
Methylation Status of Gene Bodies of Selected microRNA Genes Associated with Neoplastic Transformation in Equine Sarcoids


Abstract
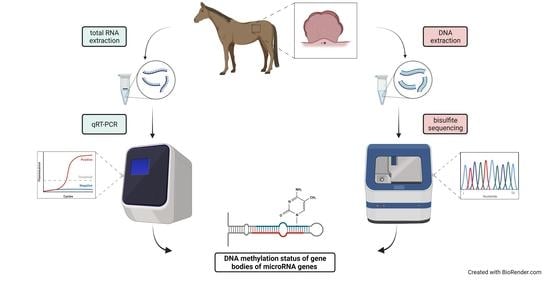
:
1. Introduction
2. Material and Methods
2.1. Research Material
2.2. BPV and microRNA Gene Expression
2.3. miR-10b Expression Validation
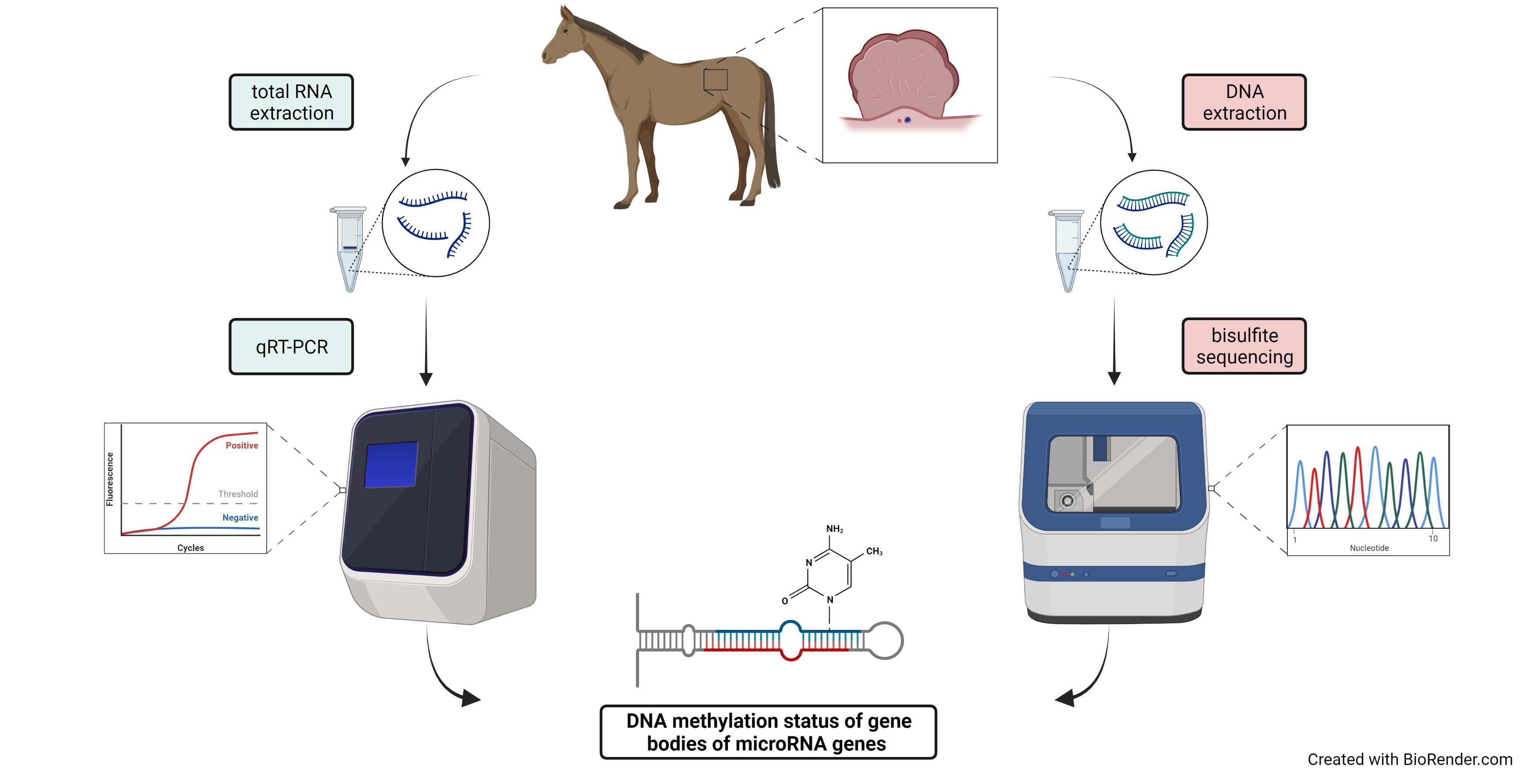
2.4. Methylation Analysis of miRNA Localized CpGs
2.5. Statistics
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Jackson, C. The incidence and pathology of tumours of domesticated animals in South Africa. Onderstepoort J. Vet. Sci. Anim. Ind. 1936, 6, 378–385. [Google Scholar]
- Bogaert, L.; Martens, A.; Depoorter, P.; Gasthuys, F. Equine sarcoids-Part 1: Clinical presentation and epidemiology. Vlaams Diergeneeskd. Tijdschr. 2008, 77, 2–9. [Google Scholar]
- Ogłuszka, M.; Starzyński, R.R.; Pierzchała, M.; Otrocka-Domagała, I.; Raś, A. Equine Sarcoids—Causes, Molecular Changes, and Clinicopathologic Features: A Review. Vet. Pathol. 2021, 58, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Broström, H. Equine sarcoids. A clinical and epidemiological study in relation to equine leucocyte antigens (ELA). Acta Vet. Scand. 1995, 36, 223–236. [Google Scholar] [CrossRef]
- Chambers, G.; Ellsmore, V.A.; O’Brien, P.M.; Reid, S.W.J.; Love, S.; Campo, M.S.; Nasir, L. Association of bovine papillomavirus with the equine sarcoid. J. Gen. Virol. 2003, 84, 1055–1062. [Google Scholar] [CrossRef]
- Semik-Gurgul, E. Molecular approaches to equine sarcoids. Equine Vet. J. 2021, 53, 221–230. [Google Scholar] [CrossRef]
- Knottenbelt, D.C. The Equine Sarcoid: Why Are There so Many Treatment Options? Vet. Clin. N. Am. Equine Pract. 2019, 35, 243–262. [Google Scholar] [CrossRef]
- Filipowicz, W.; Jaskiewicz, L.; Kolb, F.A.; Pillai, R.S. Post-transcriptional gene silencing by siRNAs and miRNAs. Curr. Opin. Struct. Biol. 2005, 15, 331–341. [Google Scholar] [CrossRef]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef]
- Elson-Schwab, I.; Lorentzen, A.; Marshall, C.J. MicroRNA-200 family members differentially regulate morphological plasticity and mode of melanoma cell invasion. PLoS ONE 2010, 5, e13176. [Google Scholar] [CrossRef]
- Muralidhar, G.; Barbolina, M. The miR-200 Family: Versatile Players in Epithelial Ovarian Cancer. Int. J. Mol. Sci. 2015, 16, 16833–16847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedroza-Torres, A.; López-Urrutia, E.; García-Castillo, V.; Jacobo-Herrera, N.; Herrera, L.A.; Peralta-Zaragoza, O.; López-Camarillo, C.; De Leon, D.C.; Fernández-Retana, J.; Cerna-Cortés, J.F.; et al. MicroRNAs in cervical cancer: Evidences for a miRNA profile deregulated by HPV and its impact on radio-resistance. Molecules 2014, 19, 6263–6281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlina, K.; Gurgul, A.; Szmatoła, T.; Koch, C.; Mählmann, K.; Witkowski, M.; Bugno-Poniewierska, M. Comprehensive characteristics of microRNA expression profile of equine sarcoids. Biochimie 2017, 137, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Jones, P.A. DNA