
Features of ZED1227: The First-In-Class Tissue Transglutaminase Inhibitor Undergoing Clinical Evaluation for the Treatment of Celiac Disease

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
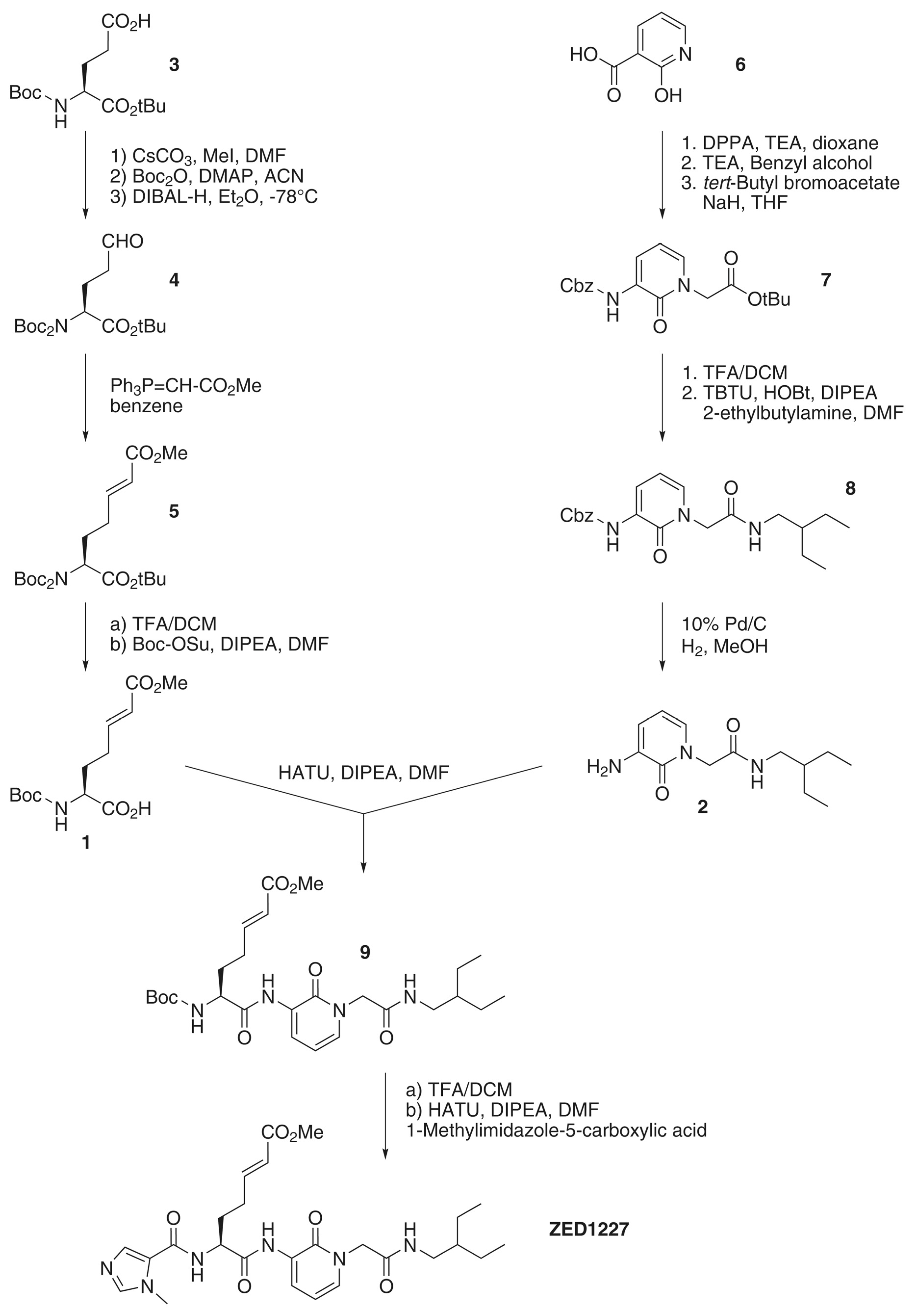
2.1. Overview on the Synthesis Route of ZED1227
2.2. Physicochemical Features and Stability
2.3. Caco-2 Cell Permeability Assay
2.4. Cytotoxicological Analysis
2.5. Inhibition of Tissue Transglutaminase and Other Isoenzymes
2.6. Reactivity of ZED1227 towards Thiols
3. Results and Discussion
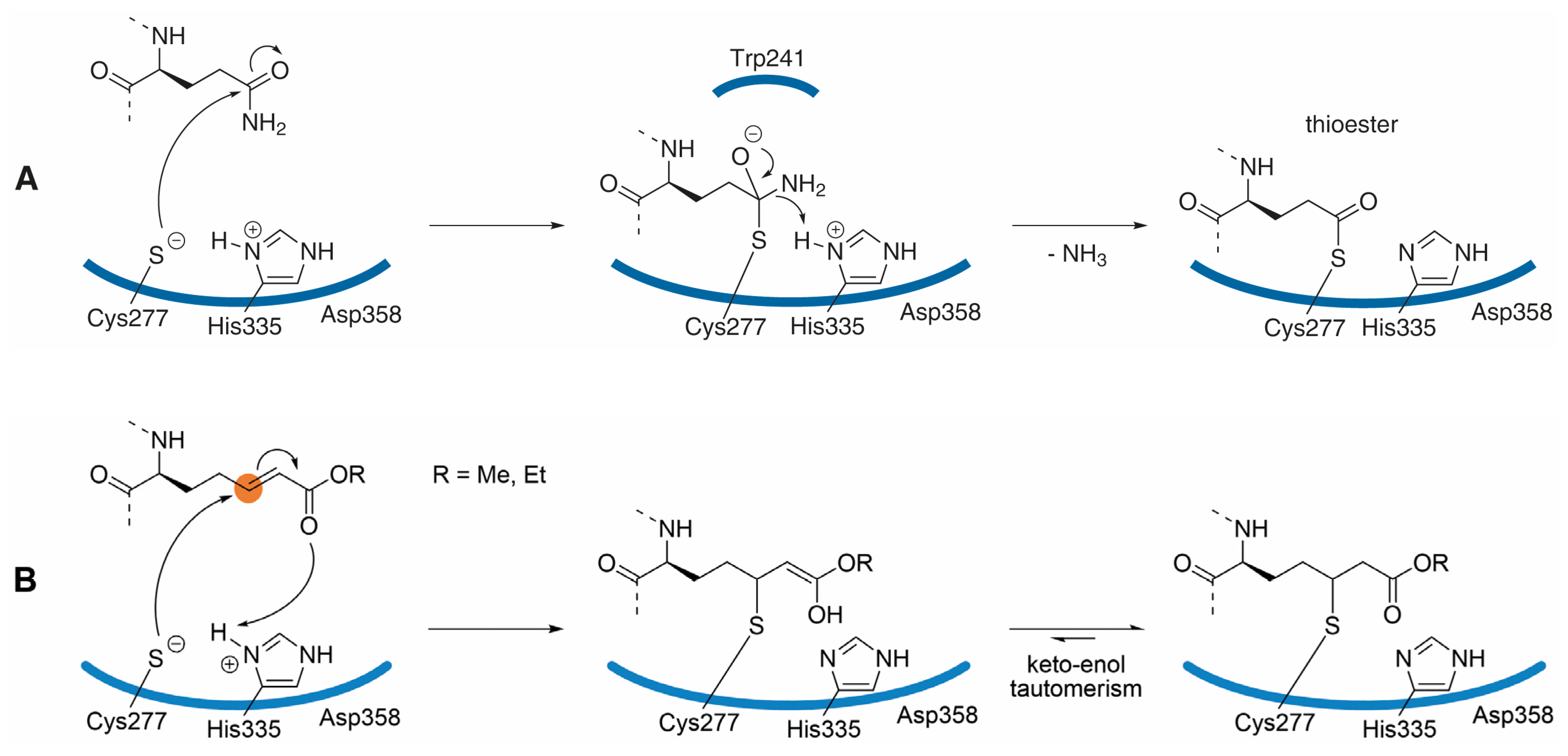
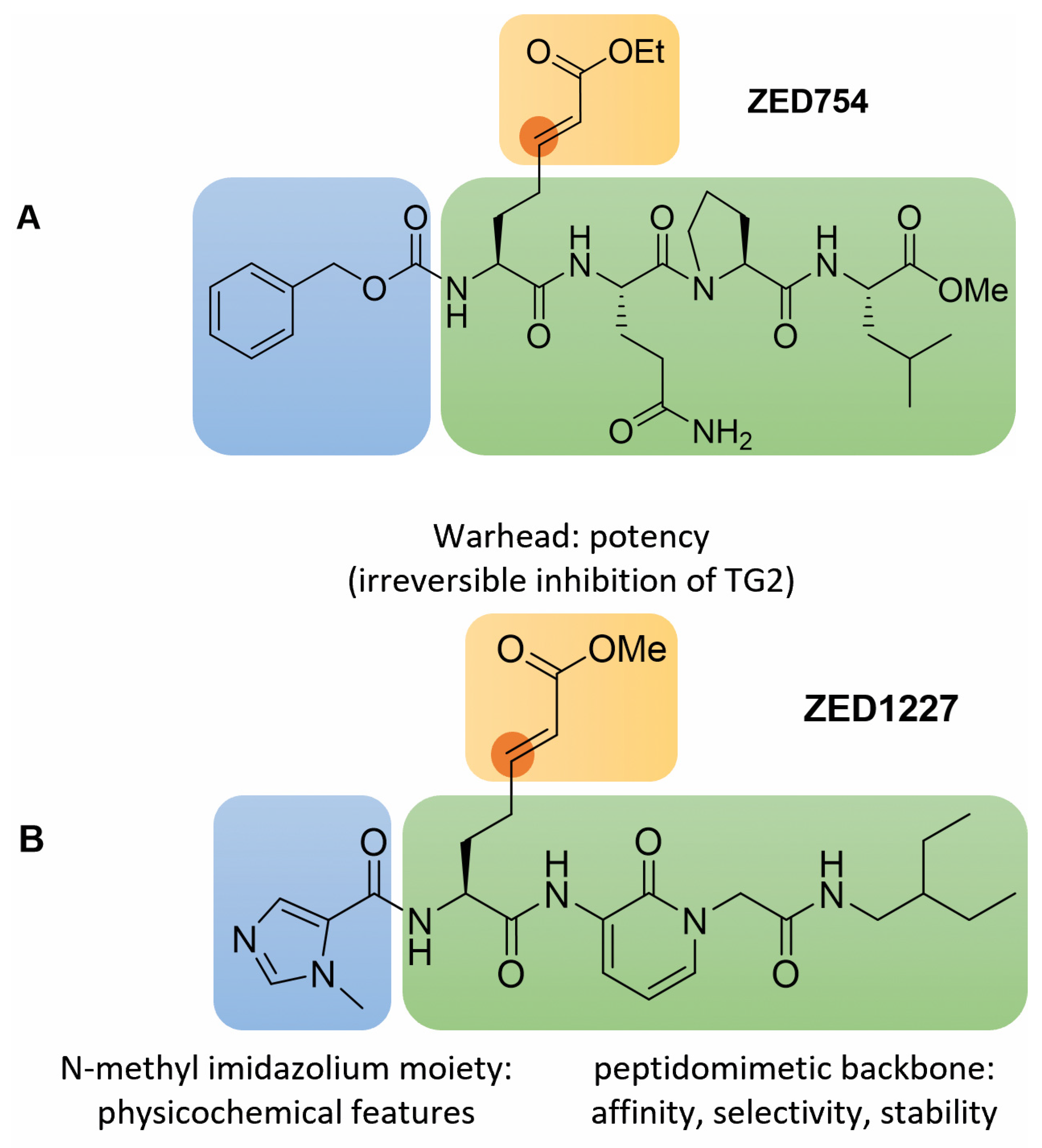
3.1. Inhibitor Design

3.2. Structural Features and In Vitro Profiling of ZED1227

3.3. Potency and Selectivity of ZED1227
3.4. Early Clinical Development
4. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
Appendix A
Appendix A.1. Supplementary Abbreviations
Appendix A.2. Synthesis
Appendix A.3. Structure Verification of ZED1227

{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| 13C NMR δ [ppm] | Position | Type | 1H NMR δ [ppm] |
| 10.75 | C32, C33 | -CH3 | 0.82 |
| 23.30 | C30, C31 | -CH2- | 1.26 |
| 28.30 | C20 | -CH2- | 2.32 |
| 29.43 | C19 | -CH2 | 1.91, 2.04 |
| 33.44 | C38 | -CH3 | 3.79 |
| 40.43 | C14 | >CH- | 1.26 |
| 41.10 | C13 | -CH2- | 3.01 |
| 51.18 | C26 | -CH3 | 3.62 |
| 51.52 | C9 | -CH2- | 4.58 |
| 53.18 | C16 | =CH- | 4.58 |
| 104.52 | C6 | =CH- | 6.25 |
| 121.00 | C22 | =CH- | 5.86 |
| 122.34 | C7 | =CH- | 8.21 |
| 125.25 | C28 | =C< | |
| 127.99 | C2 | =C< | - |
| 133.00 | C34 | =CH- | 7.72 |
| 133.50 | C5 | =CH- | 7.33 |
| 142.06 | C36 | =CH- | 7.77 |
| 148.46 | C21 | =CH- | 6.93 |
| 156.69 | C3 | >C=O | - |
| 160.34 | C27 | >C=O | - |
| 165.35 | C23 | >C=O | - |
| 166.77 | C10 | >C=O | - |
| 170.82 | C15 | >C=O | - |
| - | H11 | >NH | 8.05 |
| - | H18 | >NH | 8.63 |
| - | H1 | >NH | 9.29 |
References
- Sarkar, N.K.; Clarke, D.D.; Waelsch, H. An enzymically catalyzed incorporation of amines into proteins. Biochim. Biophys. Acta 1957, 25, 451–452. [Google Scholar] [CrossRef]
