
Extracellular Vesicular Transmission of miR-423-5p from HepG2 Cells Inhibits the Differentiation of Hepatic Stellate Cells

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Extracellular Vesicle Isolation
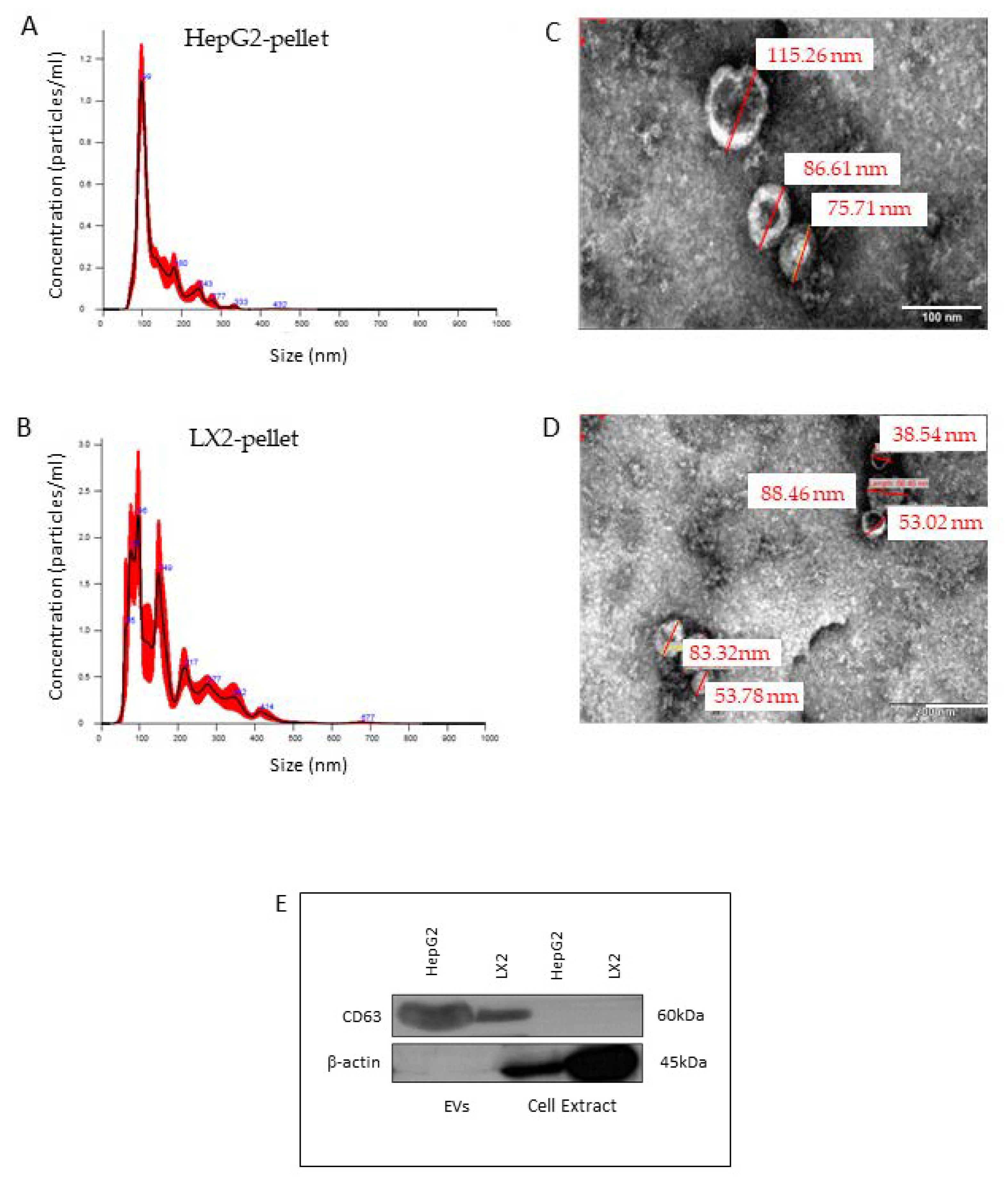
2.3. Nanosight
2.4. Electron Microscopy
2.5. Fluorescent Labeling
2.6. Primary Mouse Hepatic Stellate Cell Isolation
2.7. Primary Mouse Hepatocyte Isolation
2.8. Primary Mouse Kidney Cell Isolation
2.9. Animal Experiments
2.10. Stress Conditions
2.10.1. Treatment with ethanol (mimicking alcoholic steatohepatitis (ASH))
2.10.2. Treatment with lauric acid (mimicking NASH)
2.11. Real-Time qPCR Analysis
2.12. Western Blot Analysis
2.13. MicroRNA Transfection
2.14. Carbon Tetrachloride-Induced Liver Fibrosis Mouse Model
2.15. Masson’s Trichrome Staining
2.16. Statistical Analysis
3. Results
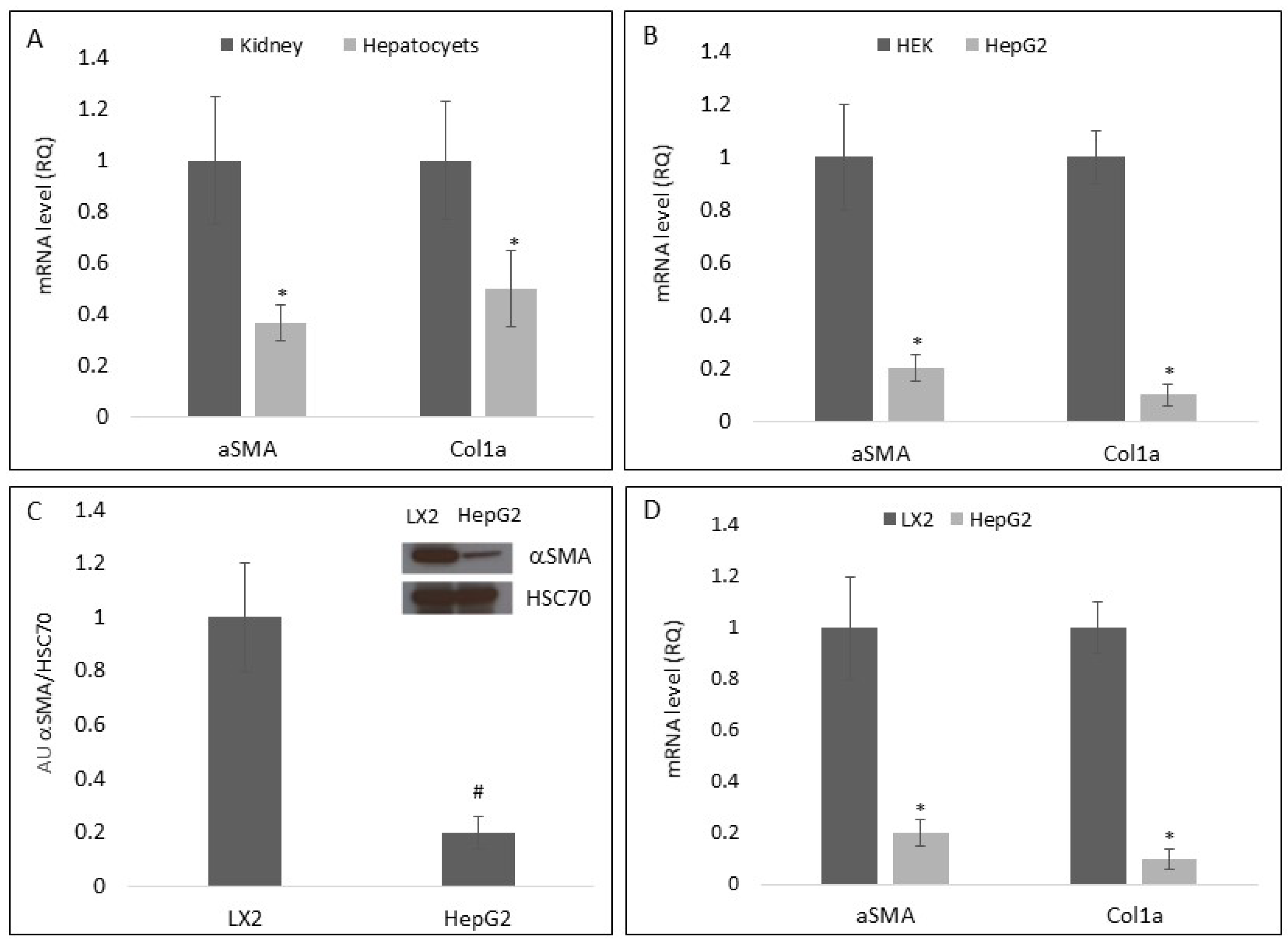
3.1. Hepatocyte-Primed Medium Reduced Hepatic Stellate Cell Differentiation
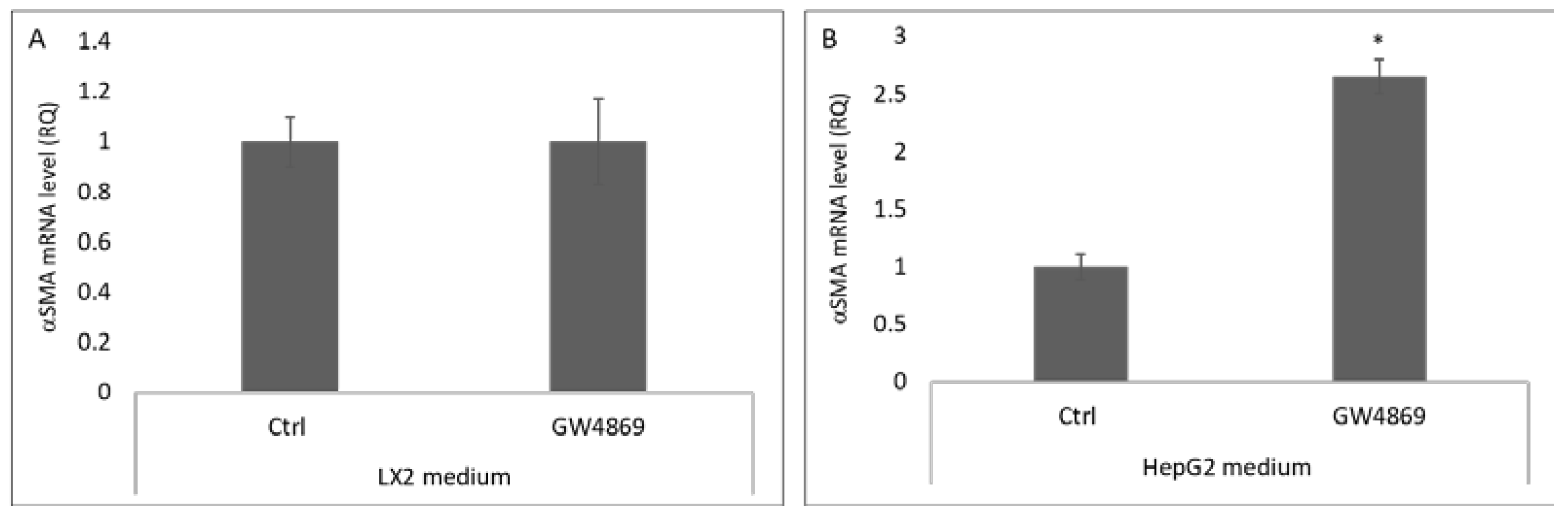
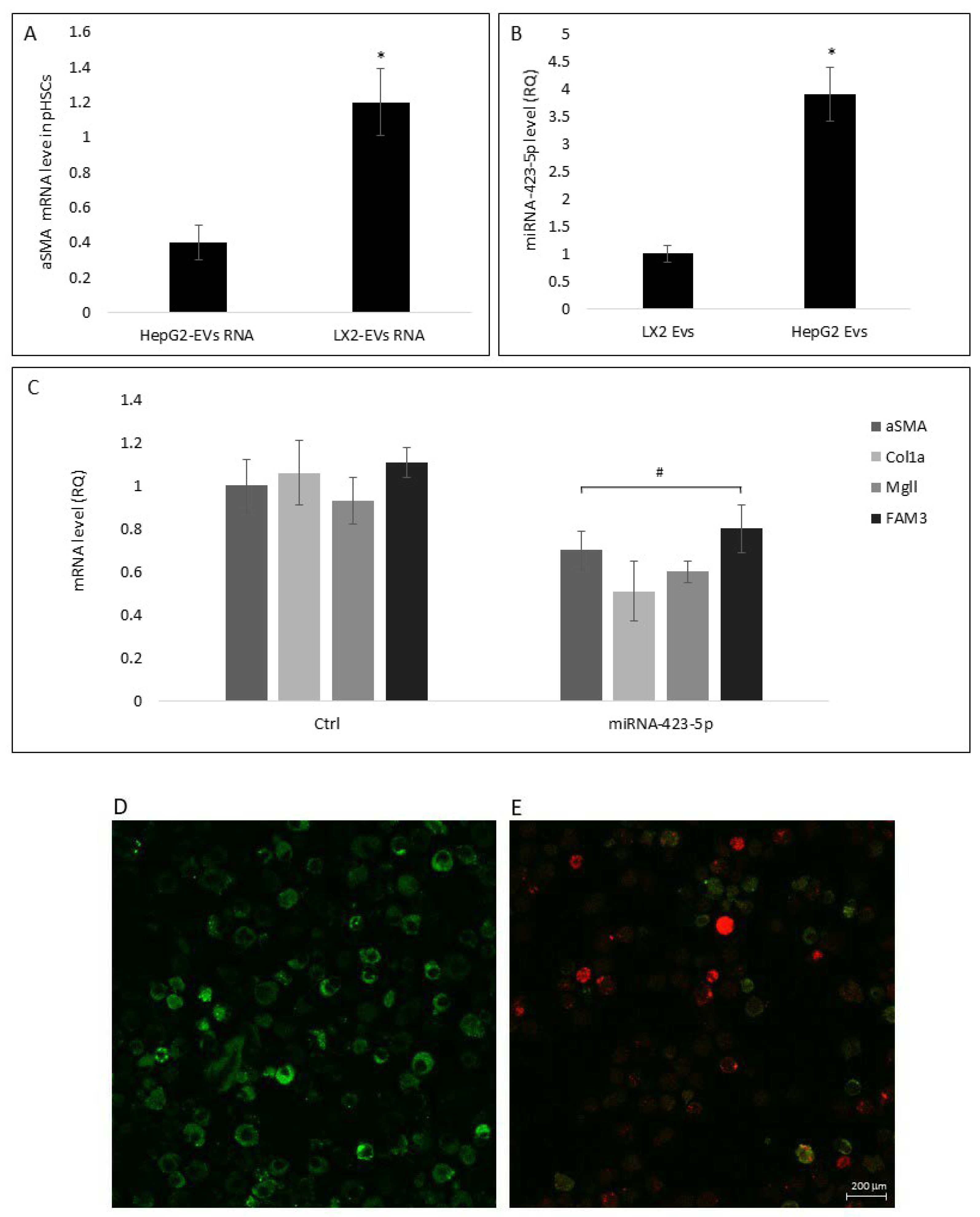
3.2. Hepatocyte Extracellular Vesicles Mediate the Inhibition of Hepatic Stellate Cell Differentiation in Primed Medium
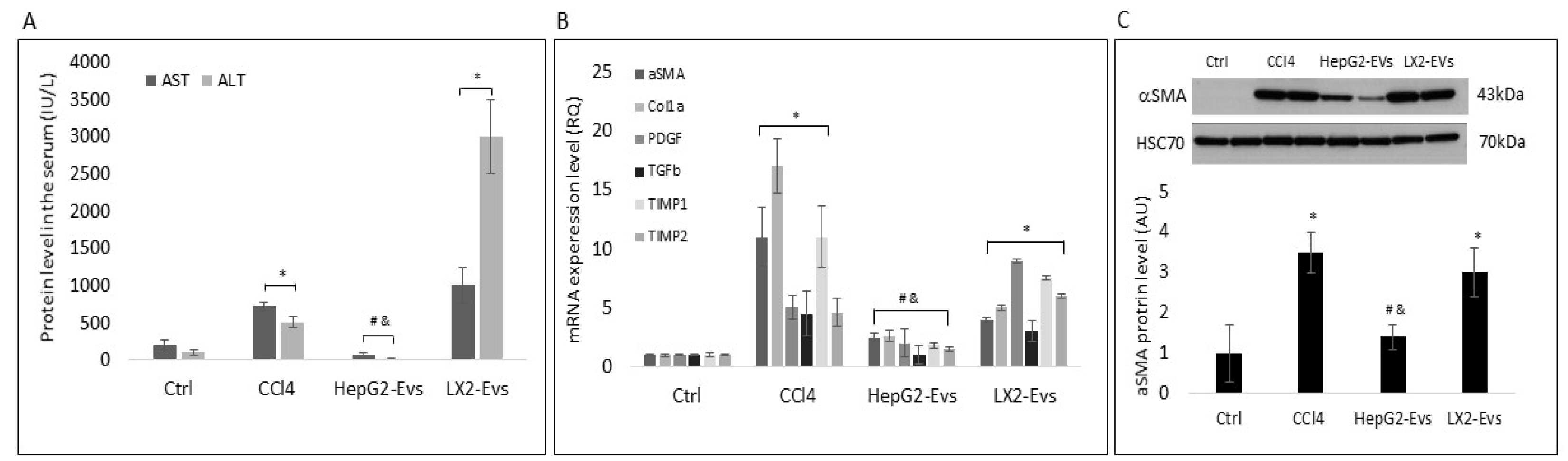
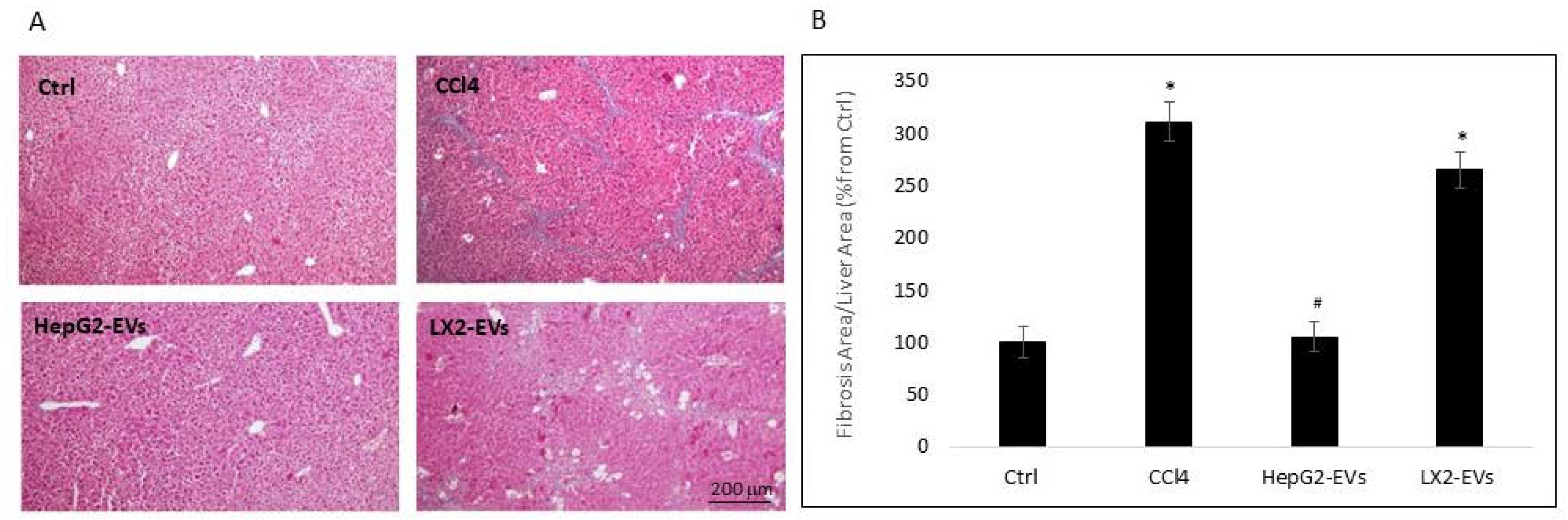
3.3. Hepatocyte Extracellular Vesicles Inhibit the Development of Liver Fibrosis In-Vivo
