
Discovery and Early Clinical Development of Selective Immunoproteasome Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Anti-Inflammatory Activity of First-Generation Proteasome Inhibitors
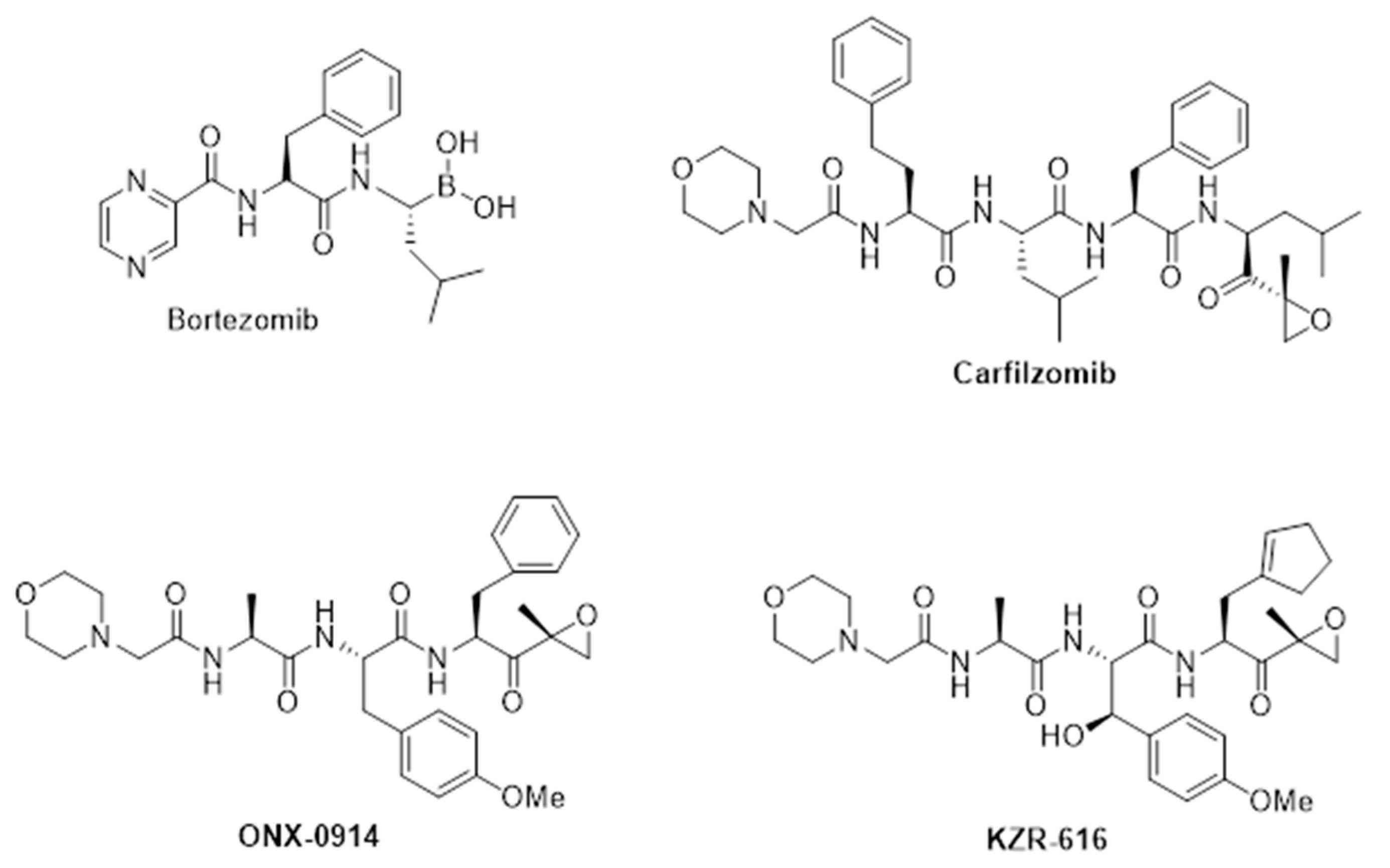
3. Therapeutic Potential of Selective Immunoproteasome Inhibitors
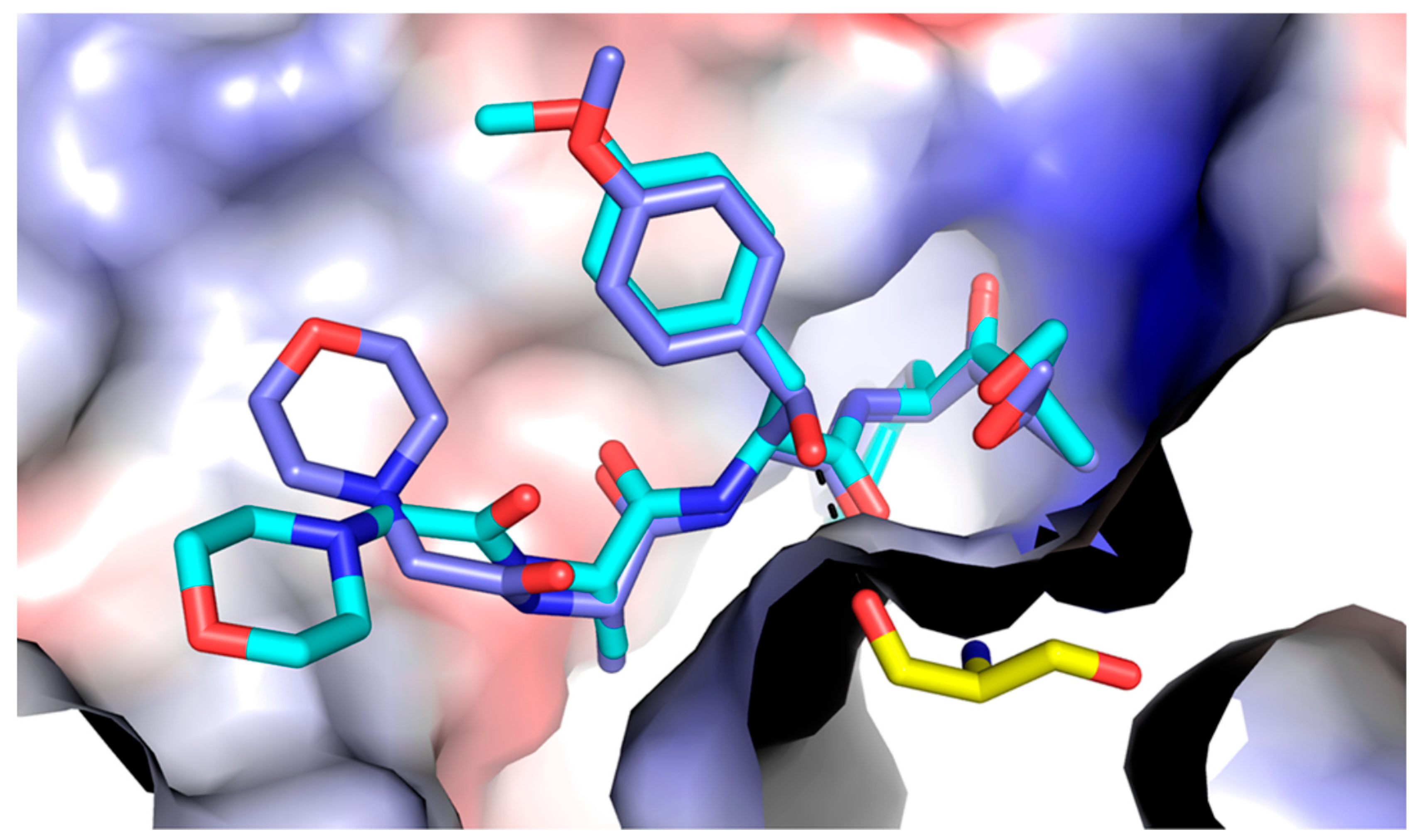
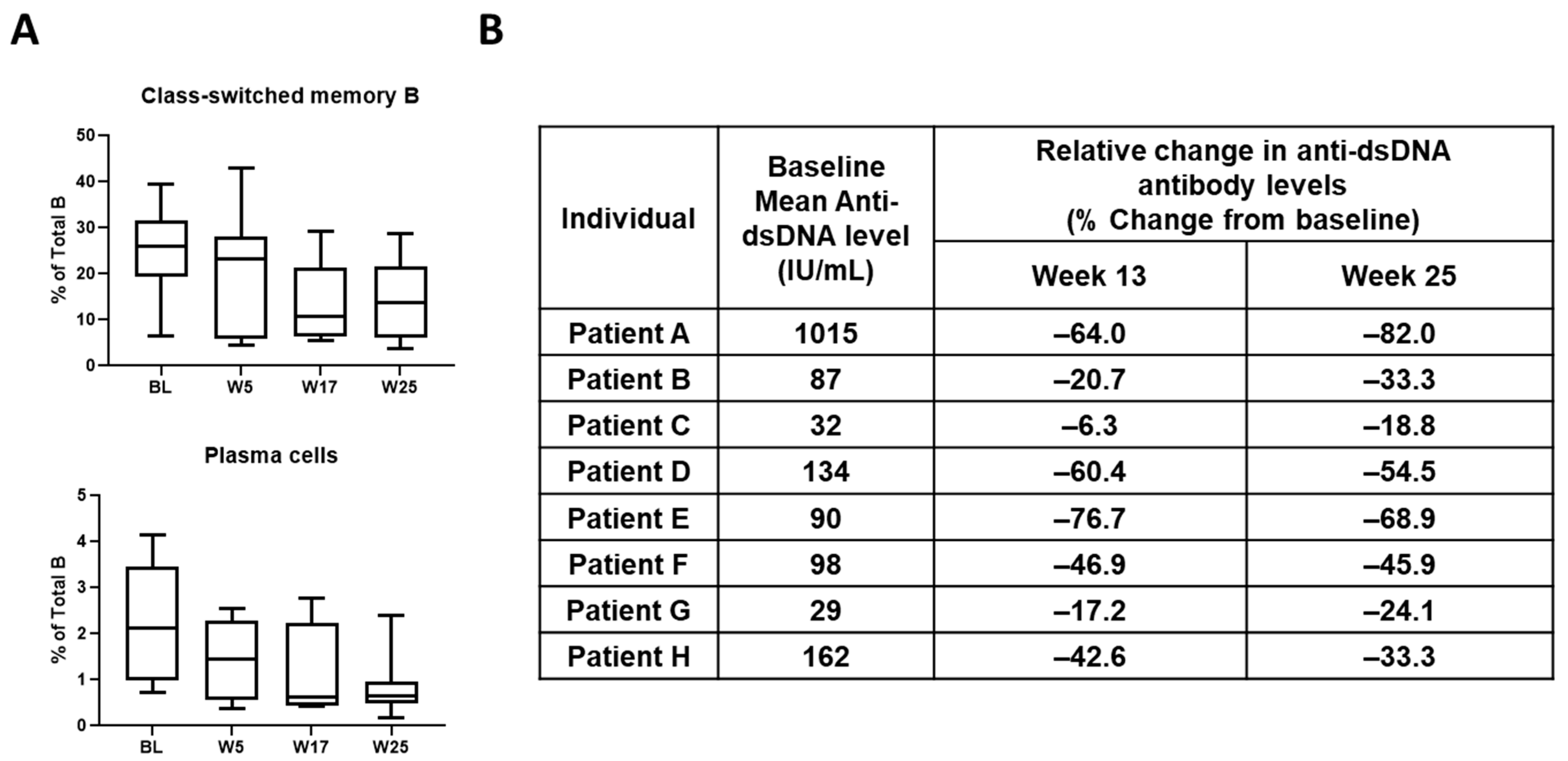
4. Discovery and Early Development of KZR-616
5. Discussion
Funding
Conflicts of Interest
References
- Ciechanover, A. Intracellular Protein Degradation: From a Vague Idea Thru the Lysosome and the Ubiquitin-Proteasome System and onto Human Diseases and Drug Targeting. Cell Death Differ. 2005, 12, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.F.; Kane, R.; Farrell, A.T.; Abraham, S.; Benson, K.; Brower, M.E.; Bradley, S.; Gobburu, J.V.; Goheer, A.; Lee, S.-L.; et al. Approval Summary for Bortezomib for Injection in the Treatment of Multiple Myeloma. Clin. Cancer Res. 2004, 10, 3954–3964. [Google Scholar] [CrossRef] [Green Version]
- Robak, P.; Robak, T. Bortezomib for the Treatment of Hematologic Malignancies: 15 Years Later. Drugs R D 2019, 19, 73–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chim, C.S.; Kumar, S.K.; Orlowski, R.Z.; Cook, G.; Richardson, P.G.; Gertz, M.A.; Giralt, S.; Mateos, M.v.; Leleu, X.; Anderson, K.C. Management of Relapsed and Refractory Multiple Myeloma: Novel Agents, Antibodies, Immunotherapies and Beyond. Leukemia 2018, 32, 252–262. [Google Scholar] [CrossRef]
- Goetzke, C.C.; Ebstein, F.; Kallinich, T. Role of Proteasomes in Inflammation. J. Clin. Med. 2021, 10, 1783. [Google Scholar] [CrossRef]
- Wu, J. On the Role of Proteasomes in Cell Biology and Proteasome Inhibition as a Novel Frontier in the Development of Immunosuppressants. Am. J. Transplant. 2002, 2, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Palombella, V.J.; Rando, O.J.; Goldberg, A.L.; Maniatis, T. The Ubiquitin-Proteasome Pathway Is Required for Processing the NF-Kappa B1 Precursor Protein and the Activation of NF-Kappa B. Cell 1994, 78, 773–785. [Google Scholar] [CrossRef]
- King, R.W.; Peters, J.-M.; Tugendreich, S.; Rolfe, M.; Hieter, P.; Kirschnert, M.W. A 20S Complex Containing CDC27 and CDC16 Catalyzes the Mitosis-Specific Conjugation of Ubiquitin to Cyclin B. Cell 1995, 81, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Adams, J. The Proteasome: A Suitable Antineoplastic Target. Nat. Rev. Cancer 2004, 4, 349–360. [Google Scholar] [CrossRef]