methylation: Past, present and future directions. Carcinogenesis 2000, 21, 461–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, R.E.; Curtin, G.M.; Doolittle, D.J.; Goodman, J.I. Progressive alterations in global and GC-rich DNA methylation during tumorigenesis. Toxicol. Sci. 2003, 75, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Arechederra, M.; Daian, F.; Yim, A.; Bazai, S.K.; Richelme, S.; Dono, R.; Saurin, A.J.; Habermann, B.H.; Maina, F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018, 9, 3164. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Murtha, M.; Esteller, M. Extraordinary Cancer Epigenomics: Thinking Outside the Classical Coding and Promoter Box. Trends Cancer 2016, 2, 572–584. [Google Scholar] [CrossRef]
- Suzuki, H.; Maruyama, R.; Yamamoto, E.; Kai, M. Epigenetic alteration and microRNA dysregulation in cancer. Front. Genet. 2013, 4, 258. [Google Scholar] [CrossRef] [Green Version]
- Semik, E.; Ząbek, T.; Gurgul, A.; Fornal, A.; Szmatoła, T.; Pawlina, K.; Wnuk, M.; Klukowska-Rötzler, J.; Koch, C.; Mählmann, K.; et al. Comparative analysis of DNA methylation patterns of equine sarcoid and healthy skin samples. Vet. Comp. Oncol. 2018, 16, 37–46. [Google Scholar] [CrossRef]
- Altamura, G.; Strazzullo, M.; Corteggio, A.; Francioso, R.; Roperto, F.; D’Esposito, M.; Borzacchiello, G. O(6)-methylguanine-DNA methyltransferase in equine sarcoids: Molecular and epigenetic analysis. BMC Vet. Res. 2012, 8, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strazzullo, M.; Corteggio, A.; Altamura, G.; Francioso, R.; Roperto, F.; D’Esposito, M.; Borzacchiello, G. Molecular and epigenetic analysis of the fragile histidine triad tumour suppressor gene in equine sarcoids. BMC Vet. Res. 2012, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semik-Gurgul, E.; Ząbek, T.; Fornal, A.; Wnuk, M.; Pawlina-Tyszko, K.; Gurgul, A.; Klukowska-Rötzler, J.; Koch, C.; Mählmann, K.; Bugno-Poniewierska, M. DNA methylation patterns of the S100A14, POU2F3 and SFN genes in equine sarcoid tissues. Res. Vet. Sci. 2018, 119, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Srivastava, S. Epigenetyka nowotworów: Implikacje dla wczesnego wykrywania i zapobiegania. Lancet Oncol. 2003, 2, 121–131. [Google Scholar]
- Burri, N.; Shaw, P.; Bouzourene, H.; Sordat, I.; Sordat, B.; Gillet, M.; Schorderet, D.; Bosman, F.T.; Chaubert, P. Methylation Silencing and Mutations of the p14ARF and p16INK4a Genes in Colon Cancer. Lab. Investig. 2001, 81, 217–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, J.F.; Frühwald, M.C.; Smiraglia, D.J.; Rush, L.J.; Robertson, G.P.; Gao, X.; Wright, F.A.; Feramisco, J.D.; Peltomäki, P.; Lang, J.C.; et al. Aberrant CpG-island methylation has non-random and tumour-type–specific patterns. Nat. Genet. 2000, 24, 132–138. [Google Scholar] [CrossRef]
