes to Mitochondrial Exhaustion of CD8(+) T Cells. Cancer Immunol. Res. 2019, 7, 476–486. [Google Scholar] [CrossRef]
- Andrews, A.M.; Tennant, M.D.; Thaxton, J.E. Stress relief for cancer immunotherapy: Implications for the ER stress response in tumor immunity. Cancer Immunol. Immunother 2021, 70, 1165–1175. [Google Scholar] [CrossRef]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, M.U.; Hernandez, S.R.; Soto-Pantoja, D.R.; Cook, K.L. Endoplasmic Reticulum Stress Pathway, the Unfolded Protein Response, Modulates Immune Function in the Tumor Microenvironment to Impact Tumor Progression and Therapeutic Response. Int. J. Mol. Sci. 2019, 21, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doering, T.A.; Crawford, A.; Angelosanto, J.M.; Paley, M.A.; Ziegler, C.G.; Wherry, E.J. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 2012, 37, 1130–1144. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Villa, M.; Sanin, D.E.; Buck, M.D.; O’Sullivan, D.; Ching, R.; Matsushita, M.; Grzes, K.M.; Winkler, F.; Chang, C.H.; et al. Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Rep. 2019, 27, 2063–2074.e5. [Google Scholar] [CrossRef] [Green Version]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E761–E770. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.R.; Imrichova, H.; Wang, H.; Chao, T.; Xiao, Z.; Gao, M.; Rincon-Restrepo, M.; Franco, F.; Genolet, R.; Cheng, W.C.; et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat. Immunol. 2020, 21, 1540–1551. [Google Scholar] [CrossRef]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef] [PubMed]
- Hope, H.C.; Salmond, R.J. The Role of Non-essential Amino Acids in T Cell Function and Anti-tumour Immunity. Arch. Immunol Ther. Exp. (Warsz) 2021, 69, 29. [Google Scholar] [CrossRef]
- Lin, R.; Zhang, H.; Yuan, Y.; He, Q.; Zhou, J.; Li, S.; Sun, Y.; Li, D.Y.; Qiu, H.B.; Wang, W.; et al. Fatty Acid Oxidation Controls CD8(+) Tissue-Resident Memory T-cell Survival in Gastric Adenocarcinoma. Cancer Immunol. Res. 2020, 8, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Tkachev, V.; Goodell, S.; Opipari, A.W.; Hao, L.Y.; Franchi, L.; Glick, G.D.; Ferrara, J.L.; Byersdorfer, C.A. Programmed death-1 controls T cell survival by regulating oxidative metabolism. J. Immunol. 2015, 194, 5789–5800. [Google Scholar] [CrossRef]
- Hildeman, D.A.; Mitchell, T.; Aronow, B.; Wojciechowski, S.; Kappler, J.; Marrack, P. Control of Bcl-2 expression by reactive oxygen species. Proc. Natl. Acad. Sci. USA 2003, 100, 15035–15040. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark Res. 2021, 9, 1. [Google Scholar] [CrossRef]
- Lu, D.; Ni, Z.; Liu, X.; Feng, S.; Dong, X.; Shi, X.; Zhai, J.; Mai, S.; Jiang, J.; Wang, Z.; et al. Beyond T Cells: Understanding the Role of PD-1/PD-L1 in Tumor-Associated Macrophages. J. Immunol. Res. 2019, 2019, 1919082. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Zhang, Y.; Wang, J.; Gu, J. Defects in Macrophage Reprogramming in Cancer Therapy: The Negative Impact of PD-L1/PD-1. Front. Immunol. 2021, 12, 690869. [Google Scholar] [CrossRef]
- Adeshakin, A.O.; Liu, W.; Adeshakin, F.O.; Afolabi, L.O.; Zhang, M.; Zhang, G.; Wang, L.; Li, Z.; Lin, L.; Cao, Q.; et al. Regulation of ROS in myeloid-derived suppressor cells through targeting fatty acid transport protein 2 enhanced anti-PD-L1 tumor immunotherapy. Cell Immunol. 2021, 362, 104286. [Google Scholar] [CrossRef] [PubMed]