- Everly, M.J.; Everly, J.J.; Susskind, B.; Brailey, P.; Arend, L.J.; Alloway, R.R.; Roy-Chaudhury, P.; Govil, A.; Mogilishetty, G.; Rike, A.H.; et al. Bortezomib Provides Effective Therapy for Antibody- and Cell-Mediated Acute Rejection. Transplantation 2008, 86, 1754–1761. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, K.; Holle, J.U.; Aries, P.M.; Gross, W.L.; Moosig, F. Successful Use of Bortezomib in a Patient with Systemic Lupus Erythematosus and Multiple Myeloma. Ann. Rheum. Dis. 2011, 70, 1344–1345. [Google Scholar] [CrossRef] [PubMed]
- Neubert, K.; Meister, S.; Moser, K.; Weisel, F.; Maseda, D.; Amann, K.; Wiethe, C.; Winkler, T.H.; Kalden, J.R.; Manz, R.A.; et al. The Proteasome Inhibitor Bortezomib Depletes Plasma Cells and Protects Mice with Lupus-like Disease from Nephritis. Nat. Med. 2008, 14, 748–755. [Google Scholar] [CrossRef]
- Alexander, T.; Sarfert, R.; Klotsche, J.; Kühl, A.A.; Rubbert-Roth, A.; Lorenz, H.M.; Rech, J.; Hoyer, B.F.; Cheng, Q.; Waka, A.; et al. The Proteasome Inhibitior Bortezomib Depletes Plasma Cells and Ameliorates Clinical Manifestations of Refractory Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2015, 74, 1474–1478. [Google Scholar] [CrossRef]
- Alexander, T.; Cheng, Q.; Klotsche, J.; Khodadadi, L.; Waka, A.; Biesen, R.; Hoyer, B.F.; Burmester, G.R.; Radbruch, A.; Hiepe, F. Proteasome Inhibition with Bortezomib Induces a Therapeutically Relevant Depletion of Plasma Cells in SLE but Does Not Target Their Precursors. Eur. J. Immunol. 2018, 48, 1573–1579. [Google Scholar] [CrossRef] [Green Version]
- de Groot, K.A.; Tsang Sjoe, M.; Niewerth, D.; Cloos, J.; Blank, J.L.; M Niessen, H.W.; Zweegman, S.; Voskuyl, A.E.; Jansen, G.; van der Heijden, J.W. Pharmacodynamic Monitoring of (Immuno)Proteasome Inhibition during Bortezomib Treatment of a Critically Ill Patient with Lupus Nephritis and Myocarditis. Lupus Sci. Med. 2015, 2, e000121. [Google Scholar] [CrossRef] [Green Version]