- Bogaert, L.; van Poucke, M.; De Baere, C.; Dewulf, J.; Peelman, L.; Ducatelle, R.; Gasthuys, F.; Martens, A. Bovine papillomavirus load and mRNA expression, cell proliferation and p53 expression in four clinical types of equine sarcoid. J. Gen. Virol. 2007, 88, 2155–2161. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Bogaert, L.; van Poucke, M.; De Baere, C.; Peelman, L.; Gasthuys, F.; Martens, A. Selection of a set of reliable reference genes for quantitative real-time PCR in normal equine skin and in equine sarcoids. BMC Biotechnol. 2006, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of Real-Time Quantitative Reverse Transcription-PCR Data: A Model-Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BLAST. Joseph Bedell, Ian Korf and Mark Yandell; OReilly & Associates: Sebastopol, CA, USA, 2003. [Google Scholar]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Flicek, P. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Leakey, T.I.; Zielinski, J.; Siegfried, R.N.; Siegel, E.R.; Fan, C.Y.; Cooney, C.A. A simple algorithm for quantifying DNA methylation levels on multiple independent CpG sites in bisulfite genomic sequencing electropherograms. Nucleic Acids Res. 2008, 36, e64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Mallona, I.; Díez-Villanueva, A.; Peinado, M.A. Methylation plotter: A web tool for dynamic visualization of DNA methylation data. Source Code Biol Med. 2014, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Lund, A.H. miR-10 in development and cancer. Cell Death Differ. 2010, 17, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Moghbeli, M. Molecular interactions of miR-338 during tumor progression and metastasis. Cell. Mol. Biol. Lett. 2021, 26, 1–24. [Google Scholar] [CrossRef]
- Wang, C.Z.; Deng, F.; Li, H.; Wang, D.D.; Zhang, W.; Ding, L.; Tang, J.H. MiR-101: A potential therapeutic target of cancers. Am. J. Transl. Res. 2018, 10, 3310–3321. [Google Scholar]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Lee, H.-C.; Park, J.-L.; Kim, M.; Kim, S.-Y.; Noh, S.-M.; Song, K.-S.; Cheon Kim, J.; Sung Kim, Y. Epigenetic regulation of microRNA-10b and targeting of oncogenic MAPRE1 in gastric cancer. Epigenetics 2011, 6, 740–751. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Xu, Y.; Pan, L.; Feng, Y.; Luo, K.; Mu, Q.; Luo, G. miR-10b Downregulated by DNA Methylation Acts as a Tumor Suppressor in HPV-Positive Cervical Cancer via Targeting Tiam1. Cell. Physiol. Biochem. 2018, 51, 1763–1777. [Google Scholar] [CrossRef]
- Sheedy, P.; Medarova, Z. The fundamental role of miR-10b in metastatic cancer. Am. J. Cancer Res. 2018, 8, 1674. [Google Scholar] [PubMed]
- Gan, L.; Denecke, B. Profiling Pre-MicroRNA and Mature MicroRNA Expressions Using a Single Microarray and Avoiding Separate Sample Preparation. Microarrays 2013, 2, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Jazdzewski, K.; Murray, E.L.; Franssila, K.; Jarzab, B.; Schoenberg, D.R.; De La Chapelle, A. Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc. Natl. Acad. Sci. USA 2008, 105, 7269–7274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlina-Tyszko, K.; Semik-Gurgul, E.; Gurgul, A.; Oczkowicz, M.; Szmatoła, T.; Bugno-Poniewierska, M. Application of the targeted sequencing approach reveals the single nucleotide polymorphism (SNP) repertoire in microRNA genes in the pig genome. Sci. Rep. 2021, 11, 9848. [Google Scholar] [CrossRef] [PubMed]