- Alter, G.; Malenfant, J.M.; Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 2004, 294, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckermann, K.E.; Hongo, R.; Ye, X.; Young, K.; Carbonell, K.; Healey, D.C.C.; Siska, P.J.; Barone, S.; Roe, C.E.; Smith, C.C.; et al. CD28 costimulation drives tumor-infiltrating T cell glycolysis to promote inflammation. JCI. Insight 2020, 5, e138729. [Google Scholar] [CrossRef] [PubMed]
- Hunig, T. Manipulation of regulatory T-cell number and function with CD28-specific monoclonal antibodies. Adv. Immunol. 2007, 95, 111–148. [Google Scholar] [CrossRef] [PubMed]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef]
- Attarwala, H. TGN1412: From Discovery to Disaster. J. Young Pharm. 2010, 2, 332–336. [Google Scholar] [CrossRef] [Green Version]
- Simpson, T.R.; Quezada, S.A.; Allison, J.P. Regulation of CD4 T cell activation and effector function by inducible costimulator (ICOS). Curr. Opin. Immunol. 2010, 22, 326–332. [Google Scholar] [CrossRef]
- Wikenheiser, D.J.; Stumhofer, J.S. ICOS Co-Stimulation: Friend or Foe? Front. Immunol. 2016, 7, 304. [Google Scholar] [CrossRef] [Green Version]
- Teijeira, A.; Garasa, S.; Etxeberria, I.; Gato-Canas, M.; Melero, I.; Delgoffe, G.M. Metabolic Consequences of T-cell Costimulation in Anticancer Immunity. Cancer Immunol. Res. 2019, 7, 1564–1569. [Google Scholar] [CrossRef]
- Zeng, H.; Cohen, S.; Guy, C.; Shrestha, S.; Neale, G.; Brown, S.A.; Cloer, C.; Kishton, R.J.; Gao, X.; Youngblood, B.; et al. mTORC1 and mTORC2 Kinase Signaling and Glucose Metabolism Drive Follicular Helper T Cell Differentiation. Immunity 2016, 45, 540–554. [Google Scholar] [CrossRef] [Green Version]
- A Phase I Open Label Study of GSK3359609 Administered Alone and in Combination with Anticancer Agents in Subjects with Selected Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT02723955.
- A Phase II, Randomized, Open-label Platform Trial Utilizing a Master Protocol to Study Novel Regimens versus Standard of Care Treatment in NSCLC Participants. Available online: https://clinicaltrials.gov/ct2/show/NCT03739710.
- Massarelli, E.; Balmanoukian, A.S.; Vieito, M.; Tourneau, C.L.; Hernandez-Guerrero, T.; Trigo, J.M.; Aljumaily, R.; Chisamore, M.J.; Rogan, D.; Sung, R.; et al. INDUCE-1: Report on safety run-in cohorts combining Inducible T-cell co-stimulatory receptor (ICOS) agonist GSK3359609 (GSK609) with platinum+5-FU chemotherapy (5-FU/plat), with or without pembrolizumab (PE), for the treatment of advanced solid tumors. J. Clin. Oncol. 2020, 38, 6544. [Google Scholar] [CrossRef]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, G.; Giunchi, L.; Ronchetti, S.; Krausz, L.T.; Bartoli, A.; Moraca, R.; Migliorati, G.; Riccardi, C. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 6216–6221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Zhang, B.; Rui, K.; Wang, S. The Role of GITR/GITRL Interaction in Autoimmune Diseases. Front. Immunol. 2020, 11, 588682. [Google Scholar] [CrossRef]