- Katt, W.P.; Antonyak, M.A.; Cerione, R.A. The diamond anniversary of tissue transglutaminase: A protein of many talents. Drug Discov. Today 2018, 23, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Lorand, L.; Iismaa, S.E. Transglutaminase diseases: From biochemistry to the bedside. FASEB J. 2019, 33, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Szondy, Z.; Korponay-Szabó, I.; Király, R.; Sarang, Z.; Tsay, G.J. Transglutaminase 2 in human diseases. BioMedicine 2017, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- See, J.; Murray, J. Gluten-Free Diet: The Medical and Nutrition Management of Celiac Disease. Nutr. Clin. Pract. 2006, 21, 1–15. [Google Scholar] [CrossRef]
- Lindfors, K.; Ciacci, C.; Kurppa, K.; Lundin, K.E.A.; Makharia, G.K.; Mearin, M.L.; Murray, J.A.; Verdu, E.F.; Kaukinen, K. Coeliac disease. Nat. Rev. Dis. Primers 2019, 5, 3. [Google Scholar] [CrossRef]
- Kivelä, L.; Caminero, A.; Leffler, D.A.; Pinto-Sanchez, M.I.; Tye-Din, J.A.; Lindfors, K. Current and emerging therapies for coeliac disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 18, 181–195. [Google Scholar] [CrossRef]
- Kamer, J.H.v.d.; Weijers, H.A.; Dicke, W.K. Coeliac Disease: An Investigation into the Injurious Constituents of Wheat in Connection with their Action on Patients with Coeliac Disease. Acta Paediatr. 1953, 42, 223–231. [Google Scholar] [CrossRef]
- Bruce, S.E.; Bjarnason, I.; Peters, T.J. Human jejunal transglutaminase: Demonstration of activity, enzyme kinetics and substrate specificity with special relation to gliadin and coeliac disease. Clin. Sci. 1985, 68, 573–579. [Google Scholar] [CrossRef]
- Dieterich, W.; Ehnis, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801. [Google Scholar] [CrossRef]
- Molberg, Ø.; Mcadam, S.N.; Körner, R.; Quarsten, H.; Kristiansen, C.; Madsen, L.; Fugger, L.; Scott, H.; Norén, O.; Roepstorff, P.; et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat. Med. 1998, 4, 713–717. [Google Scholar] [CrossRef]
