
One Size Does Not Fit All: The Past, Present and Future of Cystic Fibrosis Causal Therapies



Abstract
:1. Introduction into Cystic Fibrosis
2. One Size Does Not Fit All
2.1. CFTR—Structure, Folding and Function
2.2. CFTR Mutations
3. CFTR Causal Therapies
3.1. Improving CFTR Function: Activators, Potentiators and Co-Potentiators
3.1.1. CFTR Activators
3.1.2. CFTR Potentiators
3.1.3. CFTR Co-Potentiators
3.2. Improving the Amount of CFTR at the Plasma Membrane: Correctors, Proteostasis Modulators and Stabilizers
3.2.1. CFTR Correctors
3.2.2. Proteostasis Modulators
3.2.3. Stabilizers
3.3. Improving the Amount of Immature CFTR Protein
3.3.1. Amplifiers
3.3.2. miRNA Modulation
3.3.3. Nonsense Mediated mRNA Decay (NMD) Suppression
3.3.4. Translational Readthrough Inducing Drugs (TRIDs)
3.4. Producing Correct CFTR
3.4.1. mRNA Repair
3.4.2. mRNA Therapy
3.4.3. Targeting the DNA—Towards a Cure for CF
4. Towards the Future: Personalizing Therapies for PwCF
4.1. Theratyping & Expanding the Label for Existing Therapies
4.2. What Is in the Pipeline for the Last 15% of PwCF without Causal Treatment?
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zolin, A.; Orenti, A.; Naerlich, L.; Jung, A.; van Rens, J. ECFSPR Annual Report 2018; European Cystic Fibrosis Society: Karup, Denmark, 2020. [Google Scholar]
- CFF. Cystic Fibrosis Foundation Patient Registry; 2019 Annual Data Report; Bethesda: Rockville, MD, USA, 2020. [Google Scholar]
- De Boeck, K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020, 109, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Boon, M.; Verleden, S.E.; Bosch, B.; Lammertyn, E.J.; McDonough, J.E.; Mai, C.; Verschakelen, J.; Kemner-van de Corput, M.; Tiddens, H.A.; Proesmans, M.; et al. Morphometric Analysis of Explant Lungs in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 516–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronan, N.J.; Elborn, J.S.; Plant, B.J. Current and emerging comorbidities in cystic fibrosis. La Presse Médicale 2017, 46, e125–e138. [Google Scholar] [CrossRef] [PubMed]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Borowitz, D. CFTR, bicarbonate, and the pathophysiology of cystic fibrosis. Pediatric Pulmonol. 2015, 50 (Suppl. 40), S24–S30. [Google Scholar] [CrossRef] [Green Version]
- Abou Alaiwa, M.H.; Reznikov, L.R.; Gansemer, N.D.; Sheets, K.A.; Horswill, A.R.; Stoltz, D.A.; Zabner, J.; Welsh, M.J. pH modulates the activity and synergism of the airway surface liquid antimicrobials β-defensin-3 and LL-37. Proc. Natl. Acad. Sci. USA 2014, 111, 18703–18708. [Google Scholar] [CrossRef] [Green Version]
- Simonin, J.; Bille, E.; Crambert, G.; Noel, S.; Dreano, E.; Edwards, A.; Hatton, A.; Pranke, I.; Villeret, B.; Cottart, C.-H.; et al. Airway surface liquid acidification initiates host defense abnormalities in Cystic Fibrosis. Sci. Rep. 2019, 9, 6516. [Google Scholar] [CrossRef]
- Law, S.M.; Gray, R.D. Neutrophil extracellular traps and the dysfunctional innate immune response of cystic fibrosis lung disease: A review. J. Inflamm. 2017, 14, 29. [Google Scholar] [CrossRef]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Lung Infections Associated with Cystic Fibrosis. Clin. Microbiol. Rev. 2002, 15, 194–222. [Google Scholar] [CrossRef] [Green Version]
- Csanády, L.; Vergani, P.; Gadsby, D.C. Structure, Gating, and Regulation of the CFTR Anion Channel. Physiol. Rev. 2019, 99, 707–738. [Google Scholar] [CrossRef]
- Kleizen, B.; Van Vlijmen, T.; De Jonge, H.R.; Braakman, I. Folding of CFTR Is Predominantly Cotranslational. Mol. Cell 2005, 20, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Sharma, M.; Lukacs, G.L. The ΔF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 2005, 12, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Skach, W.R. Mechanisms of CFTR Folding at the Endoplasmic Reticulum. Front. Pharmacol. 2012, 3, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farinha, C.M.; Canato, S. From the endoplasmic reticulum to the plasma membrane: Mechanisms of CFTR folding and trafficking. Cell. Mol. Life Sci. 2017, 74, 39–55. [Google Scholar] [CrossRef]
- Mijnders, M.; Kleizen, B.; Braakman, I. Correcting CFTR folding defects by small-molecule correctors to cure cystic fibrosis. Curr. Opin. Pharmacol. 2017, 34, 83–90. [Google Scholar] [CrossRef]
- Coppinger, J.A.; Hutt, D.M.; Razvi, A.; Koulov, A.V.; Pankow, S.; Yates, J.R.; Balch, W.E. A Chaperone Trap Contributes to the Onset of Cystic Fibrosis. PLoS ONE 2012, 7, e37682. [Google Scholar] [CrossRef] [Green Version]
- Farinha, C.M.; Amaral, M.D. Most F508del-CFTR Is Targeted to Degradation at an Early Folding Checkpoint and Independently of Calnexin. Mol. Cell. Biol. 2005, 25, 5242–5252. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Matteson, J.; An, Y.; Moyer, B.; Yoo, J.-S.; Bannykh, S.; Wilson, I.A.; Riordan, J.R.; Balch, W.E. COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code. J. Cell Biol. 2004, 167, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Michelsen, K.; Yuan, H.; Schwappach, B. Hide and run. EMBO Rep. 2005, 6, 717–722. [Google Scholar] [CrossRef]
- McClure, M.L.; Barnes, S.; Brodsky, J.L.; Sorscher, E.J. Trafficking and function of the cystic fibrosis transmembrane conductance regulator: A complex network of posttranslational modifications. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 311, L719–L733. [Google Scholar] [CrossRef] [Green Version]
- Okiyoneda, T.; Barrière, H.; Bagdány, M.; Rabeh, W.M.; Du, K.; Höhfeld, J.; Young, J.C.; Lukacs, G.L. Peripheral Protein Quality Control Removes Unfolded CFTR from the Plasma Membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H.; et al. Misfolding diverts CFTR from recycling to degradation. J. Cell Biol. 2004, 164, 923–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegedűs, T.; Aleksandrov, A.; Mengos, A.; Cui, L.; Jensen, T.J.; Riordan, J.R. Role of individual R domain phosphorylation sites in CFTR regulation by protein kinase A. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2009, 1788, 1341–1349. [Google Scholar] [CrossRef] [Green Version]
- Hwang, T.-C.; Sheppard, D.N. Gating of the CFTR Cl− channel by ATP-driven nucleotide-binding domain dimerisation. J. Physiol. 2009, 587, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.C.; Kirk, K.L. The CFTR Ion Channel: Gating, Regulation, and Anion Permeation. Cold Spring Harb. Perspect. Med. 2013, 3, a009498. [Google Scholar] [CrossRef] [Green Version]
- Yeh, H.I.; Yu, Y.C.; Kuo, P.L.; Tsai, C.K.; Huang, H.T.; Hwang, T.C. Functional stability of CFTR depends on tight binding of ATP at its degenerate ATP-binding site. J. Physiol. 2021, 599, 4625–4642. [Google Scholar] [CrossRef]
- Hwang, T.-C.; Yeh, J.-T.; Zhang, J.; Yu, Y.-C.; Yeh, H.-I.; Destefano, S. Structural mechanisms of CFTR function and dysfunction. J. Gen. Physiol. 2018, 150, 539–570. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, F.; Chen, J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. USA 2018, 115, 12757–12762. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Zhang, Z.; Csanády, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95.e88. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Chen, J. Atomic Structure of the Cystic Fibrosis Transmembrane Conductance Regulator. Cell 2016, 167, 1586–1597.e9. [Google Scholar] [CrossRef] [Green Version]
- Fay, J.F.; Aleksandrov, L.A.; Jensen, T.J.; Cui, L.L.; Kousouros, J.N.; He, L.; Aleksandrov, A.A.; Gingerich, D.S.; Riordan, J.R.; Chen, J.Z. Cryo-EM Visualization of an Active High Open Probability CFTR Anion Channel. Biochemistry 2018, 57, 6234–6246. [Google Scholar] [CrossRef] [PubMed]
- Fiedorczuk, K.; Chen, J. Mechanism of CFTR correction by type I folding correctors. Cell 2022, 185, 158–168.e11. [Google Scholar] [CrossRef] [PubMed]
- Farkas, B.; Tordai, H.; Padányi, R.; Tordai, A.; Gera, J.; Paragi, G.; Hegedűs, T. Discovering the chloride pathway in the CFTR channel. Cell. Mol. Life Sci. 2020, 77, 765–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gené, G.G.; Llobet, A.; Larriba, S.; De Semir, D.; Martínez, I.; Escalada, A.; Solsona, C.; Casals, T.; Aran, J.M. N-terminal CFTR missense variants severely affect the behavior of the CFTR chloride channel. Hum. Mutat. 2008, 29, 738–749. [Google Scholar] [CrossRef]
- Sabusap, C.M.; Joshi, D.; Simhaev, L.; Oliver, K.E.; Senderowitz, H.; Van Willigen, M.; Braakman, I.; Rab, A.; Sorscher, E.J.; Hong, J.S. The CFTR P67L variant reveals a key role for N-terminal lasso helices in channel folding, maturation, and pharmacologic rescue. J. Biol. Chem. 2021, 296, 100598. [Google Scholar] [CrossRef]
- Ren, H.Y.; Grove, D.E.; De La Rosa, O.; Houck, S.A.; Sopha, P.; Van Goor, F.; Hoffman, B.J.; Cyr, D.M. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Classification of CFTR mutation classes. Lancet Respir. Med. 2016, 4, e37–e38. [Google Scholar] [CrossRef] [Green Version]
- Dörk, T.; Macek, M., Jr.; Mekus, F.; Tümmler, B.; Tzountzouris, J.; Casals, T.; Krebsová, A.; Koudová, M.; Sakmaryová, I.; Macek Sr, M.; et al. Characterization of a novel 21-kb deletion, CFTRdele2,3(21 kb), in the CFTR gene: A cystic fibrosis mutation of Slavic origin common in Central and East Europe. Hum. Genet. 2000, 106, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Sosnay, P.R.; Ramalho, A.S.; Douville, C.; Franca, A.; Gottschalk, L.B.; Park, J.; Lee, M.; Vecchio-Pagan, B.; Raraigh, K.S.; et al. Experimental Assessment of Splicing Variants Using Expression Minigenes and Comparison with In Silico Predictions. Hum. Mutat. 2014, 35, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A.; Luz, V.C.C.; Targowski, S.; Ramalho, S.S.; Farinha, C.M.; Amaral, M.D. Integrity and Stability of PTC Bearing CFTR mRNA and Relevance to Future Modulator Therapies in Cystic Fibrosis. Genes 2021, 12, 1810. [Google Scholar] [CrossRef] [PubMed]
- Ensinck, M.; De Keersmaecker, L.; Heylen, L.; Ramalho, A.S.; Gijsbers, R.; Farré, R.; De Boeck, K.; Christ, F.; Debyser, Z.; Carlon, M.S. Phenotyping of Rare CFTR Mutations Reveals Distinct Trafficking and Functional Defects. Cells 2020, 9, 754. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Kennedy, A.S.; Houck, S.; Aleksandrov, A.; Quinney, N.L.; Cyr-Scully, A.; Cholon, D.M.; Gentzsch, M.; Randell, S.H.; Ren, H.Y.; et al. DNAJB12 and Hsp70 triage arrested intermediates of N1303K-CFTR for endoplasmic reticulum-associated autophagy. Mol. Biol. Cell 2021, 32, 538–553. [Google Scholar] [CrossRef]
- Bompadre, S.G.; Li, M.; Hwang, T.-C. Mechanism of G551D-CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) Potentiation by a High Affinity ATP Analog. J. Biol. Chem. 2008, 283, 5364–5369. [Google Scholar] [CrossRef] [Green Version]
- Chiba-Falek, O.; Kerem, E.; Shoshani, T.; Aviram, M.; Augarten, A.; Bentur, L.; Tal, A.; Tullis, E.; Rahat, A.; Kerem, B. The Molecular Basis of Disease Variability among Cystic Fibrosis Patients Carrying the 3849+10 kb C→T Mutation. Genomics 1998, 53, 276–283. [Google Scholar] [CrossRef]
- Fukuda, R.; Okiyoneda, T. Peripheral Protein Quality Control as a Novel Drug Target for CFTR Stabilizer. Front. Pharmacol. 2018, 9, 1100. [Google Scholar] [CrossRef] [Green Version]
- Haardt, M.; Benharouga, M.; Lechardeur, D.; Kartner, N.; Lukacs, G.L. C-terminal Truncations Destabilize the Cystic Fibrosis Transmembrane Conductance Regulator without Impairing Its Biogenesis. J. Biol. Chem. 1999, 274, 21873–21877. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Benharouga, M.; Hu, W.; Lukacs, G.L. Conformational and Temperature-sensitive Stability Defects of the ΔF508 Cystic Fibrosis Transmembrane Conductance Regulator in Post-endoplasmic Reticulum Compartments. J. Biol. Chem. 2001, 276, 8942–8950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O'Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef]
- Lukacs, G.L.; Chang, X.B.; Bear, C.; Kartner, N.; Mohamed, A.; Riordan, J.R.; Grinstein, S. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J. Biol. Chem. 1993, 268, 21592–21598. [Google Scholar] [CrossRef]
- Dalemans, W.; Barbry, P.; Champigny, G.; Jallat, S.; Jallat, S.; Dott, K.; Dreyer, D.; Crystal, R.G.; Pavirani, A.; Lecocq, J.-P.; et al. Altered chloride ion channel kinetics associated with the ΔF508 cystic fibrosis mutation. Nature 1991, 354, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Eckford, P.D.; Bartlett, C.; Ouyang, H.; Gunawardena, T.N.; Gonska, T.; Moraes, T.J.; Bear, C.E. Functional rescue of c.3846G>A (W1282X) in patient-derived nasal cultures achieved by inhibition of nonsense mediated decay and protein modulators with complementary mechanisms of action. J. Cyst. Fibros. 2020, 19, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Destefano, S.; Gees, M.; Hwang, T.-C. Physiological and pharmacological characterization of the N1303K mutant CFTR. J. Cyst. Fibros. 2018, 17, 573–581. [Google Scholar] [CrossRef]
- FDA. Press Announcements—FDA Approves Kalydeco to Treat Rare form of Cystic Fibrosis; FDA: Silver Spring, MD, USA, 2012. [Google Scholar]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- FDA. FDA Expands Approved Use of Kalydeco to Treat Additional Mutations of Cystic Fibrosis; FDA: Silver Spring, MD, USA, 2017. [Google Scholar]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef] [Green Version]
- Vertex. Vertex Announces FDA Approvals of TRIKAFTA® (Elexacaftor/Tezacaftor/Ivacaftor and Ivacaftor),SYMDEKO® (Tezacaftor/Ivacaftor and Ivacaftor) and KALYDECO® (Ivacaftor) for Use in People WithCF with Certain Rare Mutations; Vertex: Boston, MA, USA, 2020. [Google Scholar]
- Vertex. Who KALYDECO Is for. 2022. Available online: https://www.kalydeco.com/who-kalydeco (accessed on 10 May 2022).
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [Green Version]
- FDA. Press Announcements—FDA Approves New Treatment for Cystic Fibrosis; FDA: Silver Spring, MD, USA, 2015. [Google Scholar]
- FDA. Drug Trials Snapshots: SYMDEKO|FDA. 2018. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trials-snapshots-symdeko (accessed on 10 May 2022).