- Tone, M.; Tone, Y.; Adams, E.; Yates, S.F.; Frewin, M.R.; Cobbold, S.P.; Waldmann, H. Mouse glucocorticoid-induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc. Natl. Acad. Sci. USA 2003, 100, 15059–15064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabharwal, S.S.; Rosen, D.B.; Grein, J.; Tedesco, D.; Joyce-Shaikh, B.; Ueda, R.; Semana, M.; Bauer, M.; Bang, K.; Stevenson, C.; et al. GITR Agonism Enhances Cellular Metabolism to Support CD8(+) T-cell Proliferation and Effector Cytokine Production in a Mouse Tumor Model. Cancer Immunol. Res. 2018, 6, 1199–1211. [Google Scholar] [CrossRef] [Green Version]
- Mahne, A.E.; Mauze, S.; Joyce-Shaikh, B.; Xia, J.; Bowman, E.P.; Beebe, A.M.; Cua, D.J.; Jain, R. Dual Roles for Regulatory T-cell Depletion and Costimulatory Signaling in Agonistic GITR Targeting for Tumor Immunotherapy. Cancer Res. 2017, 77, 1108–1118. [Google Scholar] [CrossRef] [Green Version]
- Durham, N.M.; Holoweckyj, N.; MacGill, R.S.; McGlinchey, K.; Leow, C.C.; Robbins, S.H. GITR ligand fusion protein agonist enhances the tumor antigen-specific CD8 T-cell response and leads to long-lasting memory. J. Immunother Cancer 2017, 5, 47. [Google Scholar] [CrossRef]
- Coe, D.; Begom, S.; Addey, C.; White, M.; Dyson, J.; Chai, J.G. Depletion of regulatory T cells by anti-GITR mAb as a novel mechanism for cancer immunotherapy. Cancer Immunol. Immunother 2010, 59, 1367–1377. [Google Scholar] [CrossRef]
- Phase I/II Trial of Ipilimumab or Nivolumab with BMS-986156 and Hypofractionated Stereotactic Radiation Therapy in Patients with Advanced Solid Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT04021043.
- Sanchez-Paulete, A.R.; Labiano, S.; Rodriguez-Ruiz, M.E.; Azpilikueta, A.; Etxeberria, I.; Bolanos, E.; Lang, V.; Rodriguez, M.; Aznar, M.A.; Jure-Kunkel, M.; et al. Deciphering CD137 (4-1BB) signaling in T-cell costimulation for translation into successful cancer immunotherapy. Eur. J. Immunol. 2016, 46, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Willoughby, J.E.; Kerr, J.P.; Rogel, A.; Taraban, V.Y.; Buchan, S.L.; Johnson, P.W.; Al-Shamkhani, A. Differential impact of CD27 and 4-1BB costimulation on effector and memory CD8 T cell generation following peptide immunization. J. Immunol. 2014, 193, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.K.; Lee, D.Y.; Lee, D.G.; Kim, Y.H.; Kim, S.H.; Oh, H.S.; Han, C.; Kwon, B.S. 4-1BB signaling activates glucose and fatty acid metabolism to enhance CD8(+) T cell proliferation. Cell Mol. Immunol. 2017, 14, 748–757. [Google Scholar] [CrossRef]
- Menk, A.V.; Scharping, N.E.; Rivadeneira, D.B.; Calderon, M.J.; Watson, M.J.; Dunstane, D.; Watkins, S.C.; Delgoffe, G.M. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018, 215, 1091–1100. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Etxeberria, I.; Glez-Vaz, J.; Teijeira, A.; Melero, I. New emerging targets in cancer immunotherapy: CD137/4-1BB costimulatory axis. ESMO Open 2020, 4, e000733. [Google Scholar] [CrossRef]
- Sznol, M.; Hodi, F.S.; Margolin, K.; McDermott, D.F.; Ernstoff, M.S.; Kirkwood, J.M.; Wojtaszek, C.; Feltquate, D.; Logan, T. Phase I study of BMS-663513, a fully human anti-CD137 agonist monoclonal antibody, in patients (pts) with advanced cancer (CA). J. Clin. Oncol. 2008, 26, 3007. [Google Scholar] [CrossRef]
