
Could Causal Discovery in Proteogenomics Assist in Understanding Gene–Protein Relations? A Perennial Fruit Tree Case Study Using Sweet Cherry as a Model


 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
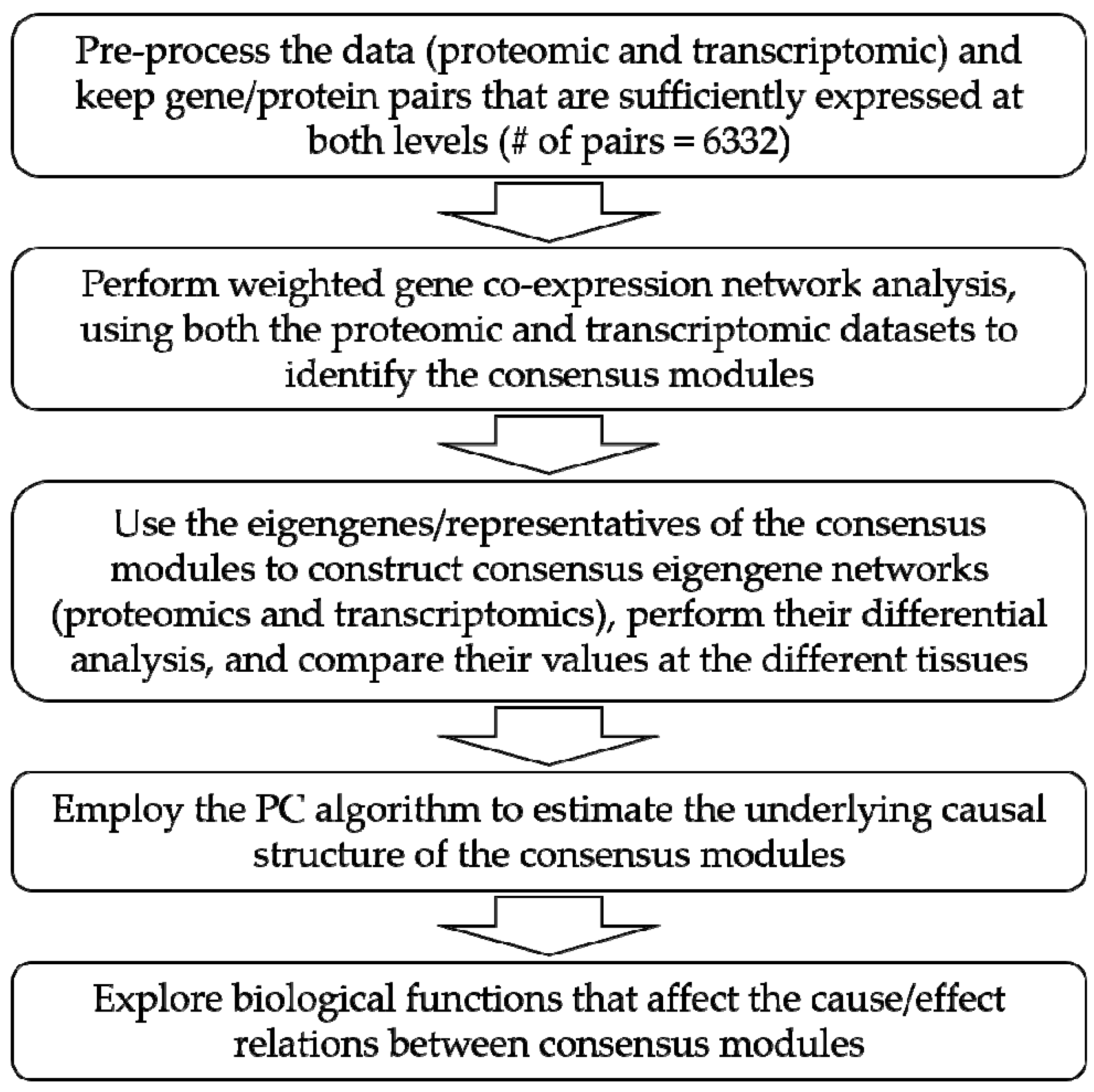
2. Materials and Methods
2.1. Data Description
2.2. PC Algorithm
2.3. Statistical Analysis
3. Results and Discussion
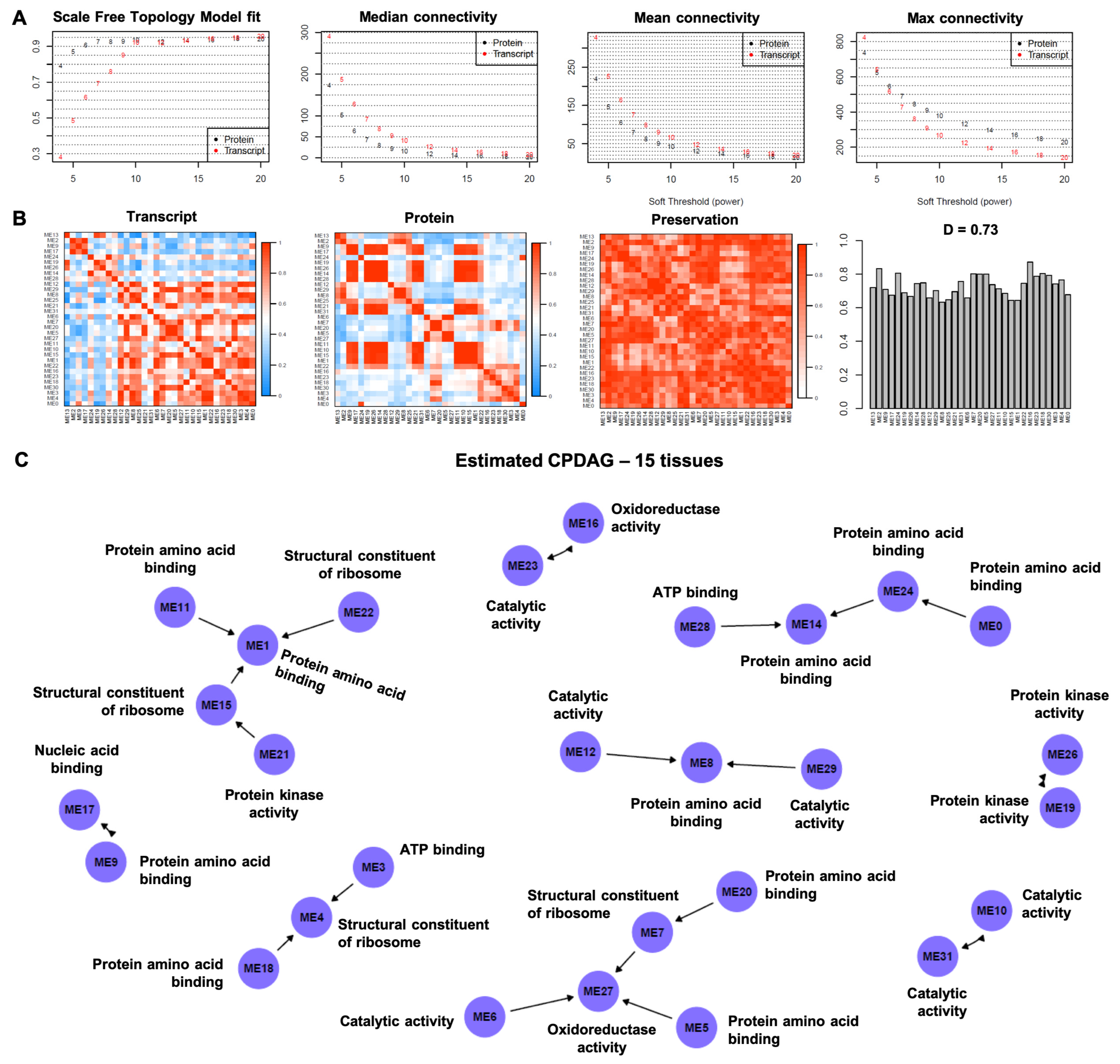
3.1. Causal Model-Based Network of Co-Expression Proteogenomic Modules in 15 Sweet Cherry Tissues
3.2. Causal Model-Based Network of Co-Expression Proteogenomic Modules across Various Sweet Cherry Fruit and Stem Developmental Stages
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buccitelli, C.; Selbach, M. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 2020, 21, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, S.; Dun, M.D.; Hondermarck, H. Proteogenomics: Emergence and promise. Cell. Mol. Life Sci. 2015, 72, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Lazar, I.M.; Karcini, A.; Ahuja, S.; Estrada-Palma, C. Proteogenomic Analysis of Protein Sequence Alterations in Breast Cancer Cells. Sci. Rep. 2019, 9, 10381. [Google Scholar] [CrossRef] [Green Version]
- Nesvizhskii, A.I. Proteogenomics: Concepts, applications and computational strategies. Nat. Methods 2014, 11, 1114–1125. [Google Scholar] [CrossRef]
- Low, T.Y.; Mohtar, M.A.; Ang, M.Y.; Jamal, R. Connecting Proteomics to Next-Generation Sequencing: Proteogenomics and Its Current Applications in Biology. Proteomics 2019, 19, 1800235. [Google Scholar] [CrossRef]
- Castellana, N.E.; Payne, S.H.; Shen, Z.; Stanke, M.; Bafna, V.; Briggs, S.P. Discovery and revision of Arabidopsis genes by proteogenomics. Proc. Natl. Acad. Sci. USA 2008, 105, 21034–21038. [Google Scholar] [CrossRef] [Green Version]
- Sheynkman, G.M.; Shortreed, M.R.; Cesnik, A.J.; Smith, L.M. Proteogenomics: Integrating Next-Generation Sequencing and Mass Spectrometry to Characterize Human Proteomic Variation. Annu. Rev. Anal. Chem. 2016, 9, 521–545. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallström, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef]