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Drumm, M.L.; Wilkinson, D.J.; Smit, L.S.; Worrell, R.T.; Strong, T.V.; Frizzell, R.A.; Dawson, D.C.; Collins, F.S. Chloride Conductance Expressed by (F508) and Other Mutant CFTRs in Xenopus Oocytes. Science 1991, 254, 1797. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.; Schey, R. Lubiprostone in constipation: Clinical evidence and place in therapy. Ther. Adv. Chronic Dis. 2015, 6, 40–50. [Google Scholar] [CrossRef]
- Bijvelds, M.J.C.; Bot, A.G.M.; Escher, J.C.; De Jonge, H.R. Activation of Intestinal Cl− Secretion by Lubiprostone Requires the Cystic Fibrosis Transmembrane Conductance Regulator. Gastroenterology 2009, 137, 976–985. [Google Scholar] [CrossRef]
- Shaughnessy, C.A.; Yadav, S.; Bratcher, P.E.; Zeitlin, P.L. Receptor-mediated activation of CFTR via prostaglandin signaling pathways in the airway. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 322, L305–L314. [Google Scholar] [CrossRef]
- Cazzola, M.; Page, C.; Calzetta, L.; Matera, M.G. Ensifentrine (RPL554): An inhaled ‘bifunctional’ dual PDE3/4 inhibitor for the treatment of asthma and chronic obstructive pulmonary disease. Pharm. Pat. Anal. 2018, 7, 249–257. [Google Scholar] [CrossRef]
- Turner, M.J.; Matthes, E.; Billet, A.; Ferguson, A.J.; Thomas, D.Y.; Randell, S.H.; Ostrowski, L.E.; Abbott-Banner, K.; Hanrahan, J.W. The dual phosphodiesterase 3 and 4 inhibitor RPL554 stimulates CFTR and ciliary beating in primary cultures of bronchial epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L59–L70. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.J.; Luo, Y.; Thomas, D.Y.; Hanrahan, J.W. The dual phosphodiesterase 3/4 inhibitor RPL554 stimulates rare class III and IV CFTR mutants. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L908–L920. [Google Scholar] [CrossRef]
- Verona Pharma. Verona Pharma Reports Positive Top-Line Data from Phase 2a Trial with RPL554 in Cystic Fibrosis Patients; FDA: Silver Spring, MD, USA, 2018. [Google Scholar]
- Gunderson, K.L.; Kopito, R.R. Effects of pyrophosphate and nucleotide analogs suggest a role for ATP hydrolysis in cystic fibrosis transmembrane regulator channel gating. J. Biol. Chem. 1994, 269, 19349–19353. [Google Scholar] [CrossRef]
- Murthy, M.; Pedemonte, N.; MacVinish, L.; Galietta, L.; Cuthbert, A. 4-Chlorobenzo[F]isoquinoline (CBIQ), a novel activator of CFTR and ΔF508 CFTR. Eur. J. Pharmacol. 2005, 516, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, X.; Li, M.; Sohma, Y.; Zou, X.; Hwang, T.C. High affinity ATP/ADP analogues as new tools for studying CFTR gating. J. Physiol. 2005, 569, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Berger, H.A.; Travis, S.M.; Welsh, M.J. Regulation of the cystic fibrosis transmembrane conductance regulator Cl− channel by specific protein kinases and protein phosphatases. J. Biol. Chem. 1993, 268, 2037–2047. [Google Scholar] [CrossRef]
- Illek, B.; Fischer, H. Flavonoids stimulate Cl conductance of human airway epithelium in vitro and in vivo. Am. J. Physiol. 1998, 275, L902–L910. [Google Scholar] [CrossRef]
- Illek, B.; Fischer, H.; Santos, G.F.; Widdicombe, J.H.; Machen, T.E.; Reenstra, W.W. cAMP-independent activation of CFTR Cl channels by the tyrosine kinase inhibitor genistein. Am. J. Physiol. 1995, 268, C886–C893. [Google Scholar] [CrossRef]
- Wang, F.; Zeltwanger, S.; Yang, I.C.H.; Nairn, A.C.; Hwang, T.-C. Actions of Genistein on Cystic Fibrosis Transmembrane Conductance Regulator Channel Gating. J. Gen. Physiol. 1998, 111, 477–490. [Google Scholar] [CrossRef] [Green Version]
- Lansdell, K.A.; Cai, Z.; Kidd, J.F.; Sheppard, D.N. Two mechanisms of genistein inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in murine cell line. J. Physiol. 2000, 524, 317–330. [Google Scholar] [CrossRef]
- Pedemonte, N.; Sonawane, N.D.; Taddei, A.; Hu, J.; Zegarra-Moran, O.; Suen, Y.F.; Robins, L.I.; Dicus, C.W.; Willenbring, D.; Nantz, M.H.; et al. Phenylglycine and Sulfonamide Correctors of Defective ΔF508 and G551D Cystic Fibrosis Transmembrane Conductance Regulator Chloride-Channel Gating. Mol. Pharmacol. 2005, 67, 1797–1807. [Google Scholar] [CrossRef] [Green Version]
- Van Goor, F.; Straley, K.S.; Cao, D.; González, J.; Hadida, S.; Hazlewood, A.; Joubran, J.; Knapp, T.; Makings, L.R.; Miller, M.; et al. Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 290, L1117–L1130. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckford, P.D.W.; Li, C.; Ramjeesingh, M.; Bear, C.E. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Potentiator VX-770 (Ivacaftor) Opens the Defective Channel Gate of Mutant CFTR in a Phosphorylation-dependent but ATP-independent Manner. J. Biol. Chem. 2012, 287, 36639–36649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jih, K.-Y.; Hwang, T.-C. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc. Natl. Acad. Sci. USA 2013, 110, 4404–4409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, H.-I.; Yeh, J.-T.; Hwang, T.-C. Modulation of CFTR gating by permeant ions. J. Gen. Physiol. 2015, 145, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrnes, L.J.; Xu, Y.; Qiu, X.; Hall, J.D.; West, G.M. Sites associated with Kalydeco binding on human Cystic Fibrosis Transmembrane Conductance Regulator revealed by Hydrogen/Deuterium Exchange. Sci. Rep. 2018, 8, 4664. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural identification of a hotspot on CFTR for potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef]
- Laselva, O.; Qureshi, Z.; Zeng, Z.-W.; Petrotchenko, E.V.; Ramjeesingh, M.; Hamilton, C.M.; Huan, L.-J.; Borchers, C.H.; Pomès, R.; Young, R.; et al. Identification of binding sites for ivacaftor on the cystic fibrosis transmembrane conductance regulator. iScience 2021, 24, 102542. [Google Scholar] [CrossRef]
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther, C.R.; Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.C.; Gentzsch, M. Potentiator ivacaftor abrogates pharmacological correction of F508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014, 6, 246ra96. [Google Scholar] [CrossRef] [Green Version]
- Phuan, P.-W.; Veit, G.; Tan, J.A.; Finkbeiner, W.E.; Lukacs, G.L.; Verkman, A.S. Potentiators of Defective ΔF508–CFTR Gating that Do Not Interfere with Corrector Action. Mol. Pharmacol. 2015, 88, 791–799. [Google Scholar] [CrossRef]
- Van der Plas, S.E.; Kelgtermans, H.; De Munck, T.; Martina, S.L.X.; Dropsit, S.; Quinton, E.; De Blieck, A.; Joannesse, C.; Tomaskovic, L.; Jans, M.; et al. Discovery of N-(3-Carbamoyl-5,5,7,7-tetramethyl-5,7-dihydro-4H-thieno[2,3-c]pyran-2-yl)-lH-pyrazole-5-carboxamide (GLPG1837), a Novel Potentiator Which Can Open Class III Mutant Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Channels to a High Extent. J. Med. Chem. 2018, 61, 1425–1435. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Da Fonte, D.F.; Avramescu, R.G.; Premchandar, A.; Bagdany, M.; Xu, H.; Bensinger, D.; Stubba, D.; Schmidt, B.; Matouk, E.; et al. Mutation-specific dual potentiators maximize rescue of CFTR gating mutants. J. Cyst. Fibros. 2020, 19, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.I.; Qiu, L.; Sohma, Y.; Conrath, K.; Zou, X.; Hwang, T.C. Identifying the molecular target sites for CFTR potentiators GLPG1837 and VX-770. J. Gen. Physiol. 2019, 151, 912–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, H.-I.; Sohma, Y.; Conrath, K.; Hwang, T.-C. A common mechanism for CFTR potentiators. J. Gen. Physiol. 2017, 149, 1105–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Khloya, P.; Seo, Y.; Kumar, S.; Lee, H.K.; Jeon, D.K.; Jo, S.; Sharma, P.K.; Namkung, W. Potentiation of ΔF508- and G551D-CFTR-Mediated Cl− Current by Novel Hydroxypyrazolines. PLoS ONE 2016, 11, e0149131. [Google Scholar] [CrossRef] [Green Version]
- Gees, M.; Musch, S.; Van der Plas, S.; Wesse, A.-S.; Vandevelde, A.; Verdonck, K.; Mammoliti, O.; Hwang, T.-C.; Sonck, K.; Stouten, P.; et al. Identification and Characterization of Novel CFTR Potentiators. Front. Pharmacol. 2018, 9, 1221. [Google Scholar] [CrossRef]
- Davies, J.C.; Van de Steen, O.; van Koningsbruggen-Rietschel, S.; Drevinek, P.; Derichs, N.; McKone, E.F.; Kanters, D.; Allamassey, L.; Namour, F.; de Kock, H.; et al. GLPG1837, a CFTR potentiator, in p.Gly551Asp (G551D)-CF patients: An open-label, single-arm, phase 2a study (SAPHIRA1). J. Cyst. Fibros. 2019, 18, 693–699. [Google Scholar] [CrossRef] [Green Version]
- Downey, D.G.; Fajac, I.; Flume, P.; O’Carroll, M.; Pressler, T.; Proesmans, M.; Quon, B.; Schwarz, C.; Sutharsan, S.; Jiang, J.; et al. WS11.5 Evaluation of combinations of the CFTR potentiator dirocaftor, corrector posenacaftor and amplifier nesolicaftor in cystic fibrosis subjects with two copies of the F508del mutation. J. Cyst. Fibros. 2020, 19, S19. [Google Scholar] [CrossRef]
- Rowe, S.M.; Jones, I.; Dransfield, M.T.; Haque, N.; Gleason, S.; Hayes, K.A.; Kulmatycki, K.; Yates, D.P.; Danahay, H.; Gosling, M.; et al. Efficacy and Safety of the CFTR Potentiator Icenticaftor (QBW251) in COPD: Results from a Phase 2 Randomized Trial. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 2399–2409. [Google Scholar] [CrossRef]
- Volkova, N.; Moy, K.; Evans, J.; Campbell, D.; Tian, S.; Simard, C.; Higgins, M.; Konstan, M.W.; Sawicki, G.S.; Elbert, A.; et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. J. Cyst. Fibros. 2020, 19, 68–79. [Google Scholar] [CrossRef]
- Van Goor, F.; Yu, H.; Burton, B.; Hoffman, B.J. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros. 2014, 13, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, J.F.; Van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Berkers, G.; de Winter-de Groot, K.M.; Janssens, H.M.; Bronsveld, I.; van der Ent, C.K.; de Jonge, H.R.; et al. Potentiator synergy in rectal organoids carrying S1251N, G551D, or F508del CFTR mutations. J. Cyst. Fibros. 2016, 15, 568–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phuan, P.-W.; Son, J.-H.; Tan, J.-A.; Li, C.; Musante, I.; Zlock, L.; Nielson, D.W.; Finkbeiner, W.E.; Kurth, M.J.; Galietta, L.J.; et al. Combination potentiator (‘co-potentiator’) therapy for CF caused by CFTR mutants, including N1303K, that are poorly responsive to single potentiators. J. Cyst. Fibros. 2018, 17, 595–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phuan, P.-W.; Tan, J.-A.; Rivera, A.A.; Zlock, L.; Nielson, D.W.; Finkbeiner, W.E.; Haggie, P.M.; Verkman, A.S. Nanomolar-potency ‘co-potentiator’ therapy for cystic fibrosis caused by a defined subset of minimal function CFTR mutants. Sci. Rep. 2019, 9, 17640. [Google Scholar] [CrossRef] [PubMed]
- Haggie, P.M.; Phuan, P.-W.; Tan, J.-A.; Xu, H.; Avramescu, R.G.; Perdomo, D.; Zlock, L.; Nielson, D.W.; Finkbeiner, W.E.; Lukacs, G.L.; et al. Correctors and Potentiators Rescue Function of the Truncated W1282X-Cystic Fibrosis Transmembrane Regulator (CFTR) Translation Product. J. Biol. Chem. 2017, 292, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ensinck, M.M.; De Keersmaecker, L.; Ramalho, A.S.; Cuyx, S.; Van Biervliet, S.; Dupont, L.; Christ, F.; Debyser, Z.; Vermeulen, F.; Carlon, M.S. Novel CFTR modulator combinations maximize rescue of G85E and N1303K in rectal organoids. ERJ Open Res. 2022, 8, 00716–02021. [Google Scholar] [CrossRef]
- Laselva, O.; Bartlett, C.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Moraes, T.J.; Bear, C.E.; Gonska, T. Rescue of multiple class II CFTR mutations by elexacaftor+tezacaftor+ivacaftor mediated in part by the dual activities of elexacaftor as both corrector and potentiator. Eur. Respir. J. 2021, 57, 2002774. [Google Scholar] [CrossRef]
- Veit, G.; Vaccarin, C.; Lukacs, G.L. Elexacaftor co-potentiates the activity of F508del and gating mutants of CFTR. J. Cyst. Fibros. 2021, 20, 895–898. [Google Scholar] [CrossRef]
- Denning, G.M.; Anderson, M.P.; Amara, J.F.; Marshall, J.; Smith, A.E.; Welsh, M.J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992, 358, 761–764. [Google Scholar] [CrossRef]
- Patrick, A.E.; Karamyshev, A.L.; Millen, L.; Thomas, P.J. Alteration of CFTR transmembrane span integration by disease-causing mutations. Mol. Biol. Cell 2011, 22, 4461–4471. [Google Scholar] [CrossRef]
- Rapino, D.; Sabirzhanova, I.; Lopes-Pacheco, M.; Grover, R.; Guggino, W.B.; Cebotaru, L. Rescue of NBD2 Mutants N1303K and S1235R of CFTR by Small-Molecule Correctors and Transcomplementation. PLoS ONE 2015, 10, e0119796. [Google Scholar] [CrossRef] [Green Version]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M.; et al. Revertants, Low Temperature, and Correctors Reveal the Mechanism of F508del-CFTR Rescue by VX-809 and Suggest Multiple Agents for Full Correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Ward, C.L.; Krouse, M.E.; Wine, J.J.; Kopito, R.R. Glycerol Reverses the Misfolding Phenotype of the Most Common Cystic Fibrosis Mutation. J. Biol. Chem. 1996, 271, 635–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Correctors Promote Maturation of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR)-processing Mutants by Binding to the Protein. J. Biol. Chem. 2007, 282, 33247–33251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.; Simon, A.; et al. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Additive effect of multiple pharmacological chaperones on maturation of CFTR processing mutants. Biochem. J. 2007, 406, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Verkman, A.S. Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harb. Perspect. Med. 2013, 3, a009761. [Google Scholar] [CrossRef] [Green Version]
- Pedemonte, N. Small-molecule correctors of defective F508-CFTR cellular processing identified by high-throughput screening. J. Clin. Investig. 2005, 115, 2564–2571. [Google Scholar] [CrossRef]
- Hirth, B.H.; Qiao, S.; Cuff, L.M.; Cochran, B.M.; Pregel, M.J.; Gregory, J.S.; Sneddon, S.F.; Kane, J.L. Discovery of 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid diamides that increase CFTR mediated chloride transport. Bioorganic Med. Chem. Lett. 2005, 15, 2087–2091. [Google Scholar] [CrossRef]
- Botelho, H.M.; Uliyakina, I.; Awatade, N.T.; Proença, M.C.; Tischer, C.; Sirianant, L.; Kunzelmann, K.; Pepperkok, R.; Amaral, M.D. Protein Traffic Disorders: An Effective High-Throughput Fluorescence Microscopy Pipeline for Drug Discovery. Sci. Rep. 2015, 5, 9038. [Google Scholar] [CrossRef] [Green Version]
- Carlile, G.W.; Robert, R.; Zhang, D.; Teske, K.A.; Luo, Y.; Hanrahan, J.W.; Thomas, D.Y. Correctors of protein trafficking defects identified by a novel high-throughput screening assay. Chembiochem 2007, 8, 1012–1020. [Google Scholar] [CrossRef]
- Sampson, H.M.; Robert, R.; Liao, J.; Matthes, E.; Carlile, G.W.; Hanrahan, J.W.; Thomas, D.Y. Identification of a NBD1-binding pharmacological chaperone that corrects the trafficking defect of F508del-CFTR. Chem. Biol. 2011, 18, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Phuan, P.-W.; Veit, G.; Tan, J.-A.; Roldan, A.; Finkbeiner, W.E.; Haggie, P.M.; Lukacs, G.L.; Verkman, A.S. ΔF508-CFTR Modulator Screen Based on Cell Surface Targeting of a Chimeric Nucleotide Binding Domain 1 Reporter. SLAS DISCOVERY Adv. Sci. Drug Discov. 2018, 23, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Robert, R.; Carlile, G.W.; Liao, J.; Balghi, H.; Lesimple, P.; Liu, N.; Kus, B.; Rotin, D.; Wilke, M.; De Jonge, H.R.; et al. Correction of the ΔPhe508 Cystic Fibrosis Transmembrane Conductance Regulator Trafficking Defect by the Bioavailable Compound Glafenine. Mol. Pharmacol. 2010, 77, 922–930. [Google Scholar] [CrossRef] [Green Version]
- Shishido, H.; Yoon, J.S.; Skach, W.R. A small molecule high throughput screening platform to profile conformational properties of nascent, ribosome-bound proteins. Sci. Rep. 2022, 12, 2509. [Google Scholar] [CrossRef]
- Grove, D.E.; Rosser, M.F.N.; Ren, H.Y.; Naren, A.P.; Cyr, D.M. Mechanisms for Rescue of Correctable Folding Defects in CFTRΔF508. Mol. Biol. Cell 2009, 20, 4059–4069. [Google Scholar] [CrossRef] [Green Version]