- Sjöström, H.; Lundin, K.; Molberg, Ø.; Körner, R.; Mcadam, S.N.; Anthonsen, D.; Quarsten, H.; Norén, O.; Roepstorff, P.; Thorsby, E.; et al. Identification of a Gliadin T-Cell Epitope in Coeliac Disease: General Importance of Gliadin Deamidation for Intestinal T-Cell Recognition. Scand. J. Immunol. 1998, 48, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Molberg, Ø.; Parrot, I.; Hausch, F.; Filiz, F.; Gray, G.M.; Sollid, L.M.; Khosla, C. Structural Basis for Gluten Intolerance in Celiac Sprue. Science 2002, 297, 2275–2279. [Google Scholar] [CrossRef] [Green Version]
- Shan, L.; Qiao, S.-W.; Arentz-Hansen, H.; Molberg, Ø.; Gray, G.M.; Sollid, L.M.; Khosla, C. Identification and Analysis of Multivalent Proteolytically Resistant Peptides from Gluten: Implications for Celiac Sprue. J. Proteome Res. 2005, 4, 1732–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleckenstein, B.; Molberg, Ø.; Qiao, S.-W.; Schmid, D.G.; von der Mülbe, F.; Elgstøen, K.; Jung, G.; Sollid, L.M. Gliadin T cell epitope selection by tissue transglutaminase in Celiac disease - Role of enzyme specificity and pH influence on the transamidation versus deamidation reactions. J. Biol. Chem. 2002, 277, 34109–34116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, S.-W.; Bergseng, E.; Molberg, Ø.; Xia, J.; Fleckenstein, B.; Khosla, C.; Sollid, L.M. Antigen Presentation to Celiac Lesion-Derived T Cells of a 33-Mer Gliadin Peptide Naturally Formed by Gastrointestinal Digestion. J. Immunol. 2004, 173, 1757–1762. [Google Scholar] [CrossRef] [Green Version]
- Bodd, M.; Tollefsen, S.; Bergseng, E.; Lundin, K.E.; Sollid, L.M. Evidence that HLA-DQ9 confers risk to celiac disease by presence of DQ9-restricted gluten-specific T cells. Hum. Immunol. 2012, 73, 376–381. [Google Scholar] [CrossRef]
- Sollid, L.M. The roles of MHC class II genes and post-translational modification in celiac disease. Immunogenetics 2017, 69, 605–616. [Google Scholar] [CrossRef]
- Schuppan, D.; Dieterich, W.; Riecken, E.O. Exposing gliadin as a tasty food for lymphocytes. Nat. Med. 1998, 4, 666–667. [Google Scholar] [CrossRef]
- Fleckenstein, B.; Qiao, S.-W.; Larsen, M.R.; Jung, G.; Roepstorff, P.; Sollid, L.M. Molecular Characterization of Covalent Complexes between Tissue Transglutaminase and Gliadin Peptides. J. Biol. Chem. 2004, 279, 17607–17616. [Google Scholar] [CrossRef] [Green Version]