- Segarra, A.; Arredondo, K.V.; Jaramillo, J.; Jatem, E.; Salcedo, M.T.; Agraz, I.; Ramos, N.; Carnicer, C.; Valtierra, N.; Ostos, E. Efficacy and Safety of Bortezomib in Refractory Lupus Nephritis: A Single-Center Experience. Lupus 2020, 29, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Z.; Huang, L.; Hou, J.; Zhou, M.; Huang, X.; Hu, W.; Liu, Z. The Short-Term Efficacy of Bortezomib Combined with Glucocorticoids for the Treatment of Refractory Lupus Nephritis. Lupus 2017, 26, 952–958. [Google Scholar] [CrossRef]
- Khandelwal, P.; Davies, S.M.; Grimley, M.S.; Jordan, M.B.; Curtis, B.R.; Jodele, S.; Marsh, R.; Filipovich, A.J. Bortezomib for Refractory Autoimmunity in Pediatrics. Biol. Blood Marrow Transplant. 2014, 20, 1641–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartono, C.; Chung, M.; Perlman, A.S.; Chevalier, J.M.; Serur, D.; Seshan, S.V.; Muthukumar, T. Bortezomib for Reduction of Proteinuria in IgA Nephropathy. Kidney Int. Rep. 2018, 3, 861–866. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Tian, D.C.; Yang, C.S.; Han, B.; Wang, J.; Yang, L.; Shi, F.D. Safety and Efficacy of Bortezomib in Patients with Highly Relapsing Neuromyelitis Optica Spectrum Disorder. JAMA Neurol. 2017, 74, 1010–1012. [Google Scholar] [CrossRef]
- Richardson, P.G.; Briemberg, H.; Jagannath, S.; Wen, P.Y.; Barlogie, B.; Berenson, J.; Singhal, S.; Siegel, D.S.; Irwin, D.; Schuster, M.; et al. Frequency, Characteristics, and Reversibility of Peripheral Neuropathy during Treatment of Advanced Multiple Myeloma with Bortezomib. J. Clin. Oncol. 2006, 24, 3113–3120. [Google Scholar] [CrossRef]
- Ishii, T.; Tanaka, Y.; Kawakami, A.; Saito, K.; Ichinose, K.; Fujii, H.; Shirota, Y.; Shirai, T.; Fujita, Y.; Watanabe, R.; et al. Multicenter Double-Blind Randomized Controlled Trial to Evaluate the Effectiveness and Safety of Bortezomib as a Treatment for Refractory Systemic Lupus Erythematosus. Mod. Rheumatol. 2018, 28, 986–992. [Google Scholar] [CrossRef]
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome Inhibitors: An Expanding Army Attacking a Unique Target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Parlati, F.; Lee, S.J.; Aujay, M.; Suzuki, E.; Levitsky, K.; Lorens, J.B.; Micklem, D.R.; Ruurs, P.; Sylvain, C.; Lu, Y.; et al. Carfilzomib Can Induce Tumor Cell Death through Selective Inhibition of the Chymotrypsin-like Activity of the Proteasome. Blood 2009, 114, 3439–3447. [Google Scholar] [CrossRef] [Green Version]