- Xanthopoulou, A.; Moysiadis, T.; Bazakos, C.; Karagiannis, E.; Karamichali, I.; Stamatakis, G.; Samiotaki, M.; Manioudaki, M.; Michailidis, M.; Madesis, P.; et al. The perennial fruit tree proteogenomics atlas: A spatial map of the sweet cherry proteome and transcriptome. Plant J. 2021; accepted. [Google Scholar] [CrossRef]
- Alkio, M.; Jonas, U.; Declercq, M.; Van Nocker, S.; Knoche, M. Transcriptional dynamics of the developing sweet cherry (Prunus avium L.) fruit: Sequencing, annotation and expression profiling of exocarp-Associated genes. Hortic. Res. 2014, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Berni, R.; Charton, S.; Planchon, S.; Legay, S.; Romi, M.; Cantini, C.; Cai, G.; Hausman, J.F.; Renaut, J.; Guerriero, G. Molecular investigation of Tuscan sweet cherries sampled over three years: Gene expression analysis coupled to metabolomics and proteomics. Hortic. Res. 2021, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, E.; Sarrou, E.; Michailidis, M.; Tanou, G.; Ganopoulos, I.; Bazakos, C.; Kazantzis, K.; Martens, S.; Xanthopoulou, A.; Molassiotis, A. Fruit quality trait discovery and metabolic profiling in sweet cherry genebank collection in Greece. Food Chem. 2021, 342, 128315. [Google Scholar] [CrossRef]
- Michailidis, M.; Karagiannis, E.; Tanou, G.; Samiotaki, M.; Tsiolas, G.; Sarrou, E.; Stamatakis, G.; Ganopoulos, I.; Martens, S.; Argiriou, A.; et al. Novel insights into the calcium action in cherry fruit development revealed by high-throughput mapping. Plant Mol. Biol. 2020, 104, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Xanthopoulou, A.; Manioudaki, M.; Bazakos, C.; Kissoudis, C.; Farsakoglou, A.M.; Karagiannis, E.; Michailidis, M.; Polychroniadou, C.; Zambounis, A.; Kazantzis, K.; et al. Whole genome re-sequencing of sweet cherry (Prunus avium L.) yields insights into genomic diversity of a fruit species. Hortic. Res. 