- Sollid, L.M.; Molberg, Ø.; Mcadam, S.; Lundin, K. Autoantibodies in coeliac disease: Tissue transglutaminase--guilt by association? Gut 1997, 41, 851–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iversen, R.; Du Pré, M.F.; Di Niro, R.; Sollid, L.M. Igs as Substrates for Transglutaminase 2: Implications for Autoantibody Production in Celiac Disease. J. Immunol. 2015, 195, 5159–5168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamnaes, J.; Iversen, R.; Du Pré, M.F.; Chen, X.; Sollid, L.M. Data from: Enhanced B-cell receptor recognition of the autoantigen transglutaminase 2 by efficient catalytic self-multimerization. PLoS ONE 2015, 10, e0134922. [Google Scholar] [CrossRef]
- Iversen, R.; Amundsen, S.F.; Kleppa, L.; du Pré, M.F.; Stamnæs, J.; Sollid, L.M. Evidence That Pathogenic Transglutaminase 2 in Celiac Disease Derives from Enterocytes. Gastroenterology 2020, 159, 788–790. [Google Scholar] [CrossRef]
- Lebreton, C.; Ménard, S.; Abed, J.; Moura, I.C.; Coppo, R.; Dugave, C.; Monteiro, R.C.; Fricot, A.; Traore, M.G.; Griffin, M.; et al. Interactions Among Secretory Immunoglobulin A, CD71, and Transglutaminase-2 Affect Permeability of Intestinal Epithelial Cells to Gliadin Peptides. Gastroenterology 2012, 143, 698–707.e4. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, M.; Griffiths, R.; Dewitt, S.; Knauper, V.; Aeschlimann, D. P2X7 receptor activation regulates rapid unconventional export of transglutaminase-2. J. Cell Sci. 2015, 128, 4615–4628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamnaes, J.; Pinkas, D.M.; Fleckenstein, B.; Khosla, C.; Sollid, L.M. Redox regulation of transglutaminase 2 activity. J. Biol. Chem. 2010, 285, 25402–25409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melkonian, A.V.; Loppinet, E.; Martin, R.; Porteus, M.; Khosla, C. An Unusual “OR” Gate for Allosteric Regulation of Mammalian Transglutaminase 2 in the Extracellular Matrix. J. Am. Chem. Soc. 2021, 143, 10537–10540. [Google Scholar] [CrossRef]