- Laselva, O.; Molinski, S.; Casavola, V.; Bear, C.E. Correctors of the Major Cystic Fibrosis Mutant Interact through Membrane-Spanning Domains. Mol. Pharmacol. 2018, 93, 612–618. [Google Scholar] [CrossRef] [Green Version]
- Pesce, E.; Bellotti, M.; Liessi, N.; Guariento, S.; Damonte, G.; Cichero, E.; Galatini, A.; Salis, A.; Gianotti, A.; Pedemonte, N.; et al. Synthesis and structure–activity relationship of aminoarylthiazole derivatives as correctors of the chloride transport defect in cystic fibrosis. Eur. J. Med. Chem. 2015, 99, 14–35. [Google Scholar] [CrossRef] [PubMed]
- Brandas, C.; Ludovico, A.; Parodi, A.; Moran, O.; Millo, E.; Cichero, E.; Baroni, D. NBD2 Is Required for the Rescue of Mutant F508del CFTR by a Thiazole-Based Molecule: A Class II Corrector for the Multi-Drug Therapy of Cystic Fibrosis. Biomolecules 2021, 11, 1417. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.A.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Correctors Enhance Maturation of ΔF508 CFTR by Promoting Interactions between the Two Halves of the Molecule. Biochemistry 2009, 48, 9882–9890. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Rescue of ΔF508 and Other Misprocessed CFTR Mutants by a Novel Quinazoline Compound. Mol. Pharm. 2005, 2, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Chiaw, P.K.; Wellhauser, L.; Huan, L.J.; Ramjeesingh, M.; Bear, C.E. A Chemical Corrector Modifies the Channel Function of F508del-CFTR. Mol. Pharmacol. 2010, 78, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Kota, P.; Aleksandrov, A.A.; Cui, L.; Jensen, T.; Dokholyan, N.V.; Riordan, J.R. Correctors of ΔF508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 2013, 27, 536–545. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem. Pharmacol. 2013, 86, 612–619. [Google Scholar] [CrossRef]
- Krainer, G.; Treff, A.; Hartmann, A.; Stone, T.A.; Schenkel, M.; Keller, S.; Deber, C.M.; Schlierf, M. A minimal helical-hairpin motif provides molecular-level insights into misfolding and pharmacological rescue of CFTR. Commun. Biol. 2018, 1, 154. [Google Scholar] [CrossRef] [PubMed]
- Krainer, G.; Schenkel, M.; Hartmann, A.; Ravamehr-Lake, D.; Deber, C.M.; Schlierf, M. CFTR transmembrane segments are impaired in their conformational adaptability by a pathogenic loop mutation and dynamically stabilized by Lumacaftor. J. Biol. Chem. 2020, 295, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Kleizen, B.; van Willigen, M.; Mijnders, M.; Peters, F.; Grudniewska, M.; Hillenaar, T.; Thomas, A.; Kooijman, L.; Peters, K.W.; Frizzell, R.; et al. Co-Translational Folding of the First Transmembrane Domain of ABC-Transporter CFTR is Supported by Assembly with the First Cytosolic Domain. J. Mol. Biol. 2021, 433, 166955. [Google Scholar] [CrossRef]
- Hudson, R.P.; Dawson, J.E.; Chong, P.A.; Yang, Z.; Millen, L.; Thomas, P.J.; Brouillette, C.G.; Forman-Kay, J.D. Direct Binding of the Corrector VX-809 to Human CFTR NBD1: Evidence of an Allosteric Coupling between the Binding Site and the NBD1:CL4 Interface. Mol. Pharmacol. 2017, 92, 124–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, T.W.; Clarke, D.M. Corrector VX-809 promotes interactions between cytoplasmic loop one and the first nucleotide-binding domain of CFTR. Biochem. Pharmacol. 2017, 136, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Eckford, P.; Ramjeesingh, M.; Molinski, S.; Pasyk, S.; Dekkers, J.F.; Li, C.; Ahmadi, S.; Ip, W.; Chung, T.E.; Du, K.; et al. VX-809 and Related Corrector Compounds Exhibit Secondary Activity Stabilizing Active F508del-CFTR after Its Partial Rescue to the Cell Surface. Chem. Biol. 2014, 21, 666–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, C.; Zhang, W.; Moon, C.S.; Actis, M.; Yarlagadda, S.; Arora, K.; Woodroofe, K.; Clancy, J.P.; Lin, S.; Ziady, A.G.; et al. Capturing the Direct Binding of CFTR Correctors to CFTR by Using Click Chemistry. ChemBioChem 2015, 16, 2017–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef] [Green Version]
- Laselva, O.; Marzaro, G.; Vaccarin, C.; Lampronti, I.; Tamanini, A.; Lippi, G.; Gambari, R.; Cabrini, G.; Bear, C.E.; Chilin, A.; et al. Molecular Mechanism of Action of Trimethylangelicin Derivatives as CFTR Modulators. Front. Pharmacol. 2018, 9, 719. [Google Scholar] [CrossRef]
- Singh, A.K.; Fan, Y.; Balut, C.; Alani, S.; Manelli, A.M.; Swensen, A.M.; Jia, Y.; Neelands, T.R.; Vortherms, T.A.; Liu, B.; et al. Biological Characterization of F508delCFTR Protein Processing by the CFTR Corrector ABBV-2222/GLPG2222. J. Pharmacol. Exp. Ther. 2020, 372, 107–118. [Google Scholar] [CrossRef]
- Bell, S.C.; Barry, P.J.; De Boeck, K.; Drevinek, P.; Elborn, J.S.; Plant, B.J.; Minić, P.; Van Braeckel, E.; Verhulst, S.; Muller, K.; et al. CFTR activity is enhanced by the novel corrector GLPG2222, given with and without ivacaftor in two randomized trials. J. Cyst. Fibros. 2019, 18, 700–707. [Google Scholar] [CrossRef] [Green Version]
- Pedemonte, N.; Bertozzi, F.; Caci, E.; Sorana, F.; Fruscia, P.D.; Tomati, V.; Ferrera, L.; Rodríguez-Gimeno, A.; Berti, F.; Pesce, E.; et al. Discovery of a picomolar potency pharmacological corrector of the mutant CFTR chloride channel. Sci. Adv. 2020, 6, eaay9669. [Google Scholar] [CrossRef] [Green Version]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Davies, J.C.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VX-659–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef]
- Baatallah, N.; Elbahnsi, A.; Mornon, J.-P.; Chevalier, B.; Pranke, I.; Servel, N.; Zelli, R.; Décout, J.-L.; Edelman, A.; Sermet-Gaudelus, I.; et al. Pharmacological chaperones improve intra-domain stability and inter-domain assembly via distinct binding sites to rescue misfolded CFTR. Cell. Mol. Life Sci. 2021, 78, 7813–7829. [Google Scholar] [CrossRef] [PubMed]
- Stratford, F.L.L.; Pereira, M.M.C.; Becq, F.; McPherson, M.A.; Dormer, R.L. Benzo(c)quinolizinium drugs inhibit degradation of ΔF508-CFTR cytoplasmic domain. Biochem. Biophys. Res. Commun. 2003, 300, 524–530. [Google Scholar] [CrossRef]
- Dormer, R.L.; DéRand, R.; McNeilly, C.M.; Mettey, Y.; Bulteau-Pignoux, L.; MéTayé, T.; Vierfond, J.-M.; Gray, M.A.; Galietta, L.J.V.; Morris, M.R.; et al. Correction of delF508-CFTR activity with benzo(c)quinolizinium compounds through facilitation of its processing in cystic fibrosis airway cells. J. Cell Sci. 2001, 114, 4073–4081. [Google Scholar] [CrossRef] [PubMed]
- Becq, F.; Mettey, Y.; Gray, M.A.; Galietta, L.J.V.; Dormer, R.L.; Merten, M.; Métayé, T.; Chappe, V.; Marvingt-Mounir, C.; Zegarra-Moran, O.; et al. Development of Substituted Benzo[c]quinolizinium Compounds as Novel Activators of the Cystic Fibrosis Chloride Channel. J. Biol. Chem. 1999, 274, 27415–27425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odolczyk, N.; Fritsch, J.; Norez, C.; Servel, N.; Da Cunha, M.F.; Bitam, S.; Kupniewska, A.; Wiszniewski, L.; Colas, J.; Tarnowski, K.; et al. Discovery of novel potent ΔF 508-CFTR correctors that target the nucleotide binding domain. EMBO Mol. Med. 2013, 5, 1484–1501. [Google Scholar] [CrossRef]
- Bitam, S.; Elbahnsi, A.; Creste, G.; Pranke, I.; Chevalier, B.; Berhal, F.; Hoffmann, B.; Servel, N.; Baatalah, N.; Tondelier, D.; et al. New insights into structure and function of bis-phosphinic acid derivatives and implications for CFTR modulation. Sci. Rep. 2021, 11, 6842. [Google Scholar] [CrossRef]
- Nieddu, E.; Pollarolo, B.; Mazzei, M.T.; Anzaldi, M.; Schenone, S.; Pedemonte, N.; Galietta, L.J.; Mazzei, M. Phenylhydrazones as Correctors of a Mutant Cystic Fibrosis Transmembrane Conductance Regulator. Arch. Der Pharm. 2016, 349, 112–123. [Google Scholar] [CrossRef]
- Carlile, G.W.; Yang, Q.; Matthes, E.; Liao, J.; Radinovic, S.; Miyamoto, C.; Robert, R.; Hanrahan, J.W.; Thomas, D.Y. A novel triple combination of pharmacological chaperones improves F508del-CFTR correction. Sci. Rep. 2018, 8, 11404. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Pacheco, M.; Bacalhau, M.; Ramalho, S.S.; Silva, I.A.L.; Ferreira, F.C.; Carlile, G.W.; Thomas, D.Y.; Farinha, C.M.; Hanrahan, J.W.; Amaral, M.D. Rescue of Mutant CFTR Trafficking Defect by the Investigational Compound MCG1516A. Cells 2022, 11, 136. [Google Scholar] [CrossRef]
- Rubenstein, R.C.; Egan, M.E.; Zeitlin, P.L. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J. Clin. Investig. 1997, 100, 2457–2465. [Google Scholar] [CrossRef] [Green Version]
- Suaud, L.; Miller, K.; Alvey, L.; Yan, W.; Robay, A.; Kebler, C.; Kreindler, J.L.; Guttentag, S.; Hubbard, M.J.; Rubenstein, R.C. ERp29 Regulates ΔF508 and Wild-type Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Trafficking to the Plasma Membrane in Cystic Fibrosis (CF) and Non-CF Epithelial Cells. J. Biol. Chem. 2011, 286, 21239–21253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suaud, L.; Miller, K.; Panichelli, A.E.; Randell, R.L.; Marando, C.M.; Rubenstein, R.C. 4-Phenylbutyrate Stimulates Hsp70 Expression through the Elp2 Component of Elongator and STAT-3 in Cystic Fibrosis Epithelial Cells. J. Biol. Chem. 2011, 286, 45083–45092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubenstein, R.C.; Zeitlin, P.L. A Pilot Clinical Trial of Oral Sodium 4-Phenylbutyrate (Buphenyl) in Δ F508-Homozygous Cystic Fibrosis Patients. Am. J. Respir. Crit. Care Med. 1998, 157, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Venable, J.; Lapointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 Cochaperone Aha1 Downregulation Rescues Misfolding of CFTR in Cystic Fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Pankow, S.; Bamberger, C.; Calzolari, D.; Martínez-Bartolomé, S.; Lavallée-Adam, M.; Balch, W.E.; Yates, J.R. ∆F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 2015, 528, 510–516. [Google Scholar] [CrossRef] [Green Version]
- Hutt, D.M.; Herman, D.; Rodrigues, A.P.C.; Noel, S.; Pilewski, J.M.; Matteson, J.; Hoch, B.; Kellner, W.; Kelly, J.W.; Schmidt, A.; et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol. 2010, 6, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Hutt, D.M.; Olsen, C.A.; Vickers, C.J.; Herman, D.; Chalfant, M.A.; Montero, A.; Leman, L.J.; Burkle, R.; Maryanoff, B.E.; Balch, W.E.; et al. Potential Agents for Treating Cystic Fibrosis: Cyclic Tetrapeptides That Restore Trafficking and Activity of ΔF508-CFTR. ACS Med. Chem. Lett. 2011, 2, 703–707. [Google Scholar] [CrossRef]
- Anglès, F.; Hutt, D.M.; Balch, W.E. HDAC inhibitors rescue multiple disease-causing CFTR variants. Hum. Mol. Genet. 2019, 28, 1982–2000. [Google Scholar] [CrossRef]
- Bergougnoux, A.; Petit, A.; Knabe, L.; Bribes, E.; Chiron, R.; De Sario, A.; Claustres, M.; Molinari, N.; Vachier, I.; Taulan-Cadars, M.; et al. The HDAC inhibitor SAHA does not rescue CFTR membrane expression in Cystic Fibrosis. Int. J. Biochem. Cell Biol. 2017, 88, 124–132. [Google Scholar] [CrossRef]
- Sondo, E.; Tomati, V.; Caci, E.; Esposito, A.I.; Pfeffer, U.; Pedemonte, N.; Galietta, L.J.V. Rescue of the mutant CFTR chloride channel by pharmacological correctors and low temperature analyzed by gene expression profiling. Am. J. Physiol.-Cell Physiol. 2011, 301, C872–C885. [Google Scholar] [CrossRef] [Green Version]
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare Dis. 2018, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Norez, C.; Noel, S.; Wilke, M.; Bijvelds, M.; Jorna, H.; Melin, P.; Dejonge, H.; Becq, F. Rescue of functional delF508-CFTR channels in cystic fibrosis epithelial cells by the α-glucosidase inhibitor miglustat. FEBS Lett. 2006, 580, 2081–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, A.; Lebecque, P.; Dingemanse, J.; Leal, T. A randomized placebo-controlled trial of miglustat in cystic fibrosis based on nasal potential difference. J. Cyst. Fibros. 2012, 11, 231–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, M.E.; Glöckner-Pagel, J.; Ambrose, C.; Cahill, P.A.; Pappoe, L.; Balamuth, N.; Cho, E.; Canny, S.; Wagner, C.A.; Geibel, J.; et al. Calcium-pump inhibitors induce functional surface expression of Delta F508-CFTR protein in cystic fibrosis epithelial cells. Nat. Med. 2002, 8, 485–492. [Google Scholar] [CrossRef]
- Norez, C.; Vandebrouck, C.; Bertrand, J.; Noel, S.; Durieu, E.; Oumata, N.; Galons, H.; Antigny, F.; Chatelier, A.; Bois, P.; et al. Roscovitine is a proteostasis regulator that corrects the trafficking defect of F508del-CFTR by a CDK-independent mechanism. Br. J. Pharmacol. 2014, 171, 4831–4849. [Google Scholar] [CrossRef]
- Shrestha, C.L.; Zhang, S.; Wisniewski, B.; Häfner, S.; Elie, J.; Meijer, L.; Kopp, B.T. (R)-Roscovitine and CFTR modulators enhance killing of multi-drug resistant Burkholderia cenocepacia by cystic fibrosis macrophages. Sci. Rep. 2020, 10, 21700. [Google Scholar] [CrossRef]
- Meijer, L.; Nelson, D.J.; Riazanski, V.; Gabdoulkhakova, A.G.; Hery-Arnaud, G.; Le Berre, R.; Loaëc, N.; Oumata, N.; Galons, H.; Nowak, E.; et al. Modulating Innate and Adaptive Immunity by (R)-Roscovitine: Potential Therapeutic Opportunity in Cystic Fibrosis. J. Innate Immun. 2016, 8, 330–349. [Google Scholar] [CrossRef]
- Meijer, L.; Hery-Arnaud, G.; Leven, C.; Nowak, E.; Hillion, S.; Renaudineau, Y.; Durieu, I.; Chiron, R.; Prevotat, A.; Fajac, I.; et al. Safety and pharmacokinetics of Roscovitine (Seliciclib) in cystic fibrosis patients chronically infected with Pseudomonas aeruginosa, a randomized, placebo-controlled study. J. Cyst. Fibros. 2021, 21, 529–536. [Google Scholar] [CrossRef]
- Vauthier, V.; Ben Saad, A.; Elie, J.; Oumata, N.; Durand-Schneider, A.-M.; Bruneau, A.; Delaunay, J.-L.; Housset, C.; Aït-Slimane, T.; Meijer, L.; et al. Structural analogues of roscovitine rescue the intracellular traffic and the function of ER-retained ABCB4 variants in cell models. Sci. Rep. 2019, 9, 6653. [Google Scholar] [CrossRef]
- Egan, M.E.; Pearson, M.; Weiner, S.A.; Rajendran, V.; Rubin, D.; Glöckner-Pagel, J.; Canny, S.; Du, K.; Lukacs, G.L.; Caplan, M.J. Curcumin, a Major Constituent of Turmeric, Corrects Cystic Fibrosis Defects. Science 2004, 304, 600–602. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Sonawane, N.D.; Salinas, D.; Qian, L.; Pedemonte, N.; Galietta, L.J.V.; Verkman, A.S. Evidence against the Rescue of Defective ΔF508-CFTR Cellular Processing by Curcumin in Cell Culture and Mouse Models. J. Biol. Chem. 2004, 279, 40629–40633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubb, B.R.; Gabriel, S.E.; Mengos, A.; Gentzsch, M.; Randell, S.H.; Van Heeckeren, A.M.; Knowles, M.R.; Drumm, M.L.; Riordan, J.R.; Boucher, R.C. SERCA Pump Inhibitors Do Not Correct Biosynthetic Arrest of ΔF508 CFTR in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2006, 34, 355–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.L.; Randak, C.O.; Ostedgaard, L.S.; Karp, P.H.; Vermeer, D.W.; Welsh, M.J. Curcumin Stimulates Cystic Fibrosis Transmembrane Conductance Regulator Cl− Channel Activity. J. Biol. Chem. 2005, 280, 5221–5226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkers, G.; Van Der Meer, R.; Van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Suen, S.W.; Heijerman, H.G.; Majoor, C.J.; Koppelman, G.H.; Roukema, J.; et al. Clinical effects of the three CFTR potentiator treatments curcumin, genistein and ivacaftor in patients with the CFTR-S1251N gating mutation. J. Cyst. Fibros. 2020, 19, 955–961. [Google Scholar] [CrossRef]
- Carlile, G.W.; Keyzers, R.A.; Teske, K.A.; Robert, R.; Williams, D.E.; Linington, R.G.; Gray, C.A.; Centko, R.M.; Yan, L.; Anjos, S.M.; et al. Correction of F508del-CFTR Trafficking by the Sponge Alkaloid Latonduine Is Modulated by Interaction with PARP. Chem. Biol. 2012, 19, 1288–1299. [Google Scholar] [CrossRef] [Green Version]
- Carlile, G.W.; Robert, R.; Matthes, E.; Yang, Q.; Solari, R.; Hatley, R.; Edge, C.M.; Hanrahan, J.W.; Andersen, R.; Thomas, D.Y.; et al. Latonduine Analogs Restore F508del–Cystic Fibrosis Transmembrane Conductance Regulator Trafficking through the Modulation of Poly-ADP Ribose Polymerase 3 and Poly-ADP Ribose Polymerase 16 Activity. Mol. Pharmacol. 2016, 90, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Centko, R.M.; Carlile, G.W.; Barne, I.; Patrick, B.O.; Blagojevic, P.; Thomas, D.Y.; Andersen, R.J. Combination of Selective PARP3 and PARP16 Inhibitory Analogues of Latonduine A Corrects F508del-CFTR Trafficking. ACS Omega 2020, 5, 25593–25604. [Google Scholar] [CrossRef]