2020, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Hoogendijk, A.J.; Pourfarzad, F.; Aarts, C.E.M.; Tool, A.T.J.; Hiemstra, I.H.; Grassi, L.; Frontini, M.; Meijer, A.B.; van den Biggelaar, M.; Kuijpers, T.W. Dynamic Transcriptome-Proteome Correlation Networks Reveal Human Myeloid Differentiation and Neutrophil-Specific Programming. Cell Rep. 2019, 29, 2505–2519.e4. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, L.; Le, T.D. Practical Approaches to Causal Relationship Exploration; Springer: Berlin/Heidelberg, Germany, 2015; ISBN 978-3-319-14432-0. [Google Scholar]
- Pearl, J. Causality; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Neapolitan, R.E. Learning Bayesian Networks; Pearson Prentice Hall: Upper Saddle River, NJ, USA, 2004. [Google Scholar]
- Spirtes, P.; Glymour, C.; Scheines, R. Causation, Prediction, and Search; MIT Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Kalisch, M.; Hauser, A.; Maathuis, M.H.; Mächler, M. An Overview of the Pcalg Package for R. 2020. Available online: https://cran.r-project.org/web/packages/pcalg/vignettes/vignette2018.pdf (accessed on 2 December 2021).
- Textor, J. Drawing and Analyzing Causal DAGs with DAGitty. arXiv 2015, arXiv:1508.04633. [Google Scholar]
- Wickham, H. Ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Core Development Team, R. A Language and Environment for Statistical Computing. Available online: http://www.r-project.org (accessed on 2 December 2021).
- Vighi, I.L.; Crizel, R.L.; Perin, E.C.; Rombaldi, C.V.; Galli, V. Crosstalk during Fruit Ripening and Stress Response among Abscisic Acid, Calcium-Dependent Protein Kinase and Phenylpropanoid. CRC Crit. Rev. Plant Sci. 2019, 38, 99–116. [Google Scholar] [CrossRef]
- Liu, J.; Hu, X.; Yu, J.; Yang, A.; Liu, Y.P. Caffeoyl shikimate esterase has a role in endocarp lignification in peach (Prunus persica L.) fruit. Korean J. Hortic. Sci. Technol. 2017, 35, 59–68. [Google Scholar] [CrossRef]