- Jin, X.; Stamnæs, J.; Klöck, C.; DiRaimondo, T.R.; Sollid, L.M.; Khosla, C. Activation of Extracellular Transglutaminase 2 by Thioredoxin. J. Biol. Chem. 2011, 286, 37866–37873. [Google Scholar] [CrossRef] [Green Version]
- Finney, S.; Seale, L.; Sawyer, R.T.; Wallis, R.B. Tridegin, a new peptidic inhibitor of factor XIIIa, from the blood-sucking leech Haementeria ghilianii. Biochem. J. 1997, 324, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, G. alpha-Halogenmethyl carbonyl compounds as very potent inhibitors of factor XIIIa in vitro. Ann. N. Y. Acad. Sci. 1981, 370, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Shebuski, R.J.; Sitko, G.R.; Claremon, A.D.; Baldwin, J.J.; Remy, D.C.; Stern, A. Inhibition of factor XIIIa in a canine model of coronary thrombosis: Effect on reperfusion and acute reocclusion after recombinant tissue-type plasminogen activator. Blood 1990, 75, 1455–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, K.F.; Doshi, K.P.; Gaul, S.L.; Claremon, D.A.; Remy, D.C.; Baldwin, J.J.; Pitzenberger, S.M.; Stern, A.M. Transglutaminase Inhibition by 2-[(2-Oxopropyl)thio]imidazolium Derivatives: Mechanism of Factor XIIIa Inactivation. Biochemistry 1994, 33, 10109–10119. [Google Scholar] [CrossRef] [PubMed]
- Hausch, F.; Halttunen, T.; Mäki, M.; Khosla, C. Design, Synthesis, and Evaluation of Gluten Peptide Analogs as Selective Inhibitors of Human Tissue Transglutaminase. Chem. Biol. 2003, 10, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.P.; Nelson, J.A.; Feldman, S.; Van Nguyen, B. Pharmacokinetic and phase I study of intravenous DON (6-diazo-5-oxo-L-norleucine) in children. Cancer Chemother. Pharmacol. 1988, 21, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Mäki, M.; Lundin, K.E.; Isola, J.; Friesing-Sosnik, T.; Taavela, J.; Popp, A.; Koskenpato, J.; Langhorst, J.; Hovde, M.; et al. A Randomized Trial of a Transglutaminase 2 Inhibitor for Celiac Disease. N. Engl. J. Med. 2021, 385, 35–45. [Google Scholar] [CrossRef]