- Chung, W.J.; Goeckeler-Fried, J.L.; Havasi, V.; Chiang, A.; Rowe, S.M.; Plyler, Z.E.; Hong, J.S.; Mazur, M.; Piazza, G.A.; Keeton, A.B.; et al. Increasing the Endoplasmic Reticulum Pool of the F508del Allele of the Cystic Fibrosis Transmembrane Conductance Regulator Leads to Greater Folding Correction by Small Molecule Therapeutics. PLoS ONE 2016, 11, e0163615. [Google Scholar] [CrossRef]
- Goeckeler-Fried, J.L.; Aldrin Denny, R.; Joshi, D.; Hill, C.; Larsen, M.B.; Chiang, A.N.; Frizzell, R.A.; Wipf, P.; Sorscher, E.J.; Brodsky, J.L. Improved correction of F508del-CFTR biogenesis with a folding facilitator and an inhibitor of protein ubiquitination. Bioorganic Med. Chem. Lett. 2021, 48, 128243. [Google Scholar] [CrossRef]
- Borgo, C.; D’Amore, C.; Capurro, V.; Tomati, V.; Sondo, E.; Cresta, F.; Castellani, C.; Pedemonte, N.; Salvi, M. Targeting the E1 ubiquitin-activating enzyme (UBA1) improves elexacaftor/tezacaftor/ivacaftor efficacy towards F508del and rare misfolded CFTR mutants. Cell. Mol. Life Sci. 2022, 79, 192. [Google Scholar] [CrossRef]
- Sondo, E.; Falchi, F.; Caci, E.; Ferrera, L.; Giacomini, E.; Pesce, E.; Tomati, V.; Mandrup Bertozzi, S.; Goldoni, L.; Armirotti, A.; et al. Pharmacological Inhibition of the Ubiquitin Ligase RNF5 Rescues F508del-CFTR in Cystic Fibrosis Airway Epithelia. Cell Chem. Biol. 2018, 25, 891–905.e898. [Google Scholar] [CrossRef] [PubMed]
- Henning, N.J.; Boike, L.; Spradlin, J.N.; Ward, C.C.; Liu, G.; Zhang, E.; Belcher, B.P.; Brittain, S.M.; Hesse, M.J.; Dovala, D.; et al. Deubiquitinase-targeting chimeras for targeted protein stabilization. Nat. Chem. Biol. 2022, 18, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Ahner, A.; Gong, X.; Frizzell, R.A. Divergent signaling via SUMO modification: Potential for CFTR modulation. Am. J. Physiol.-Cell Physiol. 2016, 310, C175–C180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, K.W.; Gong, X.; Frizzell, R.A. Cystic Fibrosis Transmembrane Conductance Regulator Folding Mutations Reveal Differences in Corrector Efficacy Linked to Increases in Immature Cystic Fibrosis Transmembrane Conductance Regulator Expression. Front. Physiol. 2021, 12, 695767. [Google Scholar] [CrossRef]
- Gong, X.; Liao, Y.; Ahner, A.; Larsen, M.B.; Wang, X.; Bertrand, C.A.; Frizzell, R.A. Different SUMO paralogues determine the fate of wild-type and mutant CFTRs: Biogenesis versus degradation. Mol. Biol. Cell 2019, 30, 4–16. [Google Scholar] [CrossRef]
- Fukuda, R.; Okiyoneda, T. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Ubiquitylation as a Novel Pharmaceutical Target for Cystic Fibrosis. Pharmaceuticals 2020, 13, 75. [Google Scholar] [CrossRef]
- Luciani, A.; Villella, V.R.; Esposito, S.; Gavina, M.; Russo, I.; Silano, M.; Guido, S.; Pettoello-Mantovani, M.; Carnuccio, R.; Scholte, B.; et al. Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on ΔF508 cystic fibrosis transmembrane conductance regulator. Autophagy 2012, 8, 1657–1672. [Google Scholar] [CrossRef] [Green Version]
- Cherqui, S. Cysteamine therapy: A treatment for cystinosis, not a cure. Kidney Int. 2012, 81, 127–129. [Google Scholar] [CrossRef] [Green Version]
- Stefano, D.D.; Villella, V.R.; Esposito, S.; Tosco, A.; Sepe, A.; Gregorio, F.D.; Salvadori, L.; Grassia, R.; Leone, C.A.; Rosa, G.D.; et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy 2014, 10, 2053–2074. [Google Scholar] [CrossRef] [Green Version]
- Tosco, A.; De Gregorio, F.; Esposito, S.; De Stefano, D.; Sana, I.; Ferrari, E.; Sepe, A.; Salvadori, L.; Buonpensiero, P.; Di Pasqua, A.; et al. A novel treatment of cystic fibrosis acting on-target: Cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ. 2016, 23, 1380–1393. [Google Scholar] [CrossRef] [Green Version]
- Romani, L.; Oikonomou, V.; Moretti, S.; Iannitti, R.G.; D'Adamo, M.C.; Villella, V.R.; Pariano, M.; Sforna, L.; Borghi, M.; Bellet, M.M.; et al. Thymosin α1 represents a potential potent single-molecule-based therapy for cystic fibrosis. Nat. Med. 2017, 23, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomati, V.; Caci, E.; Ferrera, L.; Pesce, E.; Sondo, E.; Cholon, D.M.; Quinney, N.L.; Boyles, S.E.; Armirotti, A.; Ravazzolo, R.; et al. Thymosin α-1 does not correct F508del-CFTR in cystic fibrosis airway epithelia. JCI Insight 2018, 3, e98699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthes, E.; Hanrahan, J.W.; Cantin, A.M. F508del-CFTR is not corrected by thymosin α1. Nat. Med. 2018, 24, 890–891. [Google Scholar] [CrossRef] [PubMed]
- Armirotti, A.; Tomati, V.; Matthes, E.; Veit, G.; Cholon, D.M.; Phuan, P.-W.; Braccia, C.; Guidone, D.; Gentzsch, M.; Lukacs, G.L.; et al. Bioactive Thymosin Alpha-1 Does Not Influence F508del-CFTR Maturation and Activity. Sci. Rep. 2019, 9, 10310. [Google Scholar] [CrossRef] [Green Version]
- Andersson, K.E. PDE5 inhibitors—Pharmacology and clinical applications 20 years after sildenafil discovery. Br. J. Pharmacol. 2018, 175, 2554–2565. [Google Scholar] [CrossRef] [Green Version]
- Dormer, R.L. Sildenafil (Viagra) corrects F508-CFTR location in nasal epithelial cells from patients with cystic fibrosis. Thorax 2005, 60, 55–59. [Google Scholar] [CrossRef] [Green Version]
- Lubamba, B.; Lecourt, H.; Lebacq, J.; Lebecque, P.; De Jonge, H.; Wallemacq, P.; Leal, T. Preclinical Evidence that Sildenafil and Vardenafil Activate Chloride Transport in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 506–515. [Google Scholar] [CrossRef]
- Robert, R.; Carlile, G.W.; Pavel, C.; Liu, N.; Anjos, S.M.; Liao, J.; Luo, Y.; Zhang, D.; Thomas, D.Y.; Hanrahan, J.W. Structural Analog of Sildenafil Identified as a Novel Corrector of the F508del-CFTR Trafficking Defect. Mol. Pharmacol. 2008, 73, 478–489. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.W.; Bartlett, M.C.; Shi, L.; Clarke, D.M. Corrector-mediated rescue of misprocessed CFTR mutants can be reduced by the P-glycoprotein drug pump. Biochem. Pharmacol. 2012, 83, 345–354. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Wiley, C.; Felton, L.A.; St. Clair, C.; Jones, M.; Curran-Everett, D.; Poch, K.; Nichols, D.P.; Solomon, G.M.; Saavedra, M.T.; et al. Pharmacokinetics and tolerability of oral sildenafil in adults with cystic fibrosis lung disease. J. Cyst. Fibros. 2015, 14, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Garnock-Jones, K.P. Riociguat: A Review of Its Use in Patients with Chronic Thromboembolic Pulmonary Hypertension or Pulmonary Arterial Hypertension. Drugs 2014, 74, 2065–2078. [Google Scholar] [CrossRef] [PubMed]
- Derichs, N.; Taylor-Cousar, J.L.; Davies, J.C.; Fajac, I.; Tullis, E.; Nazareth, D.; Downey, D.G.; Rosenbluth, D.; Malfroot, A.; Saunders, C.; et al. Riociguat for the treatment of Phe508del homozygous adults with cystic fibrosis. J. Cyst. Fibros. 2021, 20, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Derichs, N.; Taylor-Cousar, J.; Tullis, E.; Davies, J.; Nazareth, D.; Downey, D.G.; Rosenbluth, D.; Fajac, I.; Malfroot, A.; Bakker, M.; et al. EPS1.3 Safety, tolerability and early signs of efficacy with riociguat for the treatment of adult Phe508del homozygous cystic fibrosis patients: Study design and rationale for the Rio-CF study. J. Cyst. Fibros. 2017, 16, S36. [Google Scholar] [CrossRef]
- Bacchi, S.; Palumbo, P.; Sponta, A.; Coppolino, M.F. Clinical pharmacology of non-steroidal anti-inflammatory drugs: A review. Anti-Inflamm. Anti-Allergy Agents Med. Chem. (Former. Curr. Med. Chem.-Anti-Inflamm. Anti-Allergy Agents) 2012, 11, 52–64. [Google Scholar] [CrossRef]
- Carlile, G.W.; Robert, R.; Goepp, J.; Matthes, E.; Liao, J.; Kus, B.; Macknight, S.D.; Rotin, D.; Hanrahan, J.W.; Thomas, D.Y. Ibuprofen rescues mutant cystic fibrosis transmembrane conductance regulator trafficking. J. Cyst. Fibros. 2015, 14, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Carlile, G.W.; Yang, Q.; Matthes, E.; Liao, J.; Birault, V.; Sneddon, H.F.; Poole, D.L.; Hall, C.J.; Hanrahan, J.W.; Thomas, D.Y. The NSAID glafenine rescues class 2 CFTR mutants via cyclooxygenase 2 inhibition of the arachidonic acid pathway. Sci. Rep. 2022, 12, 4595. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Solomon, G.M.; Zeitlin, P.L.; Flume, P.A.; Casey, A.; McCoy, K.; Zemanick, E.T.; Mandagere, A.; Troha, J.M.; Shoemaker, S.A.; et al. Pharmacokinetics and safety of cavosonstat (N91115) in healthy and cystic fibrosis adults homozygous for F508DEL-CFTR. J. Cyst. Fibros. 2017, 16, 371–379. [Google Scholar] [CrossRef]
- Zaman, K.; Sawczak, V.; Zaidi, A.; Butler, M.; Bennett, D.; Getsy, P.; Zeinomar, M.; Greenberg, Z.; Forbes, M.; Rehman, S.; et al. Augmentation of CFTR maturation by S-nitrosoglutathione reductase. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2015, 310, L263–L270. [Google Scholar] [CrossRef] [Green Version]
- Zaman, K.; Bennett, D.; Fraser-Butler, M.; Greenberg, Z.; Getsy, P.; Sattar, A.; Smith, L.; Corey, D.; Sun, F.; Hunt, J.; et al. S-nitrosothiols increases cystic fibrosis transmembrane regulator expression and maturation in the cell surface. Biochem. Biophys. Res. Commun. 2014, 443, 1257–1262. [Google Scholar] [CrossRef] [Green Version]
- Nivalis Therapeutics. Nivalis Therapeutics Announces Results from Phase 2 Clinical Trial of Cavosonstat for Treatment of Cystic Fibrosis; FDA: Silver Spring, MD, USA, 2016. [Google Scholar]
- Becq, F.; Mirval, S.; Carrez, T.; Lévêque, M.; Billet, A.; Coraux, C.; Sage, E.; Cantereau, A. The rescue of F508del-CFTR by elexacaftor/tezacaftor/ivacaftor (Trikafta) in human airway epithelial cells is underestimated due to the presence of ivacaftor. Eur. Respir. J. 2021, 59, 2100671. [Google Scholar] [CrossRef]
- Matos, A.M.; Jordan, P.; Matos, P. Treatment of Polarized Cystic Fibrosis Airway Cells With HGF Prevents VX-661-Rescued F508del-CFTR Destabilization Caused by Prolonged Co-exposure to VX-770. Front. Mol. Biosci. 2021, 8, 812101. [Google Scholar] [CrossRef] [PubMed]
- Matos, A.M.; Gomes-Duarte, A.; Faria, M.; Barros, P.; Jordan, P.; Amaral, M.D.; Matos, P. Prolonged co-treatment with HGF sustains epithelial integrity and improves pharmacological rescue of Phe508del-CFTR. Sci. Rep. 2018, 8, 13026. [Google Scholar] [CrossRef] [PubMed]
- Moniz, S.; Sousa, M.; Moraes, B.J.; Mendes, A.I.; Palma, M.; Barreto, C.; Fragata, J.I.; Amaral, M.D.; Matos, P. HGF stimulation of Rac1 signaling enhances pharmacological correction of the most prevalent cystic fibrosis mutant F508del-CFTR. ACS Chem. Biol. 2013, 8, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Alshafie, W.; Chappe, F.G.; Li, M.; Anini, Y.; Chappe, V.M. VIP regulates CFTR membrane expression and function in Calu-3 cells by increasing its interaction with NHERF1 and P-ERM in a VPAC1- and PKCε-dependent manner. Am. J. Physiol.-Cell Physiol. 2014, 307, C107–C119. [Google Scholar] [CrossRef] [Green Version]
- Lobo, M.J.; Amaral, M.D.; Zaccolo, M.; Farinha, C.M. EPAC1 activation by cAMP stabilizes CFTR at the membrane by promoting its interaction with NHERF1. J. Cell Sci. 2016, 129, 2599–2612. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.D.; Pinto, F.R.; Ferreira, J.F.; Amaral, M.D.; Zaccolo, M.; Farinha, C.M. Cytoskeleton regulators CAPZA2 and INF2 associate with CFTR to control its plasma membrane levels under EPAC1 activation. Biochem. J. 2020, 477, 2561–2580. [Google Scholar] [CrossRef]
- Matos, A.M.; Pinto, F.R.; Barros, P.; Amaral, M.D.; Pepperkok, R.; Matos, P. Inhibition of calpain 1 restores plasma membrane stability to pharmacologically rescued Phe508del-CFTR variant. J. Biol. Chem. 2019, 294, 13396–13410. [Google Scholar] [CrossRef]
- Favia, M.; Guerra, L.; Fanelli, T.; Cardone, R.A.; Monterisi, S.; Di Sole, F.; Castellani, S.; Chen, M.; Seidler, U.; Reshkin, S.J.; et al. Na+/H+ Exchanger Regulatory Factor 1 Overexpression-dependent Increase of Cytoskeleton Organization Is Fundamental in the Rescue of F508del Cystic Fibrosis Transmembrane Conductance Regulator in Human Airway CFBE41o-Cells. Mol. Biol. Cell 2010, 21, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Cushing, P.R.; Vouilleme, L.; Pellegrini, M.; Boisguerin, P.; Madden, D.R. A Stabilizing Influence: CAL PDZ Inhibition Extends the Half-Life of ΔF508-CFTR. Angew. Chem. Int. Ed. 2010, 49, 9907–9911. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Rab, A.; Tang, L.P.; Bebok, Z.; Rowe, S.M.; Bartoszewski, R.; Collawn, J.F. ΔF508 CFTR Surface Stability Is Regulated by DAB2 and CHIP-Mediated Ubiquitination in Post-Endocytic Compartments. PLoS ONE 2015, 10, e0123131. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Wu, Q.; Rajagopalan, C.; Zhang, C.; Bouhamdan, M.; Wei, H.; Chen, X.; Zaman, K.; Li, C.; Sun, X.; et al. CK19 stabilizes CFTR at the cell surface by limiting its endocytic pathway degradation. FASEB J. 2019, 33, 12602–12615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farinha, C.M.; Matos, P. Rab GTPases regulate the trafficking of channels and transporters—A focus on cystic fibrosis. Small GTPases 2018, 9, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes, A.I.; Matos, P.; Moniz, S.; Luz, S.; Amaral, M.D.; Farinha, C.M.; Jordan, P. Antagonistic Regulation of Cystic Fibrosis Transmembrane Conductance Regulator Cell Surface Expression by Protein Kinases WNK4 and Spleen Tyrosine Kinase. Mol. Cell. Biol. 2011, 31, 4076–4086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, C.A.; Pinto, F.R.; Barros, P.; Matos, P.; Jordan, P. A SYK/SHC1 pathway regulates the amount of CFTR in the plasma membrane. Cell. Mol. Life Sci. 2020, 77, 4997–5015. [Google Scholar] [CrossRef]
- Oliver, K.E.; Rauscher, R.; Mijnders, M.; Wang, W.; Wolpert, M.J.; Maya, J.; Sabusap, C.M.; Kesterson, R.A.; Kirk, K.L.; Rab, A.; et al. Slowing ribosome velocity restores folding and function of mutant CFTR. J. Clin. Investig. 2019, 129, 5236–5253. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Oliver, K.; Apaja, P.M.; Perdomo, D.; Bidaud-Meynard, A.; Lin, S.-T.; Guo, J.; Icyuz, M.; Sorscher, E.J.; Hartman, J.L.; et al. Ribosomal Stalk Protein Silencing Partially Corrects the ΔF508-CFTR Functional Expression Defect. PLoS Biol. 2016, 14, e1002462. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.P.; Drew, L.; Green, O.; Villella, A.; McEwan, B.; Patel, N.; Qiu, D.; Bhalla, A.; Bastos, C.; Parks, D.; et al. CFTR Amplifiers: A New Class of CFTR Modulator that Complements the Substrate Limitations of Other CF Therapeutic Modalities. Am. J. Respir. Crit. Care Med. 2016, 193, A5574. [Google Scholar]
- Giuliano, K.A.; Wachi, S.; Drew, L.; Dukovski, D.; Green, O.; Bastos, C.; Cullen, M.D.; Hauck, S.; Tait, B.D.; Munoz, B.; et al. Use of a High-Throughput Phenotypic Screening Strategy to Identify Amplifiers, a Novel Pharmacological Class of Small Molecules That Exhibit Functional Synergy with Potentiators and Correctors. SLAS Discov. Adv. Sci. Drug Discov. 2018, 23, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Dukovski, D.; Villella, A.; Bastos, C.; King, R.; Finley, D.; Kelly, J.W.; Morimoto, R.I.; Hartl, F.U.; Munoz, B.; Lee, P.S.; et al. Amplifiers co-translationally enhance CFTR biosynthesis via PCBP1-mediated regulation of CFTR mRNA. J. Cyst. Fibros. 2020, 19, 733–741. [Google Scholar] [CrossRef] [Green Version]
- Molinski, S.V.; Ahmadi, S.; Ip, W.; Ouyang, H.; Villella, A.; Miller, J.P.; Lee, P.S.; Kulleperuma, K.; Du, K.; Di Paola, M.; et al. Orkambi® and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol. Med. 2017, 9, 1224–1243. [Google Scholar] [CrossRef]
- Proteostasis Therapeutics. Proteostasis Therapeutics Announces Positive Phase 2 Topline Results from Proprietary CFTR Modulator Combinations in F508del Homozygous Cystic Fibrosis Patients; FDA: Silver Spring, MD, USA, 2019. [Google Scholar]
- HIT-CF. HIT-CF Newsletter November 2021. 2021. Available online: https://www.ecfs.eu/sites/default/files/general-content-files/news/HIT-CF%20Newsletter%20November%202021.pdf (accessed on 10 May 2022).