- Niewerth, D.; Franke, N.E.; Jansen, G.; Assaraf, Y.G.; van Meerloo, J.; Kirk, C.J.; Degenhardt, J.; Anderl, J.; Schimmer, A.D.; Zweegman, S.; et al. Higher Ratio Immune versus Constitutive Proteasome Level as Novel Indicator of Sensitivity of Pediatric Acute Leukemia Cells to Proteasome Inhibitors. Haematologica 2013, 98, 1896–1904. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.W.; Downey-Kopyscinski, S.L.; Fields, J.L.; Rahme, G.J.; Colley, W.C.; Israel, M.A.; Maksimenko, A.V.; Fiering, S.N.; Kisselev, A.F. Activity of Immunoproteasome Inhibitor ONX-0914 in Acute Lymphoblastic Leukemia Expressing MLL–AF4 Fusion Protein. Sci. Rep. 2021, 11, 10883. [Google Scholar] [CrossRef] [PubMed]
- Tubío-Santamaría, N.; Ebstein, F.; Heidel, F.H.; Krüger, E. Immunoproteasome Function in Normal and Malignant Hematopoiesis. Cells 2021, 10, 1577. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Busch, M.; Friese-Hamim, M.; Crosignani, S.; Fuchss, T.; Musil, D.; Rohdich, F.; Sanderson, M.P.; Seenisamy, J.; Walter-Bausch, G.; et al. Structure-Based Optimization and Discovery of M3258, a Specific Inhibitor of the Immunoproteasome Subunit LMP7 (Β5i). J. Med. Chem. 2021, 64, 10230–10245. [Google Scholar] [CrossRef]
- Sanderson, M.P.; Friese-Hamim, M.; Walter-Bausch, G.; Busch, M.; Gaus, S.; Musil, D.; Rohdich, F.; Zanelli, U.; Downey-Kopyscinski, S.L.; Mitsiades, C.S.; et al. M3258 Is a Selective Inhibitor of the Immunoproteasome Subunit LMP7 (B5i) Delivering Efficacy in Multiple Myeloma Models. Mol. Cancer Ther. 2021, 20, 1378–1388. [Google Scholar] [CrossRef]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A Selective Inhibitor of the Immunoproteasome Subunit LMP7 Blocks Cytokine Production and Attenuates Progression of Experimental Arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and Constitutive Proteasome Crystal Structures Reveal Differences in Substrate and Inhibitor Specificity. Cell 2012, 148, 727–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.J.; Aujay, M.A.; Bennett, M.K.; Dajee, M.; Demo, S.D.; Fang, Y.; Ho, M.N.; Jiang, J.; Kirk, C.J.; Laidig, G.J.; et al. Design and Synthesis of an Orally Bioavailable and Selective Peptide Epoxyketone Proteasome Inhibitor (PR-047). J. Med. Chem. 2009, 52, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