- Constantinou-Kokotou, V.; Magrioti, V. Synthesis and use of N,N-di-Boc-glutamate gamma-semialdehydes and related aldehydes. Amino Acids 2003, 24, 231–243. [Google Scholar] [CrossRef]
- Kokotos, G.; Padrón, J.M.; Martín, T.; Gibbons, W.A.; Martín, V.S. A General Approach to the Asymmetric Synthesis of Unsaturated Lipidic α-Amino Acids. The First Synthesis of α-Aminoarachidonic Acid. J. Org. Chem. 1998, 63, 3741–3744. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Sarkar, A.; Brindisi, M. The Curtius rearrangement: Mechanistic insight and recent applications in natural product syntheses. Org. Biomol. Chem. 2018, 16, 2006–2027. [Google Scholar] [CrossRef]
- Bharate, S.S.; Kumar, V.; Vishwakarma, R.A. Determining Partition Coefficient (Log P), Distribution Coefficient (Log D) and Ionization Constant (pKa) in Early Drug Discovery. Comb. Chem. High Throughput Screen 2016, 19, 461–469. [Google Scholar] [CrossRef]
- Dressman, J.B.; Krämer, J. Pharmaceutical Dissolution Testing; Taylor & Francis: Boca Raton, FL, USA, 2005. [Google Scholar]
- Klein, S. The Use of Biorelevant Dissolution Media to Forecast the In Vivo Performance of a Drug. AAPS J. 2010, 12, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottaviani, G.; Gosling, D.J.; Patissier, C.; Rodde, S.; Zhou, L.; Faller, B. What is modulating solubility in simulated intestinal fluids? Eur. J. Pharm. Sci. 2010, 41, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Lorand, L.; Lockridge, O.M.; Campbell, L.K.; Myhrman, R.; Bruner-Lorand, J. Transamidating enzymes: II. A continuous fluorescent method suited for automating measurements of factor XIII in plasma. Anal. Biochem. 1971, 44, 221–231. [Google Scholar] [CrossRef]
- Pasternack, R.; Büchold, C.; Jähnig, R.; Pelzer, C.; Sommer, M.; Heil, A.; Florian, P.; Nowak, G.; Gerlach, U.; Hils, M. Novel inhibitor ZED3197 as potential drug candidate in anticoagulation targeting coagulation FXIIIa (F13a). J. Thromb. Haemost. 2019, 18, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keillor, J.W. Inhibition of Transglutaminase. In Transglutaminases; Hitomi, K., Kojima, S., Fesus, L., Eds.; Springer: Tokyo, Japan, 2015; pp. 347–372. [Google Scholar]