- Glasgow, A.M.A.; De Santi, C.; Greene, C.M. Non-coding RNA in cystic fibrosis. Biochem. Soc. Trans. 2018, 46, 619–630. [Google Scholar] [CrossRef] [PubMed]
- De Palma, F.D.E.; Raia, V.; Kroemer, G.; Maiuri, M.C. The Multifaceted Roles of MicroRNAs in Cystic Fibrosis. Diagnostics 2020, 10, 1102. [Google Scholar] [CrossRef] [PubMed]
- Viart, V.; Bergougnoux, A.; Bonini, J.; Varilh, J.; Chiron, R.; Tabary, O.; Molinari, N.; Claustres, M.; Taulan-Cadars, M. Transcription factors and miRNAs that regulate fetal to adult CFTR expression change are new targets for cystic fibrosis. Eur. Respir. J. 2015, 45, 116–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, S.; Karp, P.H.; Osterhaus, S.R.; Jiang, P.; Wohlford-Lenane, C.; Lennox, K.A.; Jacobi, A.M.; Praekh, K.; Rose, S.D.; Behlke, M.A.; et al. Post-transcriptional regulation of cystic fibrosis transmembrane conductance regulator expression and function by microRNAs. Am. J. Respir. Cell Mol. Biol. 2013, 49, 544–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutful Kabir, F.; Ambalavanan, N.; Liu, G.; Li, P.; Solomon, G.M.; Lal, C.V.; Mazur, M.; Halloran, B.; Szul, T.; Gerthoffer, W.T.; et al. MicroRNA-145 Antagonism Reverses TGF-beta Inhibition of F508del CFTR Correction in Airway Epithelia. Am. J. Respir. Crit. Care Med. 2018, 197, 632–643. [Google Scholar] [CrossRef]
- Fabbri, E.; Tamanini, A.; Jakova, T.; Gasparello, J.; Manicardi, A.; Corradini, R.; Sabbioni, G.; Finotti, A.; Borgatti, M.; Lampronti, I.; et al. A Peptide Nucleic Acid against MicroRNA miR-145-5p Enhances the Expression of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) in Calu-3 Cells. Molecules 2017, 23, 71. [Google Scholar] [CrossRef] [Green Version]
- Amato, F.; Tomaiuolo, R.; Nici, F.; Borbone, N.; Elce, A.; Catalanotti, B.; D'Errico, S.; Morgillo, C.M.; De Rosa, G.; Mayol, L.; et al. Exploitation of a very small peptide nucleic acid as a new inhibitor of miR-509-3p involved in the regulation of cystic fibrosis disease-gene expression. Biomed Res. Int. 2014, 2014, 610718. [Google Scholar] [CrossRef]
- De Santi, C.; Fernandez Fernandez, E.; Gaul, R.; Vencken, S.; Glasgow, A.; Oglesby, I.K.; Hurley, K.; Hawkins, F.; Mitash, N.; Mu, F.; et al. Precise Targeting of miRNA Sites Restores CFTR Activity in CF Bronchial Epithelial Cells. Mol. Ther. 2020, 28, 1190–1199. [Google Scholar] [CrossRef]
- Kurosaki, T.; Maquat, L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016, 129, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Linde, L.; Boelz, S.; Nissim-Rafinia, M.; Oren, Y.S.; Wilschanski, M.; Yaacov, Y.; Virgilis, D.; Neu-Yilik, G.; Kulozik, A.E.; Kerem, E.; et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J. Clin. Invest. 2007, 117, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Aksit, M.A.; Bowling, A.D.; Evans, T.A.; Joynt, A.T.; Osorio, D.; Patel, S.; West, N.; Merlo, C.; Sosnay, P.R.; Cutting, G.R.; et al. Decreased mRNA and protein stability of W1282X limits response to modulator therapy. J. Cyst. Fibros. 2019, 18, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A.; Awatade, N.T.; Felício, V.M.; Silva, I.A.; Calucho, M.; Pereira, L.; Azevedo, P.; Cavaco, J.; Barreto, C.; Bertuzzo, C.; et al. The effect of premature termination codon mutations on CFTR mRNA abundance in human nasal epithelium and intestinal organoids: A basis for read-through therapies in cystic fibrosis. Hum. Mutat. 2018, 40, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Linde, L.; Boelz, S.; Neu-Yilik, G.; Kulozik, A.E.; Kerem, B. The efficiency of nonsense-mediated mRNA decay is an inherent character and varies among different cells. Eur. J. Hum. Genet. 2007, 15, 1156–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, J.; Keeling, K.M.; Rowe, S.M. Pharmacological approaches for targeting cystic fibrosis nonsense mutations. Eur. J. Med. Chem. 2020, 200, 112436. [Google Scholar] [CrossRef] [PubMed]
- Maule, G.; Ensinck, M.; Bulcaen, M.; Carlon, M.S. Rewriting CFTR to cure cystic fibrosis. In Progress in Molecular Biology and Translational Science; Academic Press: Cambridge, MA, USA, 2021. [Google Scholar] [CrossRef]
- Venturini, A.; Borrelli, A.; Musante, I.; Scudieri, P.; Capurro, V.; Renda, M.; Pedemonte, N.; Galietta, L.J.V. Comprehensive Analysis of Combinatorial Pharmacological Treatments to Correct Nonsense Mutations in the CFTR Gene. Int. J. Mol. Sci. 2021, 22, 11972. [Google Scholar] [CrossRef]
- Valley, H.C.; Bukis, K.M.; Bell, A.; Cheng, Y.; Wong, E.; Jordan, N.J.; Allaire, N.E.; Sivachenko, A.; Liang, F.; Bihler, H.; et al. Isogenic cell models of cystic fibrosis-causing variants in natively expressing pulmonary epithelial cells. J. Cyst. Fibros. 2019, 18, 476–483. [Google Scholar] [CrossRef]
- Keenan, M.M.; Huang, L.; Jordan, N.J.; Wong, E.; Cheng, Y.; Valley, H.C.; Mahiou, J.; Liang, F.; Bihler, H.; Mense, M.; et al. Nonsense-mediated RNA Decay Pathway Inhibition Restores Expression and Function of W1282X CFTR. Am. J. Respir. Cell Mol. Biol. 2019, 61, 290–300. [Google Scholar] [CrossRef]
- Lentini, L.; Melfi, R.; Cancemi, P.; Pibiri, I.; Di Leonardo, A. Caffeine boosts Ataluren’s readthrough activity. Heliyon 2019, 5, e01963. [Google Scholar] [CrossRef] [Green Version]
- McHugh, D.R.; Cotton, C.U.; Hodges, C.A. Synergy between Readthrough and Nonsense Mediated Decay Inhibition in a Murine Model of Cystic Fibrosis Nonsense Mutations. Int. J. Mol. Sci. 2020, 22, 344. [Google Scholar] [CrossRef]
- Viotti Perisse, I.; Fan, Z.; Van Wettere, A.; Liu, Y.; Leir, S.H.; Keim, J.; Regouski, M.; Wilson, M.D.; Cholewa, K.M.; Mansbach, S.N.; et al. Sheep models of F508del and G542X cystic fibrosis mutations show cellular responses to human therapeutics. FASEB BioAdvances 2021, 3, 841–854. [Google Scholar] [CrossRef]
- De Poel, E.; Spelier, S.; Suen, S.W.F.; Kruisselbrink, E.; Graeber, S.Y.; Mall, M.A.; Weersink, E.J.M.; Van Der Eerden, M.M.; Koppelman, G.H.; Van Der Ent, C.K.; et al. Functional Restoration of CFTR Nonsense Mutations in Intestinal Organoids. J. Cyst. Fibros. 2021, 21, 246–253. [Google Scholar] [CrossRef]
- Gewandter, J.S.; Bambara, R.A.; O'Reilly, M.A. The RNA surveillance protein SMG1 activates p53 in response to DNA double-strand breaks but not exogenously oxidized mRNA. Cell Cycle 2011, 10, 2561–2567. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A. Role of SMG-1-mediated Upf1 phosphorylation in mammalian nonsense-mediated mRNA decay. Genes Cells 2013, 18, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Evans, T.A.; Pellicore, M.J.; Davis, E.; Aksit, M.A.; McCague, A.F.; Joynt, A.T.; Lu, Z.; Han, S.T.; Anzmann, A.F.; et al. Capitalizing on the heterogeneous effects of CFTR nonsense and frameshift variants to inform therapeutic strategy for cystic fibrosis. PLoS Genet. 2018, 14, e1007723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutyam, V.; Du, M.; Xue, X.; Keeling, K.M.; White, E.L.; Bostwick, J.R.; Rasmussen, L.; Liu, B.; Mazur, M.; Hong, J.S.; et al. Discovery of Clinically Approved Agents That Promote Suppression of Cystic Fibrosis Transmembrane Conductance Regulator Nonsense Mutations. Am. J. Respir. Crit. Care Med. 2016, 194, 1092–1103. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Hilarion, S.; Beghyn, T.; Jia, J.; Debreuck, N.; Berte, G.; Mamchaoui, K.; Mouly, V.; Gruenert, D.C.; Déprez, B.; Lejeune, F. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J. Rare Dis. 2012, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Nomakuchi, T.T.; Rigo, F.; Aznarez, I.; Krainer, A.R. Antisense oligonucleotide-directed inhibition of nonsense-mediated mRNA decay. Nat. Biotechnol. 2016, 34, 164–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erwood, S.; Laselva, O.; Bily, T.M.I.; Brewer, R.A.; Rutherford, A.H.; Bear, C.E.; Ivakine, E.A. Allele-Specific Prevention of Nonsense-Mediated Decay in Cystic Fibrosis Using Homology-Independent Genome Editing. Mol. Ther. Methods Clin. Dev. 2020, 17, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.S.; Avizur-Barchad, O.; Ozeri-Galai, E.; Elgrabli, R.; Schirelman, M.R.; Blinder, T.; Stampfer, C.D.; Ordan, M.; Laselva, O.; Cohen-Cymberknoh, M.; et al. Antisense oligonucleotide splicing modulation as a novel Cystic Fibrosis therapeutic approach for the W1282X nonsense mutation. J. Cyst. Fibros. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Michaels, W.E.; Pena-Rasgado, C.; Kotaria, R.; Bridges, R.J.; Hastings, M.L. Open reading frame correction using splice-switching antisense oligonucleotides for the treatment of cystic fibrosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2114886119. [Google Scholar] [CrossRef]
- Kim, Y.J.; Sivetz, N.; Layne, J.; Voss, D.M.; Yang, L.; Zhang, Q.; Krainer, A.R. Exon-skipping antisense oligonucleotides for cystic fibrosis therapy. Proc. Natl. Acad. Sci. USA 2022, 119, e2114858118. [Google Scholar] [CrossRef]
- Howard, M.; Frizzell, R.A.; Bedwell, D.M. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat. Med. 1996, 2, 467–469. [Google Scholar] [CrossRef]
- Bedwell, D.M.; Kaenjak, A.; Benos, D.J.; Bebok, Z.; Bubien, J.K.; Hong, J.; Tousson, A.; Clancy, J.P.; Sorscher, E.J. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat. Med. 1997, 3, 1280–1284. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Jones, J.R.; Lanier, J.; Keeling, K.M.; Lindsey, R.J.; Tousson, A.; Bebök, Z.; Whitsett, J.A.; Dey, C.R.; Colledge, W.H.; et al. Aminoglycoside suppression of a premature stop mutation in a Cftr–/– mouse carrying a human CFTR-G542X transgene. J. Mol. Med. 2002, 80, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Sermet-Gaudelus, I.; Renouil, M.; Fajac, A.; Bidou, L.; Parbaille, B.; Pierrot, S.; Davy, N.; Bismuth, E.; Reinert, P.; Lenoir, G.; et al. In vitroprediction of stop-codon suppression by intravenous gentamicin in patients with cystic fibrosis: A pilot study. BMC Med. 2007, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilschanski, M.; Yahav, Y.; Yaacov, Y.; Blau, H.; Bentur, L.; Rivlin, J.; Aviram, M.; Bdolah-Abram, T.; Bebok, Z.; Shushi, L.; et al. Gentamicin-Induced Correction of CFTR Function in Patients with Cystic Fibrosis andCFTRStop Mutations. N. Engl. J. Med. 2003, 349, 1433–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clancy, J.P.; Rowe, S.M.; Bebok, Z.; Aitken, M.L.; Gibson, R.; Zeitlin, P.; Berclaz, P.; Moss, R.; Knowles, M.R.; Oster, R.A.; et al. No Detectable Improvements in Cystic Fibrosis Transmembrane Conductance Regulator by Nasal Aminoglycosides in Patients with Cystic Fibrosis with Stop Mutations. Am. J. Respir. Cell Mol. Biol. 2007, 37, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Keeling, K.M.; Fan, L.; Liu, X.; Kovaçs, T.; Sorscher, E.; Bedwell, D.M. Clinical doses of amikacin provide more effective suppression of the human CFTR-G542X stop mutation than gentamicin in a transgenic CF mouse model. J. Mol. Med. 2006, 84, 573–582. [Google Scholar] [CrossRef]
- Altamura, N.; Castaldo, R.; Finotti, A.; Breveglieri, G.; Salvatori, F.; Zuccato, C.; Gambari, R.; Panin, G.C.; Borgatti, M. Tobramycin is a suppressor of premature termination codons. J. Cyst. Fibros. 2013, 12, 806–811. [Google Scholar] [CrossRef] [Green Version]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- Du, M.; Liu, X.; Welch, E.M.; Hirawat, S.; Peltz, S.W.; Bedwell, D.M. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR -G542X nonsense allele in a CF mouse model. Proc. Natl. Acad. Sci. USA 2008, 105, 2064–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerem, E.; Hirawat, S.; Armoni, S.; Yaakov, Y.; Shoseyov, D.; Cohen, M.; Nissim-Rafinia, M.; Blau, H.; Rivlin, J.; Aviram, M.; et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: A prospective phase II trial. Lancet 2008, 372, 719–727. [Google Scholar] [CrossRef]
- Wilschanski, M.; Miller, L.L.; Shoseyov, D.; Blau, H.; Rivlin, J.; Aviram, M.; Cohen, M.; Armoni, S.; Yaakov, Y.; Pugatch, T.; et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur. Respir. J. 2011, 38, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Sermet-Gaudelus, I.; Boeck, K.D.; Casimir, G.J.; Vermeulen, F.; Leal, T.; Mogenet, A.; Roussel, D.; Fritsch, J.; Hanssens, L.; Hirawat, S.; et al. Ataluren (PTC124) Induces Cystic Fibrosis Transmembrane Conductance Regulator Protein Expression and Activity in Children with Nonsense Mutation Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 1262–1272. [Google Scholar] [CrossRef]
- Kerem, E.; Konstan, M.W.; De Boeck, K.; Accurso, F.J.; Sermet-Gaudelus, I.; Wilschanski, M.; Elborn, J.S.; Melotti, P.; Bronsveld, I.; Fajac, I.; et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2014, 2, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Zomer-Van Ommen, D.D.; Vijftigschild, L.A.W.; Kruisselbrink, E.; Vonk, A.M.; Dekkers, J.F.; Janssens, H.M.; De Winter-De Groot, K.M.; Van Der Ent, C.K.; Beekman, J.M. Limited premature termination codon suppression by read-through agents in cystic fibrosis intestinal organoids. J. Cyst. Fibros. 2016, 15, 158–162. [Google Scholar] [CrossRef] [Green Version]
- McHugh, D.R.; Steele, M.S.; Valerio, D.M.; Miron, A.; Mann, R.J.; LePage, D.F.; Conlon, R.A.; Cotton, C.U.; Drumm, M.L.; Hodges, C.A. A G542X cystic fibrosis mouse model for examining nonsense mutation directed therapies. PLoS ONE 2018, 13, e0199573. [Google Scholar] [CrossRef] [Green Version]
- Pibiri, I.; Lentini, L.; Melfi, R.; Tutone, M.; Baldassano, S.; Ricco Galluzzo, P.; Di Leonardo, A.; Pace, A. Rescuing the CFTR protein function: Introducing 1,3,4-oxadiazoles as translational readthrough inducing drugs. Eur. J. Med. Chem. 2018, 159, 126–142. [Google Scholar] [CrossRef]
- Pibiri, I.; Melfi, R.; Tutone, M.; Di Leonardo, A.; Pace, A.; Lentini, L. Targeting Nonsense: Optimization of 1,2,4-Oxadiazole TRIDs to Rescue CFTR Expression and Functionality in Cystic Fibrosis Cell Model Systems. Int. J. Mol. Sci. 2020, 21, 6420. [Google Scholar] [CrossRef]
- Rowe, S.M.; Sloane, P.; Tang, L.P.; Backer, K.; Mazur, M.; Buckley-Lanier, J.; Nudelman, I.; Belakhov, V.; Bebok, Z.; Schwiebert, E.; et al. Suppression of CFTR premature termination codons and rescue of CFTR protein and function by the synthetic aminoglycoside NB54. J. Mol. Med. 2011, 89, 1149–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.; Mutyam, V.; Tang, L.; Biswas, S.; Du, M.; Jackson, L.A.; Dai, Y.; Belakhov, V.; Shalev, M.; Chen, F.; et al. Synthetic Aminoglycosides Efficiently Suppress Cystic Fibrosis Transmembrane Conductance Regulator Nonsense Mutations and Are Enhanced by Ivacaftor. Am. J. Respir. Cell Mol. Biol. 2014, 50, 805–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandasamy, J.; Atia-Glikin, D.; Shulman, E.; Shapira, K.; Shavit, M.; Belakhov, V.; Baasov, T. Increased Selectivity toward Cytoplasmic versus Mitochondrial Ribosome Confers Improved Efficiency of Synthetic Aminoglycosides in Fixing Damaged Genes: A Strategy for Treatment of Genetic Diseases Caused by Nonsense Mutations. J. Med. Chem. 2012, 55, 10630–10643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerem, E. ELX-02: An investigational read-through agent for the treatment of nonsense mutation-related genetic disease. Expert Opin Investig Drugs 2020, 29, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Leubitz, A.; Frydman-Marom, A.; Sharpe, N.; Van Duzer, J.; Campbell, K.C.M.; Vanhoutte, F. Safety, Tolerability, and Pharmacokinetics of Single Ascending Doses of ELX-02, a Potential Treatment for Genetic Disorders Caused by Nonsense Mutations, in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2019, 8, 984–994. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.K.; Mullenders, J.; Pott, J.; Boj, S.F.; Landskroner-Eiger, S.; Goddeeris, M.M. Targeting G542X CFTR nonsense alleles with ELX-02 restores CFTR function in human-derived intestinal organoids. J. Cyst. Fibros. 2021, 20, 436–442. [Google Scholar] [CrossRef]