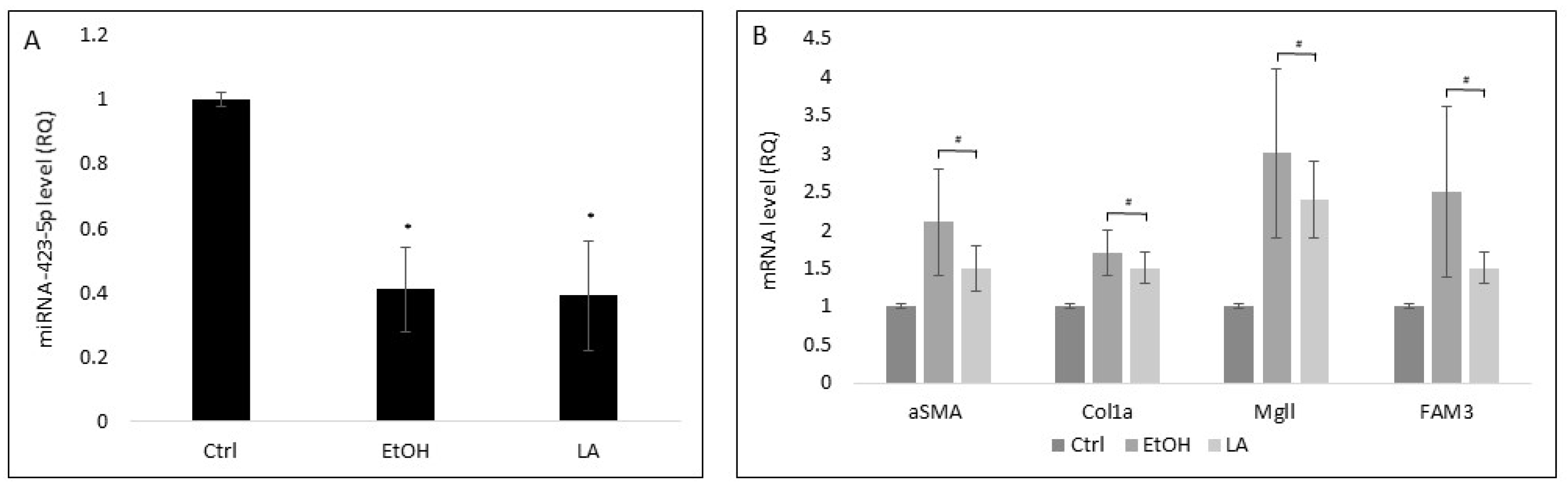
3.4. Stress Reduces miRNA-423-5p Levels in HepG2-EVs
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elpek, G.Ö. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J. Gastroenterol. 2014, 20, 7260–7276. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of Liver Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Zhang, X.; Li, W.; Feng, R.-X.; Li, L.; Yi, G.-R.; Zhang, X.-N.; Yin, C.; Yu, H.-Y.; Zhang, J.-P.; et al. Chronic Liver Injury Induces Conversion of Biliary Epithelial Cells into Hepatocytes. Cell Stem Cell 2018, 23, 114–122.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Center for Health Statistics. Summary Health Statistics: National Health Interview Survey, 2018; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2017.
- Aizarani, N.; Saviano, A.; Sagar; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grün, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Narumiya, S. Roles of hepatic stellate cells in liver inflammation: A new perspective. Inflamm. Regen. 2016, 36, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-Y.; Yuan, W.-G.; He, P.; Lei, J.-H.; Wang, C.-X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef]
- Edgar, J.R. Q&A: What are exosomes, exactly? BMC Biol. 2016, 14, 46. [Google Scholar]