- Mundt, S.; Engelhardt, B.; Kirk, C.J.; Groettrup, M.; Basler, M. Inhibition and Deficiency of the Immunoproteasome Subunit LMP7 Attenuates LCMV-Induced Meningitis. Eur. J. Immunol. 2014, 6, 226–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Althof, N.; Goetzke, C.C.; Kespohl, M.; Voss, K.; Heuser, A.; Pinkert, S.; Kaya, Z.; Klingel, K.; Beling, A. The Immunoproteasome-specific Inhibitor ONX 0914 Reverses Susceptibility to Acute Viral Myocarditis. EMBO Mol. Med. 2018, 10, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Basler, M.; Alvarez, G.; Brunner, T.; Kirk, C.J.; Groettrup, M. Immunoproteasome Inhibition Prevents Chronic Antibody-Mediated Allograft Rejection in Renal Transplantation. Kidney Int. 2018, 93, 670–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Koerner, J.; Basler, M.; Brunner, T.; Kirk, C.J.; Groettrup, M. Immunoproteasome Inhibition Induces Plasma Cell Apoptosis and Preserves Kidney Allografts by Activating the Unfolded Protein Response and Suppressing Plasma Cell Survival Factors. Kidney Int. 2019, 95, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Kalim, K.W.; Basler, M.; Kirk, C.J.; Groettrup, M. Immunoproteasome Subunit LMP7 Deficiency and Inhibition Suppresses Th1 and Th17 but Enhances Regulatory T Cell Differentiation. J. Immunol. 2012, 189, 4182–4193. [Google Scholar] [CrossRef]
- Ichikawa, H.T.; Conley, T.; Muchamuel, T.; Jiang, J.; Lee, S.; Owen, T.; Barnard, J.; Nevarez, S.; Goldman, B.I.; Kirk, C.J.; et al. Beneficial Effect of Novel Proteasome Inhibitors in Murine Lupus via Dual Inhibition of Type i Interferon and Autoantibody-Secreting Cells. Arthritis Rheum. 2012, 64, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Beck, U.; Kirk, C.J.; Groettrup, M. The Antiviral Immune Response in Mice Devoid of Immunoproteasome Activity. J. Immunol. 2011, 187, 5548–5557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, H.W.B.; Lowe, E.; Anderl, J.L.; Fan, A.; Muchamuel, T.; Bowers, S.; Moebius, D.C.; Kirk, C.; McMinn, D.L. Required Immunoproteasome Subunit Inhibition Profile for Anti-Inflammatory Efficacy and Clinical Candidate KZR-616 ((2 S,3 R)- N-((S)-3-(Cyclopent-1-En-1-Yl)-1-((R)-2-Methyloxiran-2-Yl)-1-Oxopropan-2-Yl)-3-Hydroxy-3-(4-Methoxyphenyl)-2-((S)-2-(2-Morpholinoacetamido)Propanamido)Propenamide). J. Med. Chem. 2018, 61, 11127–11143. [Google Scholar] [CrossRef]