- Xue, X.; Mutyam, V.; Thakerar, A.; Mobley, J.; Bridges, R.J.; Rowe, S.M.; Keeling, K.M.; Bedwell, D.M. Identification of the amino acids inserted during suppression of CFTR nonsense mutations and determination of their functional consequences. Hum. Mol. Genet. 2017, 26, 3116–3129. [Google Scholar] [CrossRef] [Green Version]
- Keeling, K.M.; Wang, D.; Conard, S.E.; Bedwell, D.M. Suppression of premature termination codons as a therapeutic approach. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 444–463. [Google Scholar] [CrossRef] [Green Version]
- Sharma, J.; Du, M.; Wong, E.; Mutyam, V.; Li, Y.; Chen, J.; Wangen, J.; Thrasher, K.; Fu, L.; Peng, N.; et al. A small molecule that induces translational readthrough of CFTR nonsense mutations by eRF1 depletion. Nat. Commun. 2021, 12, 4358. [Google Scholar] [CrossRef]
- Smith, E.; Dukovski, D.; Shumate, J.; Scampavia, L.; Miller, J.P.; Spicer, T.P. Identification of Compounds That Promote Readthrough of Premature Termination Codons in the CFTR. SLAS Discov. 2021, 26, 205–215. [Google Scholar] [CrossRef]
- Lueck, J.D.; Yoon, J.S.; Perales-Puchalt, A.; Mackey, A.L.; Infield, D.T.; Behlke, M.A.; Pope, M.R.; Weiner, D.B.; Skach, W.R.; McCray, P.B.; et al. Engineered transfer RNAs for suppression of premature termination codons. Nat. Commun. 2019, 10, 822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, W.; Porter, J.J.; Sipple, M.T.; Edwards, K.M.; Lueck, J.D. Efficient suppression of endogenous CFTR nonsense mutations using anticodon-engineered transfer RNAs. Mol. Ther.-Nucleic Acids 2022, 28, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, Q.; Mansfield, S.G.; Puttaraju, M.; Zhang, Y.; Zhou, W.; Cohn, J.A.; Garcia-Blanco, M.A.; Mitchell, L.G.; Engelhardt, J.F. Partial correction of endogenous ΔF508 CFTR in human cystic fibrosis airway epithelia by spliceosome-mediated RNA trans-splicing. Nat. Biotechnol. 2002, 20, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, S.G.; Kole, J.; Puttaraju, M.; Yang, C.C.; Garcia-Blanco, M.A.; Cohn, J.A.; Mitchell, L.G. Repair of CFTR mRNA by spliceosome-mediated RNA trans-splicing. Gene Ther. 2000, 7, 1885–1895. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Raychowdhury, M.K.; Tabatadze, D.R.; Cantiello, H.F. Reversal of cystic fibrosis phenotype in a cultured Δ508 cystic fibrosis transmembrane conductance regulator cell line by oligonucleotide insertion. Proc. Natl. Acad. Sci. USA 2004, 101, 8150–8155. [Google Scholar] [CrossRef] [Green Version]
- Beumer, W.; Swildens, J.; Leal, T.; Noel, S.; Anthonijsz, H.; Van Der Horst, G.; Kuiperij-Boersma, H.; Potman, M.; Van Putten, C.; Biasutto, P.; et al. Evaluation of eluforsen, a novel RNA oligonucleotide for restoration of CFTR function in in vitro and murine models of p.Phe508del cystic fibrosis. PLoS ONE 2019, 14, e0219182. [Google Scholar] [CrossRef]
- Drevinek, P.; Pressler, T.; Cipolli, M.; De Boeck, K.; Schwarz, C.; Bouisset, F.; Boff, M.; Henig, N.; Paquette-Lamontagne, N.; Montgomery, S.; et al. Antisense oligonucleotide eluforsen is safe and improves respiratory symptoms in F508DEL cystic fibrosis. J. Cyst. Fibros. 2020, 19, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Sermet-Gaudelus, I.; Clancy, J.P.; Nichols, D.P.; Nick, J.A.; De Boeck, K.; Solomon, G.M.; Mall, M.A.; Bolognese, J.; Bouisset, F.; Den Hollander, W.; et al. Antisense oligonucleotide eluforsen improves CFTR function in F508del cystic fibrosis. J. Cyst. Fibros. 2019, 18, 536–542. [Google Scholar] [CrossRef] [Green Version]
- Montiel-Gonzalez, M.F.; Vallecillo-Viejo, I.; Yudowski, G.A.; Rosenthal, J.J.C. Correction of mutations within the cystic fibrosis transmembrane conductance regulator by site-directed RNA editing. Proc. Natl. Acad. Sci. USA 2013, 110, 18285–18290. [Google Scholar] [CrossRef] [Green Version]
- Melfi, R.; Cancemi, P.; Chiavetta, R.; Barra, V.; Lentini, L.; Di Leonardo, A. Investigating REPAIRv2 as a Tool to Edit CFTR mRNA with Premature Stop Codons. Int. J. Mol. Sci. 2020, 21, 4781. [Google Scholar] [CrossRef]
- Aartsma-Rus, A. FDA Approval of Nusinersen for Spinal Muscular Atrophy Makes 2016 the Year of Splice Modulating Oligonucleotides. Nucleic Acid. Ther. 2017, 27, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Igreja, S.; Clarke, L.A.; Botelho, H.M.; Marques, L.; Amaral, M.D. Correction of a Cystic Fibrosis Splicing Mutation by Antisense Oligonucleotides. Hum. Mutat. 2016, 37, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Friedman, K.J.; Kole, J.; Cohn, J.A.; Knowles, M.R.; Silverman, L.M.; Kole, R. Correction of aberrant splicing of the cystic fibrosis transmembrane conductance regulator (CFTR) gene by antisense oligonucleotides. J. Biol. Chem. 1999, 274, 36193–36199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaels, W.E.; Bridges, R.J.; Hastings, M.L. Antisense oligonucleotide-mediated correction of CFTR splicing improves chloride secretion in cystic fibrosis patient-derived bronchial epithelial cells. Nucleic Acids Res. 2020, 48, 7454–7467. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.S.; Irony-Tur Sinai, M.; Golec, A.; Barchad-Avitzur, O.; Mutyam, V.; Li, Y.; Hong, J.; Ozeri-Galai, E.; Hatton, A.; Leibson, C.; et al. Antisense oligonucleotide-based drug development for Cystic Fibrosis patients carrying the 3849+10 kb C-to-T splicing mutation. J. Cyst. Fibros. 2021, 20, 865–875. [Google Scholar] [CrossRef] [PubMed]
- SpliSense. SpliSense Announces FDA and EMA Grant Orphan Drug Designation to SPL84-23-1 for the Treatment of Cystic Fibrosis; FDA: Silver Spring, MD, USA, 2022. [Google Scholar]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar] [CrossRef]
- Translate Bio. Translate Bio Announces Results from Second Interim Data Analysis from Ongoing Phase 1/2 Clinical Trial of MRT5005 in Patients with Cystic Fibrosis (CF); FDA: Silver Spring, MD, USA, 2021. [Google Scholar]
- Translate Bio. Translate Bio Announces Interim Results from Phase 1/2 Clinical Trial of MRT5005 in Patients with Cystic Fibrosis; FDA: Silver Spring, MD, USA, 2019. [Google Scholar]
- Arcturus Therapeutics. LUNAR-CF. Available online: https://arcturusrx.com/mrna-medicines-pipeline/#lunarCf (accessed on 10 May 2022).
- Kolonko, A.K.; Efing, J.; González-Espinosa, Y.; Bangel-Ruland, N.; Van Driessche, W.; Goycoolea, F.M.; Weber, W.-M. Capsaicin-Loaded Chitosan Nanocapsules for wtCFTR-mRNA Delivery to a Cystic Fibrosis Cell Line. Biomedicines 2020, 8, 364. [Google Scholar] [CrossRef]
- Trimble, A.T.; Donaldson, S.H. Ivacaftor withdrawal syndrome in cystic fibrosis patients with the G551D mutation. J. Cyst. Fibros. 2018, 17, e13–e16. [Google Scholar] [CrossRef] [Green Version]
- CFF. Path to a Cure: Many Routes, One Mission|CF Foundation. Available online: https://www.cff.org/Research/About-Our-Research/Path-to-a-Cure-Many-Routes-One-Mission/ (accessed on 17 October 2020).
- Cooney, A.L.; McCray, P.B., Jr.; Sinn, P.L. Cystic Fibrosis Gene Therapy: Looking Back, Looking Forward. Genes 2018, 9, 538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondhi, D.; Stiles, K.M.; De, B.P.; Crystal, R.G. Genetic Modification of the Lung Directed Toward Treatment of Human Disease. Hum. Gene Ther. 2017, 28, 3–84. [Google Scholar] [CrossRef] [PubMed]
- Alton, E.W.F.W.; Boyd, A.C.; Davies, J.C.; Gill, D.R.; Griesenbach, U.; Harrison, P.T.; Henig, N.; Higgins, T.; Hyde, S.C.; Innes, J.A.; et al. Genetic medicines for CF: Hype versus reality. Pediatric Pulmonol. 2016, 51, S5–S17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidović, D.; Carlon, M.S.; Da Cunha, M.F.; Dekkers, J.F.; Hollenhorst, M.I.; Bijvelds, M.J.C.; Ramalho, A.S.; Van Den Haute, C.; Ferrante, M.; Baekelandt, V.; et al. rAAV-CFTRΔR Rescues the Cystic Fibrosis Phenotype in Human Intestinal Organoids and Cystic Fibrosis Mice. Am. J. Respir. Crit. Care Med. 2016, 193, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Cooney, A.L.; Abou Alaiwa, M.H.; Shah, V.S.; Bouzek, D.C.; Stroik, M.R.; Powers, L.S.; Gansemer, N.D.; Meyerholz, D.K.; Welsh, M.J.; Stoltz, D.A.; et al. Lentiviral-mediated phenotypic correction of cystic fibrosis pigs. JCI Insight 2016, 1, e88730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alton, E.W.F.W.; Beekman, J.M.; Boyd, A.C.; Brand, J.; Carlon, M.S.; Connolly, M.M.; Chan, M.; Conlon, S.; Davidson, H.E.; Davies, J.C.; et al. Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax 2017, 72, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Steines, B.; Dickey, D.D.; Bergen, J.; Excoffon, K.J.D.A.; Weinstein, J.R.; Li, X.; Yan, Z.; Alaiwa, M.H.A.; Shah, V.S.; Bouzek, D.C.; et al. CFTR gene transfer with AAV improves early cystic fibrosis pig phenotypes. JCI Insight 2016, 1, e88728. [Google Scholar] [CrossRef]
- Alton, E.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 684–691. [Google Scholar] [CrossRef] [Green Version]
- Spirovant Sciences. FDA Grants Spirovant Sciences Orphan Drug and Rare Pediatric Disease Designations for SPIRO-2101 for Treatment of Cystic Fibrosis; FDA: Silver Spring, MD, USA, 2020. [Google Scholar]
- Krystal Biotech. Krystal Biotech’s KB407 Granted Orphan Drug Designation by the FDA to Treat Patients With Cystic Fibrosis; FDA: Silver Spring, MD, USA, 2020. [Google Scholar]
- Boehringer Ingelheim. Boehringer Ingelheim and Partners to Accelerate Development of First-In-Class Gene Therapy for Patients with Cystic Fibrosis; FDA: Silver Spring, MD, USA, 2021. [Google Scholar]
- Ledford, H.; Callaway, E. Pioneers of revolutionary CRISPR gene editing win chemistry Nobel. Nature 2020, 586, 346–347. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Maule, G.; Casini, A.; Montagna, C.; Ramalho, A.S.; De Boeck, K.; Debyser, Z.; Carlon, M.S.; Petris, G.; Cereseto, A. Allele specific repair of splicing mutations in cystic fibrosis through AsCas12a genome editing. Nat. Commun. 2019, 10, 3556. [Google Scholar] [CrossRef] [PubMed]
- Geurts, M.H.; De Poel, E.; Pleguezuelos-Manzano, C.; Oka, R.; Carrillo, L.; Andersson-Rolf, A.; Boretto, M.; Brunsveld, J.E.; Van Boxtel, R.; Beekman, J.M.; et al. Evaluating CRISPR-based prime editing for cancer modeling and CFTR repair in organoids. Life Sci. Alliance 2021, 4, e202000940. [Google Scholar] [CrossRef] [PubMed]
- Geurts, M.H.; de Poel, E.; Amatngalim, G.D.; Oka, R.; Meijers, F.M.; Kruisselbrink, E.; van Mourik, P.; Berkers, G.; de Winter-de Groot, K.M.; Michel, S.; et al. CRISPR-Based Adenine Editors Correct Nonsense Mutations in a Cystic Fibrosis Organoid Biobank. Cell Stem Cell 2020, 26, 503–510 e507. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, S.; Salahudeen, A.A.; Sellers, Z.M.; Bravo, D.T.; Choi, S.S.; Batish, A.; Le, W.; Baik, R.; de la, O.S.; Kaushik, M.P.; et al. High-Efficiency, Selection-free Gene Repair in Airway Stem Cells from Cystic Fibrosis Patients Rescues CFTR Function in Differentiated Epithelia. Cell Stem Cell 2020, 26, 161–171 e14. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Crane, A.M.; Anirudhan, V.; Barilla, C.; Matthias, N.; Randell, S.H.; Rab, A.; Sorscher, E.J.; Kerschner, J.L.; Yin, S.; et al. Highly Efficient Gene Editing of Cystic Fibrosis Patient-Derived Airway Basal Cells Results in Functional CFTR Correction. Mol. Ther. 2020, 28, 1684–1695. [Google Scholar] [CrossRef]
- Ensinck, M.; Mottais, A.; Detry, C.; Leal, T.; Carlon, M.S. On the Corner of Models and Cure: Gene Editing in Cystic Fibrosis. Front. Pharmacol. 2021, 12, 662110. [Google Scholar] [CrossRef]
- Moss, R.B.; Flume, P.A.; Elborn, J.S.; Cooke, J.; Rowe, S.M.; McColley, S.A.; Rubenstein, R.C.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: A double-blind, randomised controlled trial. Lancet Respir. Med. 2015, 3, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Fidler, M.C.; Buckley, A.; Sullivan, J.C.; Statia, M.; Boj, S.F.; Vries, R.G.J.; Munck, A.; Higgins, M.; Moretto Zita, M.; Negulescu, P.; et al. G970R-CFTR Mutation (c.2908G>C) Results Predominantly in a Splicing Defect. Clin. Transl. Sci. 2020, 14, 656–663. [Google Scholar] [CrossRef]
- Sosnay, P.R.; Siklosi, K.R.; Van Goor, F.; Kaniecki, K.; Yu, H.; Sharma, N.; Ramalho, A.S.; Amaral, M.D.; Dorfman, R.; Zielenski, J.; et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet. 2013, 45, 1160–1167. [Google Scholar] [CrossRef] [Green Version]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; Vandevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Van Mourik, P.; Michel, S.; Vonk, A.M.; Beekman, J.M.; Van Der Ent, C.K.; De Keyser, H.; Lammertyn, E.; Van Koningsbruggen-Rietschel, S.; Naehrlich, L.; Pool, J.; et al. Rationale and design of the HIT-CF organoid study: Stratifying cystic fibrosis patients based on intestinal organoid response to different CFTR-modulators. Transl. Med. Commun. 2020, 5, 9. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Wiegerinck, C.L.; De Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; De Winter-De Groot, K.M.; Brandsma, A.M.; De Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; De Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; Van De Graaf, E.A.; Nieuwenhuis, E.E.S.; Houwen, R.H.J.; et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016, 8, 344ra84. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, A.S.; Fürstová, E.; Vonk, A.M.; Ferrante, M.; Verfaillie, C.; Dupont, L.; Boon, M.; Proesmans, M.; Beekman, J.M.; Sarouk, I.; et al. Correction of CFTR function in intestinal organoids to guide treatment of cystic fibrosis. Eur. Respir. J. 2021, 57, 1902426. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Papaspyropoulos, A.; Zomer-Van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, T.; Burton, B.; Clark, H.; Van Goor, F. Use of Primary Cultures of Human Bronchial Epithelial Cells Isolated from Cystic Fibrosis Patients for the Pre-clinical Testing of CFTR Modulators. In Cystic Fibrosis; Humana Press: Totowa, NJ, USA, 2011; pp. 39–54. [Google Scholar]
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 7375. [Google Scholar] [CrossRef]
- Sette, G.; Lo Cicero, S.; Blaconà, G.; Pierandrei, S.; Bruno, S.M.; Salvati, V.; Castelli, G.; Falchi, M.; Fabrizzi, B.; Cimino, G.; et al. Theratyping cystic fibrosis in vitro in ALI culture and organoid models generated from patient-derived nasal epithelial conditionally reprogrammed stem cells. Eur. Respir. J. 2021, 58, 2100908. [Google Scholar] [CrossRef]
- Vertex. Who Symdeko Is for. 2022. Available online: https://www.symdeko.com/who-symdeko-is-for (accessed on 10 May 2022).