- Kuhn, N.; Maldonado, J.; Ponce, C.; Arellano, M.; Time, A.; Multari, S.; Martens, S.; Carrera, E.; Donoso, J.M.; Sagredo, B.; et al. RNAseq reveals different transcriptomic responses to GA3 in early and midseason varieties before ripening initiation in sweet cherry fruits. Sci. Rep. 2021, 11, 1–20. [Google Scholar] [CrossRef]
- Li, H.; Tian, J.; Yao, Y.Y.; Zhang, J.; Song, T.T.; Li, K.T.; Yao, Y.C. Identification of leucoanthocyanidin reductase and anthocyanidin reductase genes involved in proanthocyanidin biosynthesis in Malus crabapple plants. Plant Physiol. Biochem. 2019, 139, 141–151. [Google Scholar] [CrossRef]
- Prasanna, V.; Prabha, T.N.; Tharanathan, R.N. Fruit ripening phenomena-an overview. Crit. Rev. Food Sci. Nutr. 2007, 47, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Nakata, P.A.; Okita, T.W. Studies to enhance starch biosynthesis by manipulation of ADP-glucose pyrophosphorylase genes. Biotechnol. Agric. Mol. Cell. Biol. Potato 1994, 12, 31–44. [Google Scholar]
- Zhang, Z.; Wang, N.; Jiang, S.; Xu, H.; Wang, Y.; Wang, C.; Li, M.; Liu, J.; Qu, C.; Liu, W.; et al. Analysis of the xyloglucan endotransglucosylase/hydrolase gene family during apple fruit ripening and softening. J. Agric. Food Chem. 2017, 65, 429–434. [Google Scholar] [CrossRef]
- Gilkerson, J.; Perez-Ruiz, J.M.; Chory, J.; Callis, J. The plastid-localized pfkB-type carbohydrate kinases FRUCTOKINASE-LIKE 1 and 2 are essential for growth and development of Arabidopsis thaliana. BMC Plant Biol. 2012, 12, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geigenberger, P. Regulation of starch biosynthesis in response to a fluctuating environment. Plant Physiol. 2011, 155, 1566–1577. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Fan, Z.; Zhang, Q.; Wang, C.; Gao, Y.; Deng, Y.; Zhu, B.; Zhu, H.; Chen, J.; Shan, W.; et al. BEL1-LIKE HOMEODOMAIN 11 regulates chloroplast development and chlorophyll synthesis in tomato fruit. Plant J. 2018, 94, 1126–1140. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Jin, J.; Xu, Y.; Liu, W.; Yang, G.; Bu, H.; Li, T.; Wang, A. Ethylene-activated MdPUB24 mediates ubiquitination of MdBEL7 to promote chlorophyll degradation in apple fruit. Plant J. 2021, 108, 169–182. [Google Scholar] [CrossRef]