- Segal, N.H.; Gopal, A.K.; Bhatia, S.; Kohrt, H.E.; Levy, R.; Pishvaian, M.J.; Houot, R.; Bartlett, N.; Nghiem, P.; Kronenberg, S.A.; et al. A phase 1 study of PF-05082566 (anti-4-1BB) in patients with advanced cancer. J. Clin. Oncol. 2014, 32, 3007. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Sznol, M.; Hu-Lieskovan, S.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Di Gravio, D.; Huang, B.; Gambhire, D.; Chen, Y.; et al. Phase Ib Study of Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Combination with Pembrolizumab (MK-3475) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5349–5357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.; Chui, S.Y.; Emens, L.A. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. Reply. N. Engl. J. Med. 2019, 380, 987–988. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulovic, S.; Demey, W.; Ullen, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef]
- Wang, X.; Yang, X.; Zhang, C.; Wang, Y.; Cheng, T.; Duan, L.; Tong, Z.; Tan, S.; Zhang, H.; Saw, P.E.; et al. Tumor cell-intrinsic PD-1 receptor is a tumor suppressor and mediates resistance to PD-1 blockade therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 6640–6650. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Glunde, K.; Raman, V.; Mori, N.; Bhujwalla, Z.M. RNA interference-mediated choline kinase suppression in breast cancer cells induces differentiation and reduces proliferation. Cancer Res. 2005, 65, 11034–11043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco-Torres, J.; Penet, M.F.; Mironchik, Y.; Krishnamachary, B.; Bhujwalla, Z.M. The PD-L1 metabolic interactome intersects with choline metabolism and inflammation. Cancer Metab. 2021, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Talty, R.; Bosenberg, M. The role of ferroptosis in melanoma. Pigment. Cell Melanoma Res. 2021, 35, 18–25. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.W.; You, Y.; Hsu, J.M.; Nie, L.; Chen, Y.; Wang, Y.C.; Liu, C.; et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020, 22, 1264–1275. [Google Scholar] [CrossRef]
- Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020, 369, 1633–1637. [Google Scholar] [CrossRef]
- Feng, J.; Yang, H.; Zhang, Y.; Wei, H.; Zhu, Z.; Zhu, B.; Yang, M.; Cao, W.; Wang, L.; Wu, Z. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene 2017, 36, 5829–5839. [Google Scholar] [CrossRef]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.I.; Celis, E.; Lennox, B.; et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Al-Khami, A.A.; Zheng, L.; Del Valle, L.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 2017, 6, e1344804. [Google Scholar] [CrossRef]
- Henrich, F.C.; Singer, K.; Poller, K.; Bernhardt, L.; Strobl, C.D.; Limm, K.; Ritter, A.P.; Gottfried, E.; Volkl, S.; Jacobs, B.; et al. Suppressive effects of tumor cell-derived 5’-deoxy-5’-methylthioadenosine on human T cells. Oncoimmunology 2016, 5, e1184802. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Bu, X.; Gao, Y.; Guo, J.; Hu, J.; Jiang, C.; Zhang, Z.; Xu, K.; Duan, J.; He, S.; et al. Energy status dictates PD-L1 protein abundance and anti-tumor immunity to enable checkpoint blockade. Mol. Cell 2021, 81, 2317–2331.e6. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Liang, Y.; Chen, Y.; Wang, L.; Li, D.; Liang, Z.; Wang, X.; Tian, D.; Yang, X.; Niu, H. Glutamine Deprivation Induces PD-L1 Expression via Activation of EGFR/ERK/c-Jun Signaling in Renal Cancer. Mol. Cancer Res. 2020, 18, 324–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cioccoloni, G.; Aquino, A.; Notarnicola, M.; Caruso, M.G.; Bonmassar, E.; Zonfrillo, M.; Caporali, S.; Faraoni, I.; Villiva, C.; Fuggetta, M.P.; et al. Fatty acid synthase inhibitor orlistat impairs cell growth and down-regulates PD-L1 expression of a human T-cell leukemia line. J. Chemother 2020, 32, 30–40. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Lartigau, E.; Randrianarivelo, H.; Avril, M.F.; Margulis, A.; Spatz, A.; Eschwege, F.; Guichard, M. Intratumoral oxygen tension in metastatic melanoma. Melanoma Res. 1997, 7, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Sivridis, E.; Kouskoukis, C.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Hypoxia-inducible factors 1alpha and 2alpha are related to vascular endothelial growth factor expression and a poorer prognosis in nodular malignant melanomas of the skin. Melanoma Res. 2003, 13, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Dales, J.P.; Beaufils, N.; Silvy, M.; Picard, C.; Pauly, V.; Pradel, V.; Formisano-Treziny, C.; Bonnier, P.; Giusiano, S.; Charpin, C.; et al. Hypoxia inducible factor 1alpha gene (HIF-1alpha) splice variants: Potential prognostic biomarkers in breast cancer. BMC Med. 2010, 8, 44. [Google Scholar] [CrossRef] [Green Version]
- Schindl, M.; Schoppmann, S.F.; Samonigg, H.; Hausmaninger, H.; Kwasny, W.; Gnant, M.; Jakesz, R.; Kubista, E.; Birner, P.; Oberhuber, G.; et al. Overexpression of hypoxia-inducible factor 1alpha is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clin. Cancer Res. 2002, 8, 1831–1837. [Google Scholar]
- Neumann, A.K.; Yang, J.; Biju, M.P.; Joseph, S.K.; Johnson, R.S.; Haase, V.H.; Freedman, B.D.; Turka, L.A. Hypoxia inducible factor 1 alpha regulates T cell receptor signal transduction. Proc. Natl. Acad. Sci. USA 2005, 102, 17071–17076. [Google Scholar] [CrossRef] [Green Version]
- Robbins, J.R.; Lee, S.M.; Filipovich, A.H.; Szigligeti, P.; Neumeier, L.; Petrovic, M.; Conforti, L. Hypoxia modulates early events in T cell receptor-mediated activation in human T lymphocytes via Kv1.3 channels. J. Physiol. 2005, 564, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Conforti, L.; Petrovic, M.; Mohammad, D.; Lee, S.; Ma, Q.; Barone, S.; Filipovich, A.H. Hypoxia regulates expression and activity of Kv1.3 channels in T lymphocytes: A possible role in T cell proliferation. J. Immunol. 2003, 170, 695–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, C.C.; Kojima, H.; Lukashev, D.; Armstrong, J.; Farber, M.; Apasov, S.G.; Sitkovsky, M.V. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J. Immunol. 2001, 167, 6140–6149. [Google Scholar] [CrossRef]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Phillips, J.B.; Da Silva, L.M.; Zhou, M.; Fodstad, O.; Owen, L.B.; Tan, M. Interplay between Immune Checkpoint Proteins and Cellular Metabolism. Cancer Res. 2017, 77, 1245–1249. [Google Scholar] [CrossRef] [Green Version]
- Malczewski, A.B.; Ketheesan, N.; Coward, J.I.G.; Navarro, S. Enhancing Checkpoint Inhibitor Therapy in Solid Tissue Cancers: The Role of Diet, the Microbiome & Microbiome-Derived Metabolites. Front. Immunol. 2021, 12, 624434. [Google Scholar] [CrossRef]