- Sato, K.; Meng, F.; Glaser, S.; Alpini, G. Exosomes in liver pathology. J. Hepatol. 2016, 65, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, R.; Kemper, S.; Cong, M.; You, H.; Brigstock, D.R. Therapeutic effects of serum extracellular vesicles in liver fibrosis. J. Extracell. Vesicles 2018, 7, 1461505. [Google Scholar] [CrossRef]
- Hsu, S.-H.; Ghoshal, K. MicroRNAs in Liver Health and Disease. Curr. Pathobiol. Rep. 2013, 1, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Essandoh, K.; Yang, L.; Wang, X.; Huang, W.; Qin, D.; Hao, J.; Wang, Y.; Zingarelli, B.; Peng, T.; Fan, G.-C. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2362–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, M.K.; Mun, J.Y. Sample Preparation and Imaging of Exosomes by Transmission Electron Microscopy. J. Vis. Exp. 2018, 131, e56482. [Google Scholar] [CrossRef] [PubMed]
- Pochareddy, S.; Edenberg, H.J. Chronic Alcohol Exposure Alters Gene Expression in HepG2 Cells. Alcohol. Clin. Exp. Res. 2012, 36, 1021–1033. [Google Scholar] [CrossRef] [Green Version]
- Kanuri, G.; Bergheim, I. In Vitro and in Vivo Models of Non-Alcoholic Fatty Liver Disease (NAFLD). Int. J. Mol. Sci. 2013, 14, 11963–11980. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, A.; Feldstein, A.E. Extracellular vesicles in non-alcoholic and alcoholic fatty liver diseases. Liver Res. 2018, 2, 30–34. [Google Scholar] [CrossRef]