- Vertex. Who Trikafta Is for. Available online: https://www.trikafta.com/who-trikafta-is-for (accessed on 13 September 2021).
- Shishido, H.; Yoon, J.S.; Yang, Z.; Skach, W.R. CFTR trafficking mutations disrupt cotranslational protein folding by targeting biosynthetic intermediates. Nat. Commun. 2020, 11, 4258. [Google Scholar] [CrossRef]
- Noel, S.; Sermet-Gaudelus, I.; Sheppard, D.N. N1303K: Leaving no stone unturned in the search for transformational therapeutics. J. Cyst. Fibros. 2018, 17, 555–557. [Google Scholar] [CrossRef] [Green Version]
- Laselva, O.; Bartlett, C.; Popa, A.; Ouyang, H.; Gunawardena, T.N.A.; Gonska, T.; Moraes, T.J.; Bear, C.E. Emerging preclinical modulators developed for F508del-CFTR have the potential to be effective for ORKAMBI resistant processing mutants. J. Cyst. Fibros. 2020, 20, 106–119. [Google Scholar] [CrossRef]
- Huang, Y.; Paul, G.; Lee, J.; Yarlagadda, S.; McCoy, K.; Naren, A.P. Elexacaftor/Tezacaftor/Ivacaftor Improved Clinical Outcomes in an N1303K-CFTR Patient Based on in vitro Experimental Evidence. Am. J. Respir. Crit. Care Med. 2021, 204, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Hull, J.; Shackleton, S.; Harris, A. Abnormal mRNA splicing resulting from three different mutations in the CFTR gene. Hum. Mol. Genet. 1993, 2, 689–692. [Google Scholar] [CrossRef] [PubMed]
- SpliSense. SPL84-23-1 Treatment for 3849+10 kb C-to-T CF mutation. 2022. Available online: https://splisense.com/pipeline/spl84-23-1-program/ (accessed on 10 April 2022).
- Sanz, D.J.; Hollywood, J.A.; Scallan, M.F.; Harrison, P.T. Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PLoS ONE 2017, 12, e0184009. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, S.; Traore, S.; Cooney, A.L.; Brommel, C.M.; Kulhankova, K.; Sinn, P.L.; Newby, G.; Liu, D.R.; McCray, P.B. Functional correction of CFTR mutations in human airway epithelial cells using adenine base editors. Nucleic Acids Res. 2021, 49, 10558–10572. [Google Scholar] [CrossRef]
- Bednarski, C.; Tomczak, K.; Vom Hovel, B.; Weber, W.M.; Cathomen, T. Targeted Integration of a Super-Exon into the CFTR Locus Leads to Functional Correction of a Cystic Fibrosis Cell Line Model. PLoS ONE 2016, 11, e0161072. [Google Scholar] [CrossRef]
- Mall, M.A. ENaC inhibition in cystic fibrosis: Potential role in the new era of CFTR modulator therapies. Eur. Respir. J. 2020, 56, 2000946. [Google Scholar] [CrossRef] [PubMed]
- Quesada, R.; Dutzler, R. Alternative chloride transport pathways as pharmacological targets for the treatment of cystic fibrosis. J. Cyst. Fibros. 2020, 19 (Suppl. 1), S37–S41. [Google Scholar] [CrossRef] [Green Version]
- Hampton, T. With First CRISPR Trials, Gene Editing Moves Toward the Clinic. JAMA 2020, 323, 1537–1539. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, M.P.; Krishnakumar, R.; Timlin, J.A.; Carney, J.P.; Butler, K.S. Gene editing and CRISPR in the clinic: Current and future perspectives. Biosci. Rep. 2020, 40, BSR20200127. [Google Scholar] [CrossRef] [Green Version]