- Li, B.; Yan, J.; Jia, W. Feronia/fer-like receptor kinases integrate and modulate multiple signaling pathways in fruit development and ripening. Plant Signal. Behav. 2017, 12, e1366397. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Gallusci, P.; Lang, Z. Fruit development and epigenetic modifications. New Phytol. 2020, 228, 839–844. [Google Scholar] [CrossRef]
- Giovannoni, J.; Nguyen, C.; Ampofo, B.; Zhong, S.; Fei, Z. The Epigenome and Transcriptional Dynamics of Fruit Ripening. Annu. Rev. Plant Biol. 2017, 68, 61–84. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganopoulou, M.; Michailidis, M.; Angelis, L.; Ganopoulos, I.; Molassiotis, A.; Xanthopoulou, A.; Moysiadis, T. Could Causal Discovery in Proteogenomics Assist in Understanding Gene–Protein Relations? A Perennial Fruit Tree Case Study Using Sweet Cherry as a Model. Cells 2022, 11, 92. https://doi.org/10.3390/cells11010092
Ganopoulou M, Michailidis M, Angelis L, Ganopoulos I, Molassiotis A, Xanthopoulou A, Moysiadis T. Could Causal Discovery in Proteogenomics Assist in Understanding Gene–Protein Relations? A Perennial Fruit Tree Case Study Using Sweet Cherry as a Model. Cells. 2022; 11(1):92. https://doi.org/10.3390/cells11010092
Chicago/Turabian StyleGanopoulou, Maria, Michail Michailidis, Lefteris Angelis, Ioannis Ganopoulos, Athanassios Molassiotis, Aliki Xanthopoulou, and Theodoros Moysiadis. 2022. "Could Causal Discovery in Proteogenomics Assist in Understanding Gene–Protein Relations? A Perennial Fruit Tree Case Study Using Sweet Cherry as a Model" Cells 11, no. 1: 92. https://doi.org/10.3390/cells11010092
APA StyleGanopoulou, M., Michailidis, M., Angelis, L., Ganopoulos, I., Molassiotis, A., Xanthopoulou, A., & Moysiadis, T. (2022). Could Causal Discovery in Proteogenomics Assist in Understanding Gene–Protein Relations? A Perennial Fruit Tree Case Study Using Sweet Cherry as a Model. Cells, 11(1), 92. https://doi.org/10.3390/cells11010092