- Yang, W.; Wang, J.; Chen, Z.; Chen, J.; Meng, Y.; Chen, L.; Chang, Y.; Geng, B.; Sun, L.; Dou, L.; et al. NFE2 Induces miR-423-5p to Promote Gluconeogenesis and Hyperglycemia by Repressing the Hepatic FAM3A-ATP-Akt Pathway. Diabetes 2017, 66, 1819–1832. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Mulvihill, M.M.; Mukhopadhyay, P.; Xu, H.; Erdélyi, K.; Hao, E.; Holovac, E.; Haskó, G.; Cravatt, B.F.; Nomura, D.K.; et al. Monoacylglycerol Lipase Controls Endocannabinoid and Eicosanoid Signaling and Hepatic Injury in Mice. Gastroenterology 2013, 144, 808–817.e15. [Google Scholar] [CrossRef] [Green Version]
- Habib, A.; Chokr, D.; Wan, J.; Hegde, P.; Mabire, M.; Siebert, M.; Ribeiro-Parenti, L.; Le Gall, M.; Lettéron, P.; Pilard, N.; et al. Inhibition of monoacylglycerol lipase, an anti-inflammatory and antifibrogenic strategy in the liver. Gut 2018, 68, 522–532. [Google Scholar] [CrossRef]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68, 435–451. [Google Scholar] [CrossRef]
- Mantovani, A.; Scorletti, E.; Mosca, A.; Alisi, A.; Byrne, C.D.; Targher, G. Complications, morbidity and mortality of nonalcoholic fatty liver disease. Metabolism 2020, 111, 154170. [Google Scholar] [CrossRef] [PubMed]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Ciferri, M.; Quarto, R.; Tasso, R. Extracellular Vesicles as Biomarkers and Therapeutic Tools: From Pre-Clinical to Clinical Applications. Biology 2021, 10, 359. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.; Matsuda, A.; Yan, I.K.; Parasramka, M.; Patel, T. Extracellular vesicles in liver diseases. Am. J. Physiol.-Gastrointest. Liver Physiol. 2017, 312, G194–G200. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, R.; Velazquez, V.M.; Brigstock, D.R. Fibrogenic Signaling Is Suppressed in Hepatic Stellate Cells through Targeting of Connective Tissue Growth Factor (CCN2) by Cellular or Exosomal MicroRNA-199a-5p. Am. J. Pathol. 2016, 186, 2921–2933. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, R.; Kemper, S.; Charrier, A.; Brigstock, D.R. Suppression of fibrogenic signaling in hepatic stellate cells by Twist1-dependent microRNA-214 expression: Role of exosomes in horizontal transfer of Twist1. Am. J. Physiol. Liver Physiol. 2015, 309, G491–G499. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Brenner, D.A.; Kisseleva, T. Combatting Fibrosis: Exosome-Based Therapies in the Regression of Liver Fibrosis. Hepatol. Commun. 2018, 3, 180–192. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Kemper, S.; Brigstock, D.R. Pathways of production and delivery of hepatocyte exosomes. J. Cell Commun. Signal. 2017, 12, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Devhare, P.B.; Sasaki, R.; Shrivastava, S.; Di Bisceglie, A.M.; Ray, R.; Ray, R.B. Exosome-mediated intercellular communication between hepatitis C virus-infected hepatocytes and hepatic stellate cells. J. Virol. 2017, 91, e02225-16. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.M.; Abdelmohsen, K.; Mustapic, M.; Kapogiannis, D.; Gorospe, M. RNA in extracellular vesicles. Wiley Interdiscip. Rev. RNA 2017, 8, e1413. [Google Scholar] [CrossRef]
- Hyun, J.; Wang, S.; Kim, J.; Rao, K.M.; Park, S.Y.; Chung, I.; Ha, C.-S.; Kim, S.-W.; Yun, Y.H.; Jung, Y. MicroRNA-378 limits activation of hepatic stellate cells and liver fibrosis by suppressing Gli3 expression. Nat. Commun. 2016, 7, 10993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Benz, F.; Cardenas, D.V.; Vucur, M.; Gautheron, J.; Schneider, A.; Hellerbrand, C.; Pottier, N.; Alder, J.; Tacke, F.; et al. miR-30c and miR-193 are a part of the TGF-β-dependent regulatory network controlling extracellular matrix genes in liver fibrosis. J. Dig. Dis. 2015, 16, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Peng, L.; Gong, X.; Zhang, X.; Sun, R.; Du, J. miR-423-5p Inhibits Osteosarcoma Proliferation and Invasion Through Directly Targeting STMN1. Cell. Physiol. Biochem. 2018, 50, 2249–2259. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zeng, X.; Huang, Y.; Chen, S.; Lin, F.; Yang, G.; Yang, N. miR-423-5p serves as a diagnostic indicator and inhibits the proliferation and invasion of ovarian cancer. Exp. Ther. Med. 2018, 15, 4723–4730. [Google Scholar] [CrossRef] [PubMed]