- Basler, M.; Lindstrom, M.M.; LaStant, J.J.; Bradshaw, J.M.; Owens, T.D.; Schmidt, C.; Maurits, E.; Tsu, C.; Overkleeft, H.S.; Kirk, C.J.; et al. Co-inhibition of Immunoproteasome Subunits LMP2 and LMP7 Is Required to Block Autoimmunity. EMBO Rep. 2018, 19, e46512. [Google Scholar] [CrossRef]
- Huber, E.M.; Groll, M. A Nut for Every Bolt: Subunit-Selective Inhibitors of the Immunoproteasome and Their Therapeutic Potential. Cells 2021, 10, 1929. [Google Scholar] [CrossRef]
- Arastu-Kapur, S.; Anderl, J.L.; Kraus, M.; Parlati, F.; Shenk, K.D.; Lee, S.J.; Muchamuel, T.; Bennett, M.K.; Driessen, C.; Ball, A.J.; et al. Nonproteasomal Targets of the Proteasome Inhibitors Bortezomib and Carfilzomib: A Link to Clinical Adverse Events. Clin. Cancer Res. 2011, 17, 2734–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federspiel, J.D.; Codreanu, S.G.; Goyal, S.; Albertolle, M.E.; Lowe, E.; Teague, J.; Wong, H.; Guengerich, F.P.; Liebler, D.C. Protein Adduction by Carfilzomib Specificity of Protein Covalent Modification by the Electrophilic Proteasome Inhibitor Carfilzomib in Human Cells. Mol. Cell. Proteom. 2016, 15, 3233–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Johnson, H.; Anderl, J.L.; Muchamuel, T.; McMinn, D.; Morisseau, C.; Hammock, B.D.; Kirk, C.; Wang, J. Role of Epoxide Hydrolases and Cytochrome P450s on Metabolism of KZR-616, a First-in-Class Selective Inhibitor of the Immunoproteasome. Drug Metab. Dispos. 2021, 49, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Strelow, J.M. A Perspective on the Kinetics of Covalent and Irreversible Inhibition. J. Biomol. Screen. 2017, 22, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The Resurgence of Covalent Drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Quach, H.; White, D.; Spencer, A.; Ho, P.J.; Bhutani, D.; White, M.; Inamdar, S.; Morris, C.; Ou, Y.; Gyger, M. Pharmacokinetics and Safety of Carfilzomib in Patients with Relapsed Multiple Myeloma and End-Stage Renal Disease (ESRD): An Open-Label, Single-Arm, Phase I Study. Cancer Chemother. Pharmacol. 2017, 79, 1067–1076. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yang, J.; Kirk, C.; Fang, Y.; Alsina, M.; Badros, A.; Papadopoulos, K.; Wong, A.; Woo, T.; Bomba, D.; et al. Clinical Pharmacokinetics, Metabolism, and Drug-Drug Interaction of Carfilzomib. Drug Metab. Dispos. 2013, 41, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Fang, Y.; Fan, R.A.; Kirk, C.J. Proteasome Inhibitors and Their Pharmacokinetics, Pharmacodynamics, and Metabolism. Int. J. Mol. Sci. 2021, 22, 11595. [Google Scholar] [CrossRef]