- Lussier, D.M.; Woolf, E.C.; Johnson, J.L.; Brooks, K.S.; Blattman, J.N.; Scheck, A.C. Enhanced immunity in a mouse model of malignant glioma is mediated by a therapeutic ketogenic diet. BMC Cancer 2016, 16, 310. [Google Scholar] [CrossRef]
- Ferrere, G.; Tidjani Alou, M.; Liu, P.; Goubet, A.G.; Fidelle, M.; Kepp, O.; Durand, S.; Iebba, V.; Fluckiger, A.; Daillere, R.; et al. Ketogenic diet and ketone bodies enhance the anticancer effects of PD-1 blockade. JCI Insight 2021, 6, e145207. [Google Scholar] [CrossRef]
- Eriau, E.; Paillet, J.; Kroemer, G.; Pol, J.G. Metabolic Reprogramming by Reduced Calorie Intake or Pharmacological Caloric Restriction Mimetics for Improved Cancer Immunotherapy. Cancers 2021, 13, 1260. [Google Scholar] [CrossRef]
- Pietrocola, F.; Kroemer, G. Caloric restriction promotes the stemness and antitumor activity of T lymphocytes. Oncoimmunology 2019, 8, e1616153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bays, H.E. Adiposopathy is “sick fat” a cardiovascular disease? J. Am. Coll. Cardiol. 2011, 57, 2461–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Fuster, J.J.; Walsh, K. Adipokines: A link between obesity and cardiovascular disease. J. Cardiol. 2014, 63, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Zorena, K.; Jachimowicz-Duda, O.; Ślęzak, D.; Robakowska, M.; Mrugacz, M. Adipokines and Obesity. Potential Link to Metabolic Disorders and Chronic Complications. Int. J. Mol. Sci. 2020, 21, 3570. [Google Scholar] [CrossRef]
- Pingili, A.K.; Chaib, M.; Sipe, L.M.; Miller, E.J.; Teng, B.; Sharma, R.; Yarbro, J.R.; Asemota, S.; Al Abdallah, Q.; Mims, T.S.; et al. Immune checkpoint blockade reprograms systemic immune landscape and tumor microenvironment in obesity-associated breast cancer. Cell Rep. 2021, 35, 109285. [Google Scholar] [CrossRef] [PubMed]
- Spyrou, N.; Vallianou, N.; Kadillari, J.; Dalamaga, M. The interplay of obesity, gut microbiome and diet in the immune check point inhibitors therapy era. Semin Cancer Biol. 2021, 73, 356–376. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia 2017, 19, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.; Cheng, S.; Kou, Y.; Wang, Z.; Jin, R.; Hu, H.; Zhang, X.; Gong, J.F.; Li, J.; Lu, M.; et al. The Gut Microbiome Is Associated with Clinical Response to Anti-PD-1/PD-L1 Immunotherapy in Gastrointestinal Cancer. Cancer Immunol. Res. 2020, 8, 1251–1261. [Google Scholar] [CrossRef]
- Yu, S.; Wu, X.; Shi, Z.; Huynh, M.; Jena, P.K.; Sheng, L.; Zhou, Y.; Han, D.; Wan, Y.Y.; Hwang, S.T. Diet-induced obesity exacerbates imiquimod-mediated psoriasiform dermatitis in anti-PD-1 antibody-treated mice: Implications for patients being treated with checkpoint inhibitors for cancer. J. Dermatol. Sci. 2020, 97, 194–200. [Google Scholar] [CrossRef]
- Jin, Y.; Dong, H.; Xia, L.; Yang, Y.; Zhu, Y.; Shen, Y.; Zheng, H.; Yao, C.; Wang, Y.; Lu, S. The Diversity of Gut Microbiome is Associated With Favorable Responses to Anti-Programmed Death 1 Immunotherapy in Chinese Patients With NSCLC. J. Thorac. Oncol. 2019, 14, 1378–1389. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Sundrud, M.S.; Skepner, J.; Yamagata, T. Targeting Th17 cells in autoimmune diseases. Trends Pharmacol. Sci. 2014, 35, 493–500. [Google Scholar] [CrossRef]