- Stiuso, P.; Potenza, N.; Lombardi, A.; Ferrandino, I.; Monaco, A.; Zappavigna, S.; Vanacore, D.; Mosca, N.; Castiello, F.; Porto, S.; et al. MicroRNA-423-5p Promotes Autophagy in Cancer Cells and Is Increased in Serum From Hepatocarcinoma Patients Treated With Sorafenib. Mol. Ther.-Nucleic Acids 2015, 4, e233. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.-H.; Li, J.-W.; Sun, H.-T.; He, S.-Q.; Pang, J. Sulforaphane inhibits the activation of hepatic stellate cell by miRNA-423-5p targeting suppressor of fused. Hum. Cell 2019, 32, 403–410. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Reverse | Forward |
|---|---|---|
| 18s | CAATCCAATCGGTAGTAGCG | GTAACCCGTTGAACCCCATT |
| αSMA | GTCAGGCAGTTCGTAGCTCTTCT | CTACTGCCGAGCGTGAGATTG |
| Col1a | GAGGCACAGACGGCTGAGTAG | CTGACTGGAAGAGCGGAGAGTAC |
| TIMP1 | TGGTATCTGCTCTGGTGTGTCTCT | TGATTTCCCCGCCAACTC |
| TIMP2 | TTCTGCCTTTCCTGCAATTAGATACT | CACGGCCCCCTCTTCAG |
| PDGF | GACTCATAATCTTCAGCTCGGACAT | CACCATGAAAGTGGCTGTCAA |
| TGFβ | ACCTTTGCCAATGCTTTCTTGTA | TCACTAGATCGCCCTTTCATTTC |
| FAM3A | GATACAGCCTTCCATCTCCAG | GATCTAGCCTTCCGTGACAG |