- Lickliter, J.; Anderl, J.; Kirk, C.J.; Want, J.; Bomba, D. KZR-616, a Selective Inhibitor of the Immunoproteasome, Shows a Promising Safety and Target Inhibition Profile in a Phase I, Double-Blind, Single (SAD) and Multiple Ascending Dose (MAD) Study in Healthy Volunteers. Arthritis Rheumatol. 2017, 1413–1414. [Google Scholar] [CrossRef]
- Lee, S.J.; Levitsky, K.; Parlati, F.; Bennett, M.K.; Arastu-Kapur, S.; Kellerman, L.; Woo, T.F.; Wong, A.F.; Papadopoulos, K.P.; Niesvizky, R.; et al. Clinical Activity of Carfilzomib Correlates with Inhibition of Multiple Proteasome Subunits: Application of a Novel Pharmacodynamic Assay. Br. J. Haematol. 2016, 173, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Waller, E.K.; Richardson, P.G.; Jagannath, S.; Orlowski, R.Z.; Giver, C.R.; Jaye, D.L.; Francis, D.; Giusti, S.; Torre, C.; et al. Risk Factors and Kinetics of Thrombocytopenia Associated with Bortezomib for Relapsed, Refractory Multiple Myeloma. Blood 2005, 106, 3777–3784. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Pylypenko, H.; Grosicki, S.; Karamanesht, I.; Leleu, X.; Grishunina, M.; Rekhtman, G.; Masliak, Z.; Robak, T.; Shubina, A.; et al. Subcutaneous versus Intravenous Administration of Bortezomib in Patients with Relapsed Multiple Myeloma: A Randomised, Phase 3, Non-Inferiority Study. Lancet Oncol. 2011, 12, 431–471. [Google Scholar] [CrossRef]
- Muchamuel, T.; Anderl, J.; Fan, R.A.; Johnson, H.; McMinn, D.; Kirk, C. Selective Inhibition of the Immunoproteasome with KZR-616 Blocks Multiple Cell Signaling Pathways, Plasma Cell Signatures and Myeloid Cell Associated Damage in the NZB/W Lupus Nephritis Model. Arthritis Rheumatol. 2019. Available online: https://www.kezarlifesciences.com/our-science/medical-scientific-presentations/detail/6183/selective-inhibition-of-the-immunoproteasome-with-kzr-616-blocks-multiple-cell-signaling-pathways-plasma-cell-signatures-and-myeloid-cell-associated-damage-in-the-nzbw-lupus-nephritis-model (accessed on 11 December 2021).