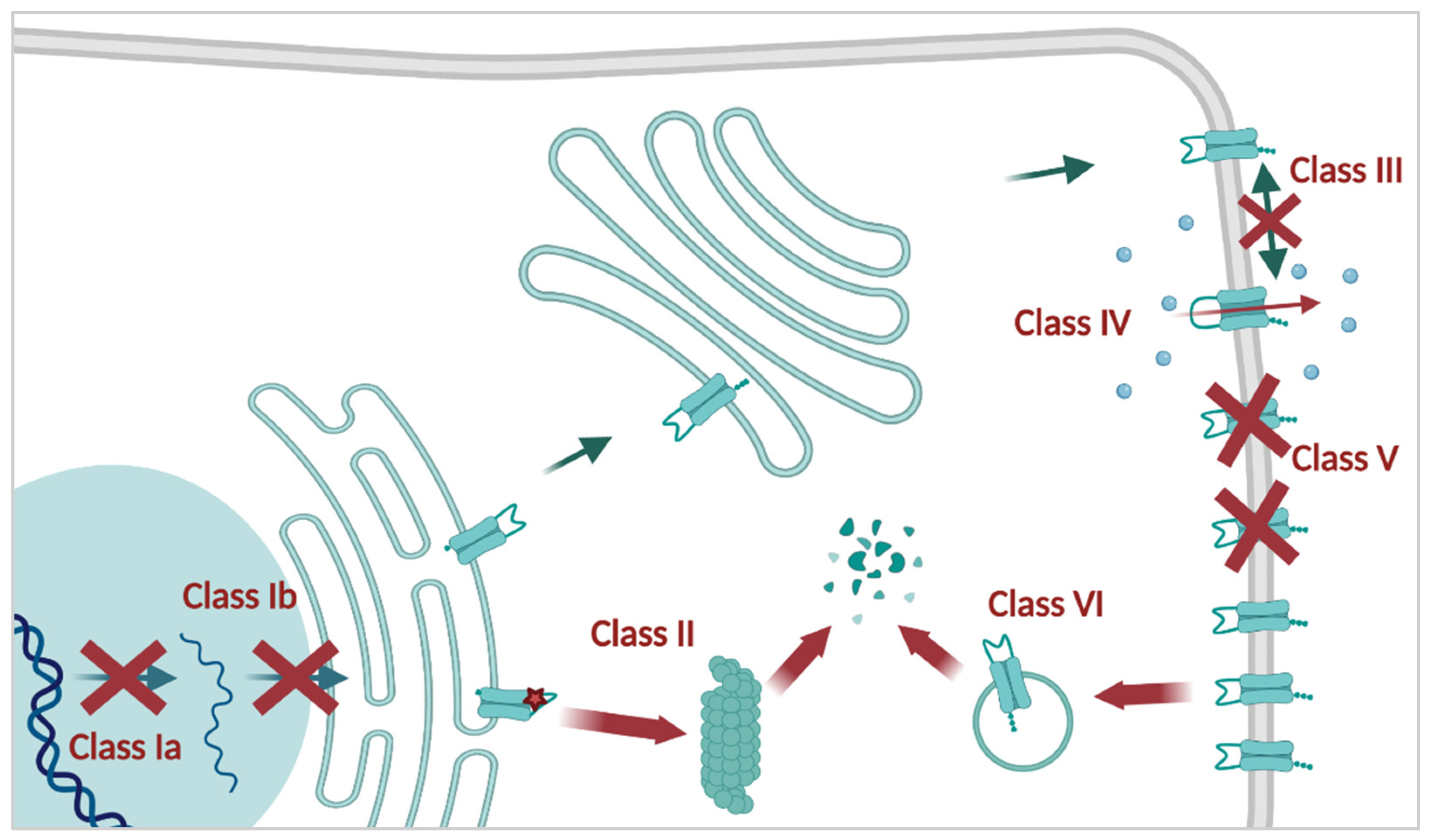{kind=link}
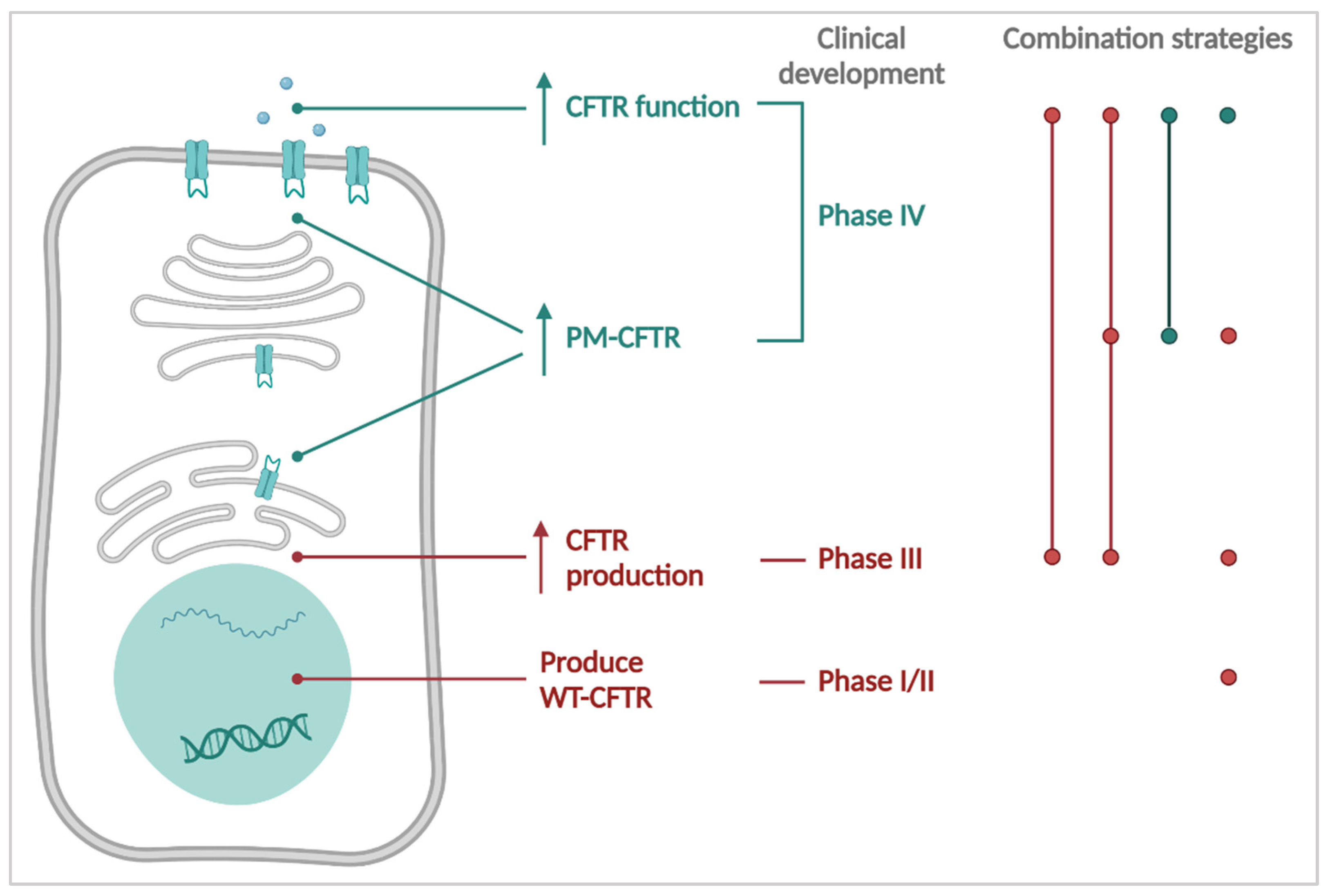{kind=link}
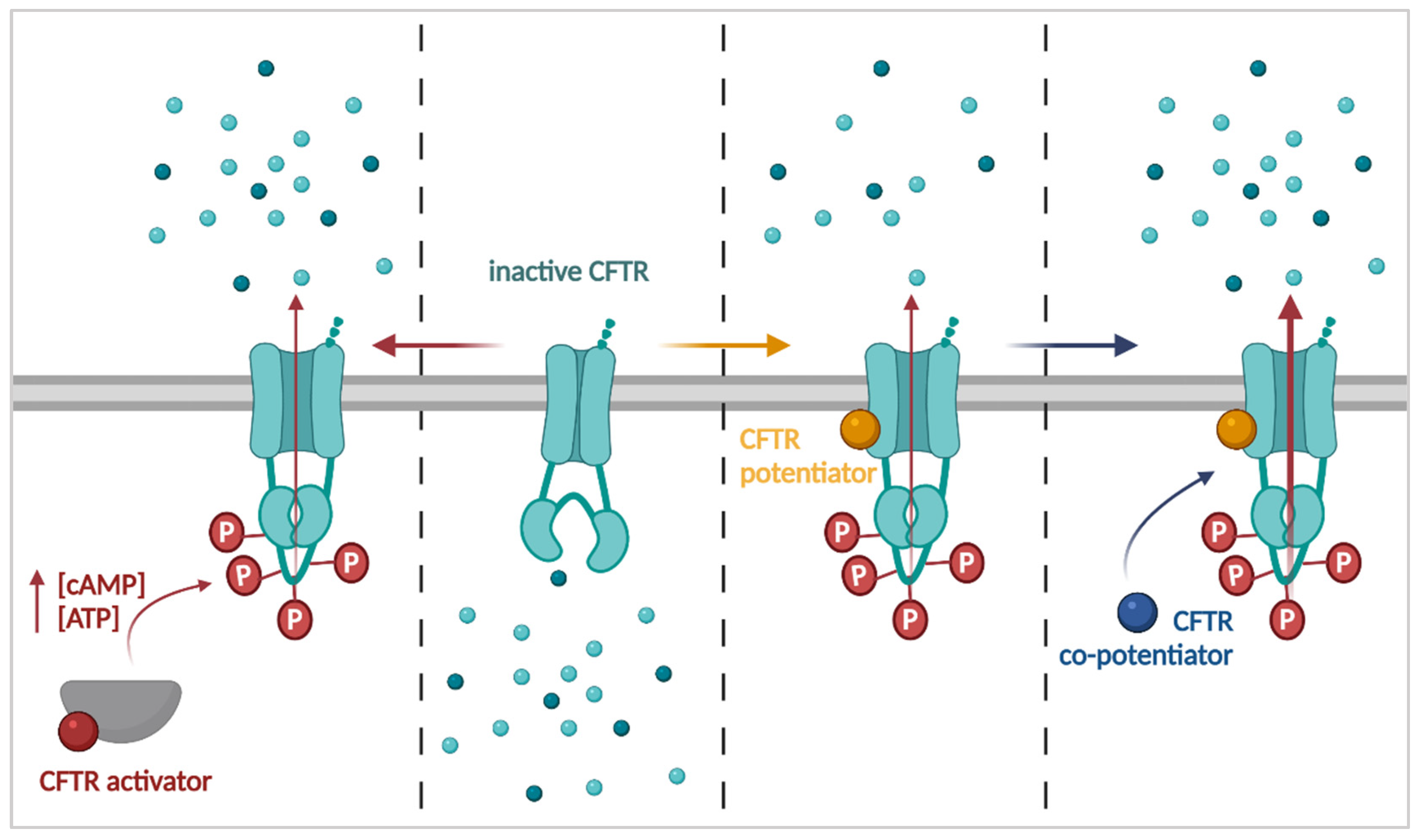{kind=link}
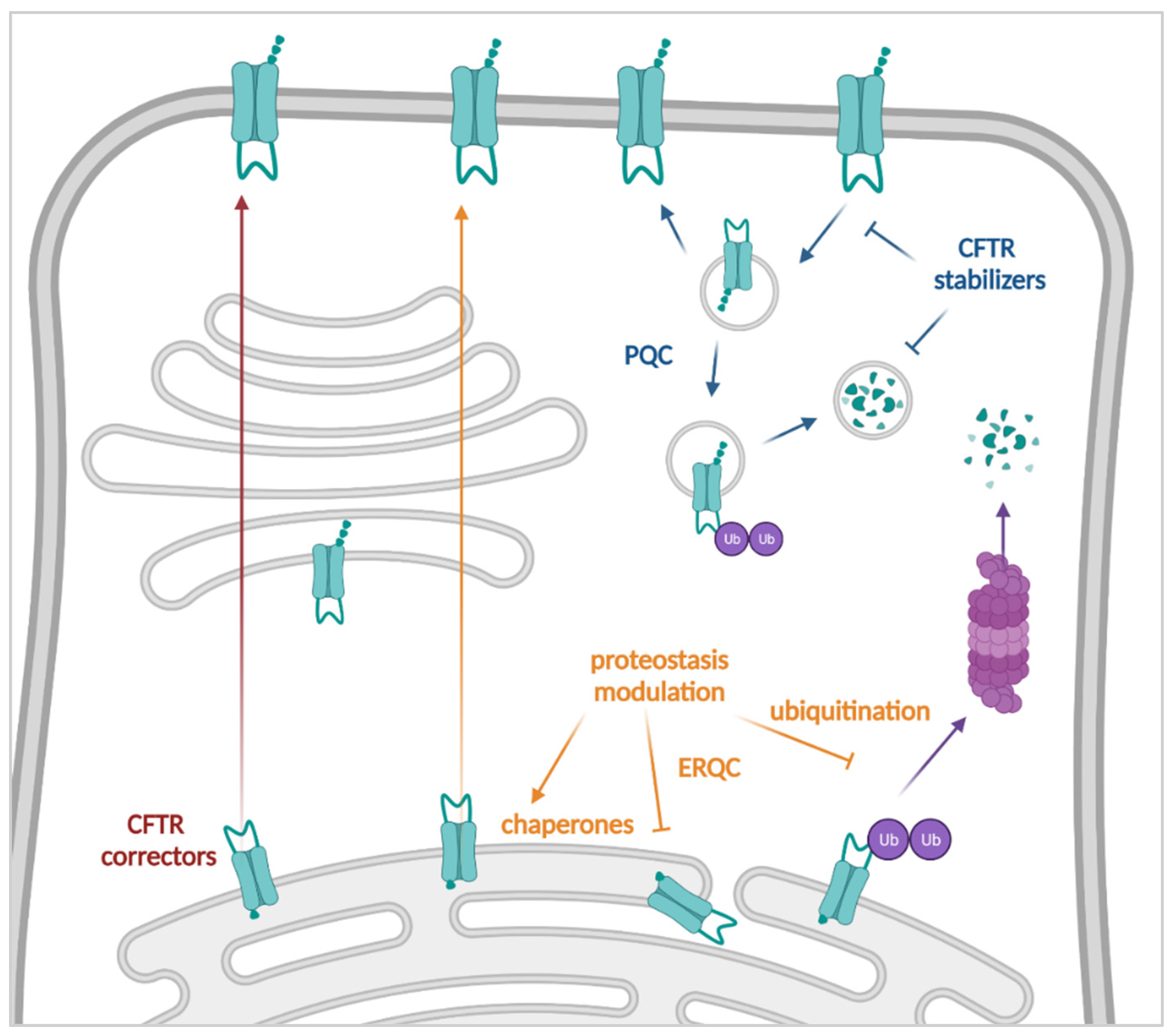{kind=link}
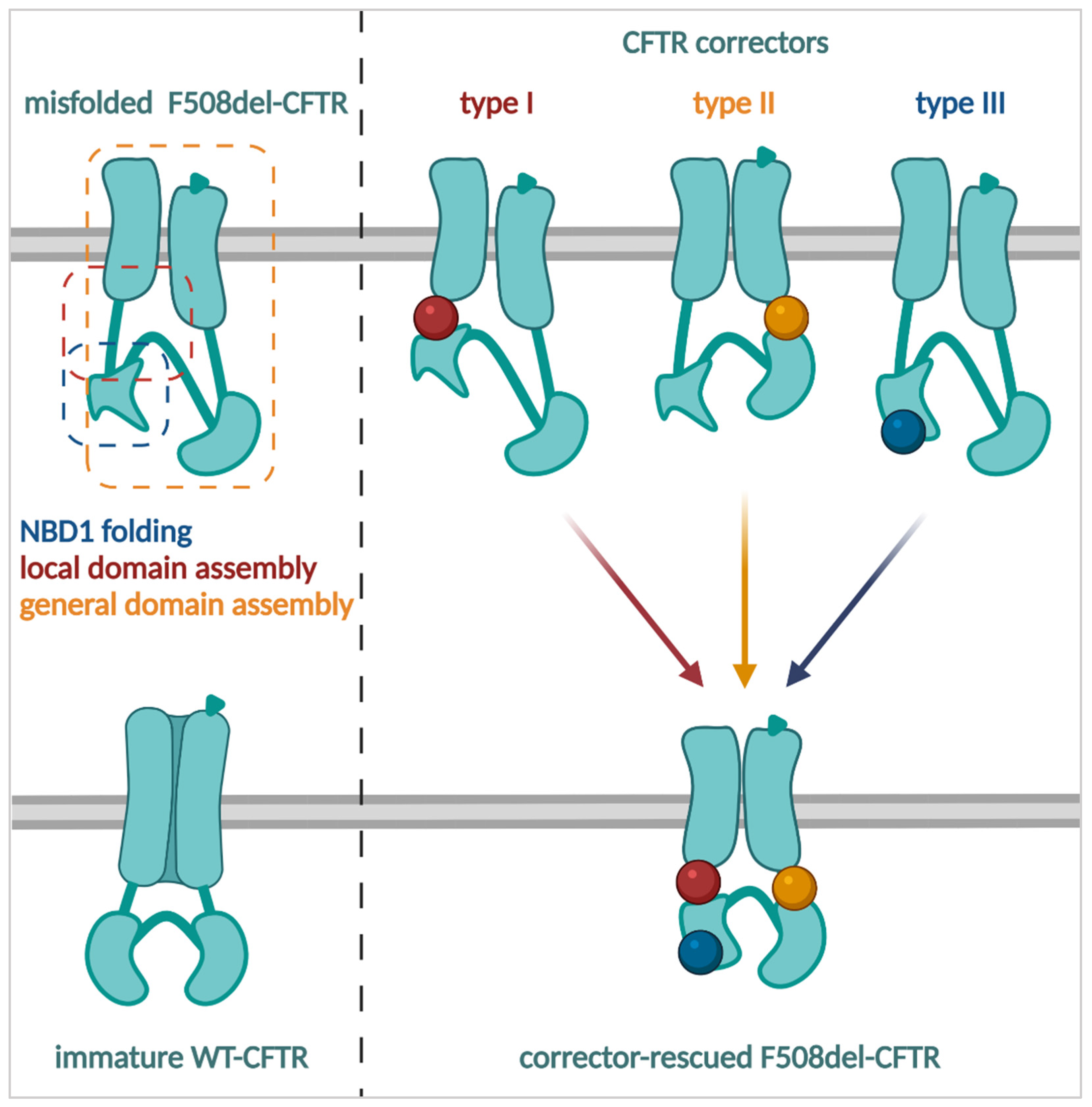{kind=link}
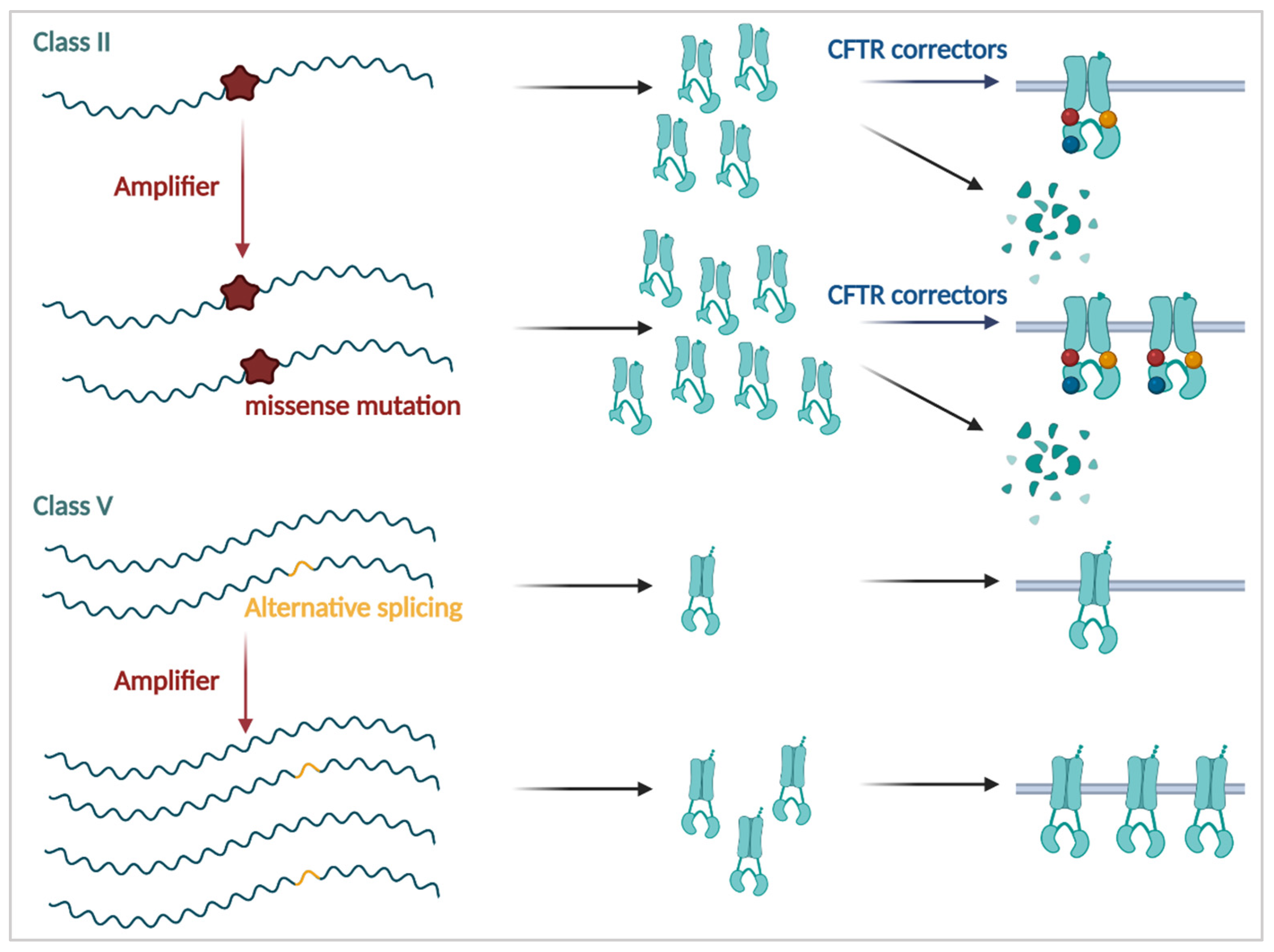{kind=link}
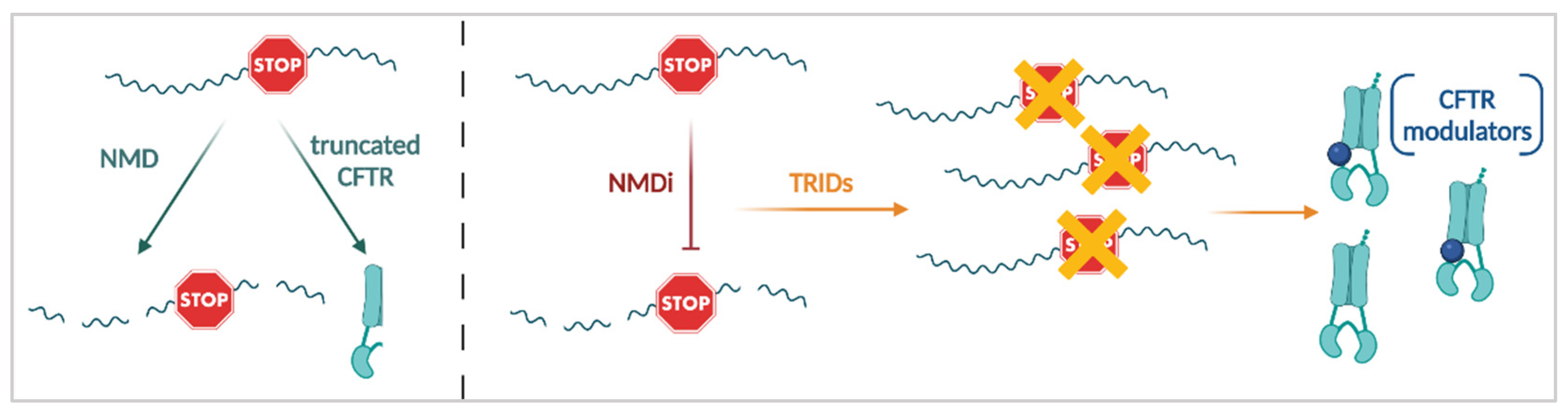{kind=link}
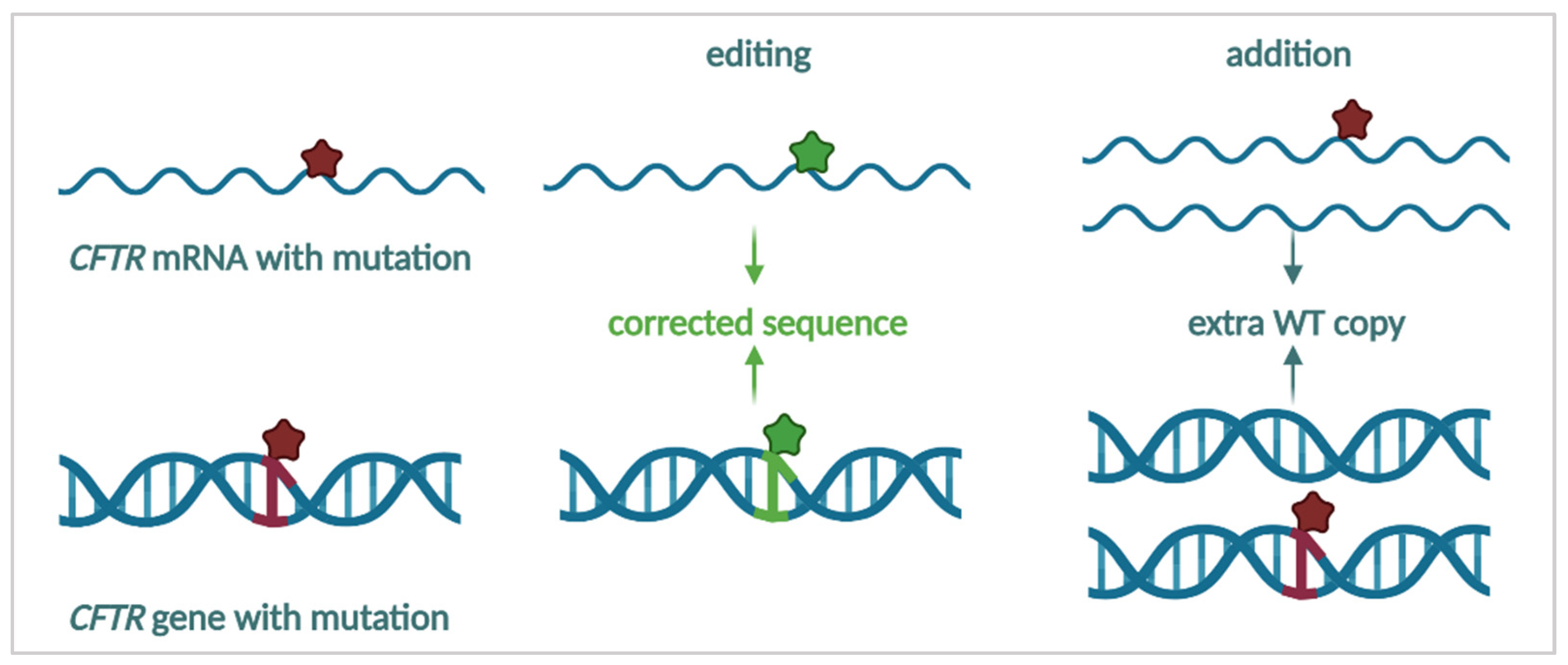{kind=link}
| Name | cDNA Name | Allele Frequency (%) | CFTR2 Alleles | Approved Treatment |
|---|---|---|---|---|
| CFTRdele2,3 | c.54-5940_273 + 10250del21kb | 0.3 | 417 | NA |
| P67L | c.200C>T | 0.2 | 239 | K/S/T |
| G85E | c.254G>A | 0.4 | 616 | T |
| E92K | c.274G>A | <0.1 | 49 | S/T |
| R117H * | c.350G>A | 1.3 | 1854 | K/S/T |
| Y122X | c.366T>A | 0.1 | 88 | NA |
| 621+1G>T | c.489+1G>T | 0.9 | 1323 | NA |
| G178R | c.532G>A | 0.1 | 87 | K/S/T |
| R334W | c.1000C>T | 0.3 | 429 | NA |
| W496X | c.1487G>A | <0.1 | 3 | NA |
| I507del | c.1519-1521delACT | 0.5 | 651 | NA |
| F508del | c.1521-1523delCTT | 69.7 | 99061 | O/S/T |
| 1717-1G>A | c.1585-1G>A | 0.9 | 1216 | NA |
| G542X | c.1624G>T | 2.5 | 3610 | NA |
| S549R | c.1645A>C | 0.1 | 93 | K/S/T |
| S549N | c.1646G>A | 0.1 | 203 | K/S/T |
| G551S | c.1651G>A | <0.1 | 19 | K/S/T |
| G551D | c.1652G>A | 2.1 | 2986 | K/S/T |
| R553X | c.1657C>T | 0.9 | 1323 | NA |
| 2789+5G>A | c.2657+5G>A | 0.7 | 1027 | K/S |
| G970R | c.2908G>C | <0.1 | 12 | NA |
| R1162X | c.3484C>T | 0.5 | 651 | NA |
| I1234V | c.3700A>G | <0.1 | 33 | NA |
| 3849+10kbC>T | c.3718-2477C>T | 0.8 | 1158 | K/S |
| G1244E | c.3731G>A | 0.1 | 106 | K/S/T |
| S1251N | c.3752G>A | 0.1 | 120 | K/S/T |
| S1255P | c.3763T>C | <0.1 | 10 | K/S/T |
| W1282X | c.3846G>A | 1.2 | 1726 | NA |
| N1303K | c.3909C>G | 1.6 | 2246 | NA |
| Q1313X | c.3937C>T | <0.1 | 30 | NA |
| G1349D | c.4046G>A | <0.1 | 22 | K/S/T |
| Q1412X | c.4234C>T | <0.1 | 4 | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ensinck, M.M.; Carlon, M.S. One Size Does Not Fit All: The Past, Present and Future of Cystic Fibrosis Causal Therapies. Cells 2022, 11, 1868. https://doi.org/10.3390/cells11121868
Ensinck MM, Carlon MS. One Size Does Not Fit All: The Past, Present and Future of Cystic Fibrosis Causal Therapies. Cells. 2022; 11(12):1868. https://doi.org/10.3390/cells11121868
Chicago/Turabian StyleEnsinck, Marjolein M., and Marianne S. Carlon. 2022. "One Size Does Not Fit All: The Past, Present and Future of Cystic Fibrosis Causal Therapies" Cells 11, no. 12: 1868. https://doi.org/10.3390/cells11121868
APA StyleEnsinck, M. M., & Carlon, M. S. (2022). One Size Does Not Fit All: The Past, Present and Future of Cystic Fibrosis Causal Therapies. Cells, 11(12), 1868. https://doi.org/10.3390/cells11121868