- Wissner, A.; Overbeek, E.; Reich, M.F.; Floyd, M.B.; Johnson, B.D.; Mamuya, N.; Rosfjord, E.C.; Discafani, C.; Davis, R.; Shi, X.; et al. Synthesis and Structure−Activity Relationships of 6,7-Disubstituted 4-Anilinoquinoline-3-carbonitriles. The Design of an Orally Active, Irreversible Inhibitor of the Tyrosine Kinase Activity of the Epidermal Growth Factor Receptor (EGFR) and the Human Epidermal Growth Factor Receptor-2 (HER-2). J. Med. Chem. 2002, 46, 49–63. [Google Scholar] [CrossRef]
- Pinkas, D.M.; Strop, P.; Brunger, A.T.; Khosla, C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007, 5, e327. [Google Scholar] [CrossRef]
- Matthews, D.A.; Dragovich, P.S.; Webber, S.E.; Fuhrman, S.A.; Patick, A.K.; Zalman, L.S.; Hendrickson, T.F.; Love, R.A.; Prins, T.J.; Marakovits, J.T.; et al. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. USA 1999, 96, 11000–11007. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, L.C.; Yee, V.C.; Bishop, P.D.; Le Trong, I.; Teller, D.C.; Stenkamp, R.E. Transglutaminase factor XIII uses proteinase-like catalytic triad to crosslink macromolecules. Protein Sci. 1994, 3, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Santos, M.M.; Moreira, R. Michael Acceptors as Cysteine Protease Inhibitors. Mini-Rev. Med. Chem. 2007, 7, 1040–1050. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- De Vita, E. 10 years into the resurgence of covalent drugs. Futur. Med. Chem. 2021, 13, 193–210. [Google Scholar] [CrossRef]
- De Laurenzi, V.; Melino, G. Gene Disruption of Tissue Transglutaminase. Mol. Cell. Biol. 2001, 21, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wodtke, R.; Pietsch, M.; Löser, R. Solution-phase synthesis of the fluorogenic TGase 2 acyl donor Z-Glu(HMC)-Gly-OH and its use for inhibitor and amine substrate characterisation. Anal. Biochem. 2020, 595, 113612. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]

| Molecular Mass [g/mol] | 528.6 |
| Solubility at pH 7.4 Solubility at pH 1.1 | 100 μM/0.05 g/L 48,900 μM/25.9 g/L |
| LogD (octanol/PBS pH 7.4) | 2.0 |