- Furie, R.; Parikh, S.; Wang, J.; Bomba, D.; Leff, R.; Kirk, C.; Henig, N. KZR-616, A Selective Immunoproteasome Inhibitor for the Treatment of Systemic Lupus Erythematosus: Results from the Completed Dose Escalation Phase 1b Portion of the MISSION Study. Ann. Rheum. Dis. 2021, 80, 595. [Google Scholar] [CrossRef]
- Fan, R.A.; Anderl, J.; Tuch, B.; Bomba, D.; Goel, N.; Kirk, C. Profiling of Gene Expression, Immune Cell Subtypes, and Circulating Protein Biomarkers in Systemic Lupus Erythematosus Patients Treated with the Selective Immunoproteasome Inhibitor, KZR-616. Arthritis Rheumatol. 2019. Available online: https://acrabstracts.org/abstract/profiling-of-gene-expression-immune-cell-subtypes-and-circulating-protein-biomarkers-in-systemic-lupus-erythematosus-patients-treated-with-the-selective-immunoproteasome-inhibitor-kzr-616/ (accessed on 11 December 2021).
- del Rio Oliva, M.; Basler, M.; Bomba, D.; Lam, D.; Brandl, J.; Kirk, C.; Groettrup, M. KZR-616, a First-in-Class Selective Inhibitor of the Immunoproteasome, Ameliorates Polymyositis in a Murine Model. Arthritis Rheumatol. 2020. Available online: https://acrabstracts.org/abstract/kzr-616-a-first-in-class-selective-inhibitor-of-the-immunoproteasome-ameliorates-polymyositis-in-a-murine-model/ (accessed on 11 December 2021).
- Karreci, E.S.; Fan, H.; Uehara, M.; Mihali, A.B.; Singh, P.K.; Kurdi, A.T.; Solhjou, Z.; Riella, L.V.; Ghobrial, I.; Laragione, T.; et al. Brief Treatment with a Highly Selective Immunoproteasome Inhibitor Promotes Long-Term Cardiac Allograft Acceptance in Mice. Proc. Natl. Acad. Sci. USA. 2016, 113, E8425–E8432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, H.; Angelo, N.G.; Warren, J.D.; Nathan, C.F.; Lin, G. Oxathiazolones Selectively Inhibit the Human Immunoproteasome over the Constitutive Proteasome. ACS Med. Chem. Lett. 2014, 5, 405–410. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirk, C.J.; Muchamuel, T.; Wang, J.; Fan, R.A. Discovery and Early Clinical Development of Selective Immunoproteasome Inhibitors. Cells 2022, 11, 9. https://doi.org/10.3390/cells11010009
Kirk CJ, Muchamuel T, Wang J, Fan RA. Discovery and Early Clinical Development of Selective Immunoproteasome Inhibitors. Cells. 2022; 11(1):9. https://doi.org/10.3390/cells11010009
Chicago/Turabian StyleKirk, Christopher J., Tony Muchamuel, Jinhai Wang, and R. Andrea Fan. 2022. "Discovery and Early Clinical Development of Selective Immunoproteasome Inhibitors" Cells 11, no. 1: 9. https://doi.org/10.3390/cells11010009
APA StyleKirk, C. J., Muchamuel, T., Wang, J., & Fan, R. A. (2022). Discovery and Early Clinical Development of Selective Immunoproteasome Inhibitors. Cells, 11(1), 9. https://doi.org/10.3390/cells11010009