- Gargaro, M.; Manni, G.; Scalisi, G.; Puccetti, P.; Fallarino, F. Tryptophan Metabolites at the Crossroad of Immune-Cell Interaction via the Aryl Hydrocarbon Receptor: Implications for Tumor Immunotherapy. Int. J. Mol. Sci. 2021, 22, 4644. [Google Scholar] [CrossRef]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [Green Version]
- Muhammad Yusoff, F.; Wong, K.K.; Mohd Redzwan, N. Th1, Th2, and Th17 cytokines in systemic lupus erythematosus. Autoimmunity 2020, 53, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Huang, L.; Lv, P.; Li, X.; Liu, G.; Chen, Y.; Wang, Z.; Qian, X.; Shen, Y.; Li, Y.; et al. The role of Th17 cells in psoriasis. Immunol. Res. 2020, 68, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Matteson, E.L.; Goronzy, J.J.; Weyand, C.M. T-cell metabolism in autoimmune disease. Arthritis Res. Ther. 2015, 17, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shyer, J.A.; Flavell, R.A.; Bailis, W. Metabolic signaling in T cells. Cell Res. 2020, 30, 649–659. [Google Scholar] [CrossRef]
- Guma, M.; Tiziani, S.; Firestein, G.S. Metabolomics in rheumatic diseases: Desperately seeking biomarkers. Nat. Rev. Rheumatol. 2016, 12, 269–281. [Google Scholar] [CrossRef]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Piotrowski, J.T.; Gomez, T.S.; Schoon, R.A.; Mangalam, A.K.; Billadeau, D.D. WASH knockout T cells demonstrate defective receptor trafficking, proliferation, and effector function. Mol. Cell Biol. 2013, 33, 958–973. [Google Scholar] [CrossRef] [Green Version]
- Caza, T.N.; Talaber, G.; Perl, A. Metabolic regulation of organelle homeostasis in lupus T cells. Clin. Immunol. 2012, 144, 200–213. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Xie, C.; Han, J.; Ye, Y.; Weiel, J.; Li, Q.; Blanco, I.; Ahn, C.; Olsen, N.; Putterman, C.; et al. Metabolic disturbances associated with systemic lupus erythematosus. PLoS ONE 2012, 7, e37210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, P.; Anthony, D.C. Metabolomics in multiple sclerosis disease course and progression. Mult. Scler. 2020, 26, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Tsoukalas, D.; Fragoulakis, V.; Sarandi, E.; Docea, A.O.; Papakonstaninou, E.; Tsilimidos, G.; Anamaterou, C.; Fragkiadaki, P.; Aschner, M.; Tsatsakis, A.; et al. Targeted Metabolomic Analysis of Serum Fatty Acids for the Prediction of Autoimmune Diseases. Front. Mol. Biosci. 2019, 6, 120. [Google Scholar] [CrossRef] [Green Version]
- Haghikia, A.; Jorg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, H.; Kim, J.; Kim, D.; Lee, J.E.; Chung, Y. Cellular and Molecular Links between Autoimmunity and Lipid Metabolism. Mol. Cells 2019, 42, 747–754. [Google Scholar] [CrossRef]
- Yim, C.; Mansell, K.; Hussein, N.; Arnason, T. Current cancer therapies and their influence on glucose control. World J. Diabetes 2021, 12, 1010–1025. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Varayathu, H.; Sarathy, V.; Thomas, B.E.; Mufti, S.S.; Naik, R. Combination Strategies to Augment Immune Check Point Inhibitors Efficacy—Implications for Translational Research. Front. Oncol. 2021, 11, 559161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Chen, S.; Li, Z.; Chen, J.; Li, W. The effect of concomitant use of statins, NSAIDs, low-dose aspirin, metformin and beta-blockers on outcomes in patients receiving immune checkpoint inhibitors: A systematic review and meta-analysis. Oncoimmunology 2021, 10, 1957605. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H.; Yang, W.H.; Xia, W.; Wei, Y.; Chan, L.C.; Lim, S.O.; Li, C.W.; Kim, T.; Chang, S.S.; Lee, H.H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.Y.; Wu, C.Y.; Powell, J.D.; Lu, K.L. Manipulation of Metabolic Pathways and Its Consequences for Anti-Tumor Immunity: A Clinical Perspective. Int. J. Mol. Sci. 2020, 21, 4030. [Google Scholar] [CrossRef]
- Kim, Y.; Vagia, E.; Viveiros, P.; Kang, C.Y.; Lee, J.Y.; Gim, G.; Cho, S.; Choi, H.; Kim, L.; Park, I.; et al. Overcoming acquired resistance to PD-1 inhibitor with the addition of metformin in small cell lung cancer (SCLC). Cancer Immunol. Immunother 2021, 70, 961–965. [Google Scholar] [CrossRef]
- Afzal, M.Z.; Mercado, R.R.; Shirai, K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. J. Immunother Cancer 2018, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Veeramachaneni, R.; Yu, W.; Newton, J.M.; Kemnade, J.O.; Skinner, H.D.; Sikora, A.G.; Sandulache, V.C. Metformin generates profound alterations in systemic and tumor immunity with associated antitumor effects. J. Immunother Cancer 2021, 9, e002773. [Google Scholar] [CrossRef]
- Lecciso, M.; Ocadlikova, D.; Sangaletti, S.; Trabanelli, S.; De Marchi, E.; Orioli, E.; Pegoraro, A.; Portararo, P.; Jandus, C.; Bontadini, A.; et al. ATP Release from Chemotherapy-Treated Dying Leukemia Cells Elicits an Immune Suppressive Effect by Increasing Regulatory T Cells and Tolerogenic Dendritic Cells. Front. Immunol. 2017, 8, 1918. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharia, Y.; McWilliams, R.R.; Rixe, O.; Drabick, J.; Shaheen, M.F.; Grossmann, K.F.; Kolhe, R.; Pacholczyk, R.; Sadek, R.; Tennant, L.L.; et al. Phase II trial of the IDO pathway inhibitor indoximod plus pembrolizumab for the treatment of patients with advanced melanoma. J. Immunother Cancer 2021, 9, e002057. [Google Scholar] [CrossRef] [PubMed]
- Coutzac, C.; Jouniaux, J.M.; Paci, A.; Schmidt, J.; Mallardo, D.; Seck, A.; Asvatourian, V.; Cassard, L.; Saulnier, P.; Lacroix, L.; et al. Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat. Commun. 2020, 11, 2168. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stirling, E.R.; Bronson, S.M.; Mackert, J.D.; Cook, K.L.; Triozzi, P.L.; Soto-Pantoja, D.R. Metabolic Implications of Immune Checkpoint Proteins in Cancer. Cells 2022, 11, 179. https://doi.org/10.3390/cells11010179
Stirling ER, Bronson SM, Mackert JD, Cook KL, Triozzi PL, Soto-Pantoja DR. Metabolic Implications of Immune Checkpoint Proteins in Cancer. Cells. 2022; 11(1):179. https://doi.org/10.3390/cells11010179
Chicago/Turabian StyleStirling, Elizabeth R., Steven M. Bronson, Jessica D. Mackert, Katherine L. Cook, Pierre L. Triozzi, and David R. Soto-Pantoja. 2022. "Metabolic Implications of Immune Checkpoint Proteins in Cancer" Cells 11, no. 1: 179. https://doi.org/10.3390/cells11010179
APA StyleStirling, E. R., Bronson, S. M., Mackert, J. D., Cook, K. L., Triozzi, P. L., & Soto-Pantoja, D. R. (2022). Metabolic Implications of Immune Checkpoint Proteins in Cancer. Cells, 11(1), 179. https://doi.org/10.3390/cells11010179