| Mgll | GTCAACCTCCGACTTGTTCC | TGATTTCACCTCTGGTCCTTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Safran, M.; Masoud, R.; Sultan, M.; Tachlytski, I.; Chai Gadot, C.; Pery, R.; Balint-Lahat, N.; Pappo, O.; Buzaglo, N.; Ben-Ari, Z. Extracellular Vesicular Transmission of miR-423-5p from HepG2 Cells Inhibits the Differentiation of Hepatic Stellate Cells. Cells 2022, 11, 1715. https://doi.org/10.3390/cells11101715
Safran M, Masoud R, Sultan M, Tachlytski I, Chai Gadot C, Pery R, Balint-Lahat N, Pappo O, Buzaglo N, Ben-Ari Z. Extracellular Vesicular Transmission of miR-423-5p from HepG2 Cells Inhibits the Differentiation of Hepatic Stellate Cells. Cells. 2022; 11(10):1715. https://doi.org/10.3390/cells11101715
Chicago/Turabian StyleSafran, Michal, Rula Masoud, Maya Sultan, Irena Tachlytski, Chofit Chai Gadot, Ron Pery, Nora Balint-Lahat, Orit Pappo, Nahum Buzaglo, and Ziv Ben-Ari. 2022. "Extracellular Vesicular Transmission of miR-423-5p from HepG2 Cells Inhibits the Differentiation of Hepatic Stellate Cells" Cells 11, no. 10: 1715. https://doi.org/10.3390/cells11101715
APA StyleSafran, M., Masoud, R., Sultan, M., Tachlytski, I., Chai Gadot, C., Pery, R., Balint-Lahat, N., Pappo, O., Buzaglo, N., & Ben-Ari, Z. (2022). Extracellular Vesicular Transmission of miR-423-5p from HepG2 Cells Inhibits the Differentiation of Hepatic Stellate Cells. Cells, 11(10), 1715. https://doi.org/10.3390/cells11101715