| Stability in PBS/saline (24 h, 37 °C) | 10% at pH 7.4 89% at pH 5.0 |
| Stability in artificial gastric and intestinal fluid (24 h, 37 °C) | 95%, simulated gastric fluid (pH 1.2) 82%, simulated intestinal fluid (pH 6.5) |
| A-B permeability (Caco-2-assay) | Papp < 1 × 10−6 cm/s |
| Cytotoxicity Proliferation: Caco-2 cells Huh7 cells Viability: Caco-2 cells Huh7 cells | no effect up to 1,000 μM no effect up to 1,000 μM no effect up to 1,000 μM no effect up to 1,000 μM |
| Reactivity towards excess glutathione (25 °C) | 98% parent drug compound recovery (after 48 h) |
| ZED1227 | hTG2 | hTG1 | hTG3 | hTG6 | hFXIII-A2 |
|---|---|---|---|---|---|
| App. IC50 | 53 nM | 24,863 nM | >50,000 nM | 6,441 nM | >50,000 nM |
| Selectivity | 469 | >900 | 122 | >900 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Büchold, C.; Hils, M.; Gerlach, U.; Weber, J.; Pelzer, C.; Heil, A.; Aeschlimann, D.; Pasternack, R. Features of ZED1227: The First-In-Class Tissue Transglutaminase Inhibitor Undergoing Clinical Evaluation for the Treatment of Celiac Disease. Cells 2022, 11, 1667. https://doi.org/10.3390/cells11101667
Büchold C, Hils M, Gerlach U, Weber J, Pelzer C, Heil A, Aeschlimann D, Pasternack R. Features of ZED1227: The First-In-Class Tissue Transglutaminase Inhibitor Undergoing Clinical Evaluation for the Treatment of Celiac Disease. Cells. 2022; 11(10):1667. https://doi.org/10.3390/cells11101667
Chicago/Turabian StyleBüchold, Christian, Martin Hils, Uwe Gerlach, Johannes Weber, Christiane Pelzer, Andreas Heil, Daniel Aeschlimann, and Ralf Pasternack. 2022. "Features of ZED1227: The First-In-Class Tissue Transglutaminase Inhibitor Undergoing Clinical Evaluation for the Treatment of Celiac Disease" Cells 11, no. 10: 1667. https://doi.org/10.3390/cells11101667
APA StyleBüchold, C., Hils, M., Gerlach, U., Weber, J., Pelzer, C., Heil, A., Aeschlimann, D., & Pasternack, R. (2022). Features of ZED1227: The First-In-Class Tissue Transglutaminase Inhibitor Undergoing Clinical Evaluation for the Treatment of Celiac Disease. Cells, 11(10), 1667. https://doi.org/10.3390/cells11101667