- Davalos, V.; Moutinho, C.; Villanueva, A.; Boque, R.; Silva, P.; Carneiro, F.; Esteller, M. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 2011, 31, 2062–2074. [Google Scholar] [CrossRef]
- Ceppi, P.; Mudduluru, G.; Kumarswamy, R.; Rapa, I.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Loss of miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype in non-small cell lung cancer. Mol. Cancer Res. 2010, 8, 1207–1216. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Maruyama, R.; Yamamoto, E.; Kai, M. DNA methylation and microRNA dysregulation in cancer. Mol. Oncol. 2012, 6, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Cao, Q.; Mani, R.S.; Shankar, S.; Wang, X.; Ateeq, B.; Laxman, B.; Cao, X.; Jing, X.; Ramnarayanan, K.; et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 2008, 322, 1695–1699. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Xiang, T.; Ren, G.; Tan, C.; Liu, R.; Xu, X.; Wu, Z. miR-101 is down-regulated by the hepatitis B virus x protein and induces aberrant DNA methylation by targeting DNA methyltransferase 3A. Cell Signal. 2013, 25, 439–446. [Google Scholar] [CrossRef]
- Huang, D.; Wang, X.; Zhuang, C.; Shi, W.; Liu, M.; Tu, Q.; Zhang, D.; Hu, L. Reciprocal negative feedback loop between EZH2 and MIR-101-1 contributes to MIR-101 deregulation in hepatocellular carcinoma. Oncol. Rep. 2016, 35, 1083–1090. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Sequence (5′–3′) | Reverse Sequence (5′–3′) | Product Size (bp) |
|---|---|---|---|
| BPV | TGCAGTTGTCTTTGCAGGAG | AGCACCGTTTAGGTTCTGACAT | 104 |
| ACTB | CCAGCACGATGAAGATCAAG | GTGGACAATGAGGCCAGAAT | 88 |
| UBB | GCAAGACCATCACCCTGGA | CTAACAGCCACCCCTGAGAC | 206 |
| eca-mir-101-1 | TCACAGTGCTGATGCTGTCA | TAGGGGAGGCACAATATGGA | 178 |
| eca-mir-200a | CTTACCGGACAGTGCTGGAT | CCGATGTGGCTGAACTGAC | 169 |
| eca-mir-10b | ATTGCCACCAAGTCCTTCAG | TGAAGTTTTTGCATCGACCA | 237 |
| eca-mir-338 | CGGAAGAAATGGTGATGGAC | AGCTGCCCTCTTCAACAAAA | 132 |
| Gene Name | Forward Sequence (5′–3′) | Reverse Sequence (5′–3′) | Product Size (bp) | Number of Analyzed CpG Site |
|---|---|---|---|---|
| eca-mir-101-1 | GAGGTTAGGGAGATAGTAAGTTTAGG | ACCTTTAAAACTAACAACATCAACA | 384 | 10 |
| eca-mir-200a | TTATTTTGGAGAGAGTAGGGG | CCTAACCCTAATAATCTATCCCA | 419 | 18 |
| eca-mir-10b | GGTTGGTAGTAGTTTGGGTATTTG | CCAAAATCTAACCCTTTAACCC | 367 | 7 |
| eca-mir-338 | GAGGGATGGTTTTGTTTTG | TACATCTACCACACAACTACTATACCA | 314 | 14 |
| mir-101-1 | mir-10b | mir-200a | mir-338 | |
|---|---|---|---|---|
| Relative average expression level in the sarcoid samples | 0.14 | 0.12 | 0.18 | 0.21 |
| Relative average expression level in the control samples | 0.27 | 0.27 | 0.50 | 0.50 |
| Fold change | −1.92 (p value = 0.031) | −2.27 (p value = 0.050) | −2.78 (p value = 0.004) | −2.38 (p value = 0.211) |
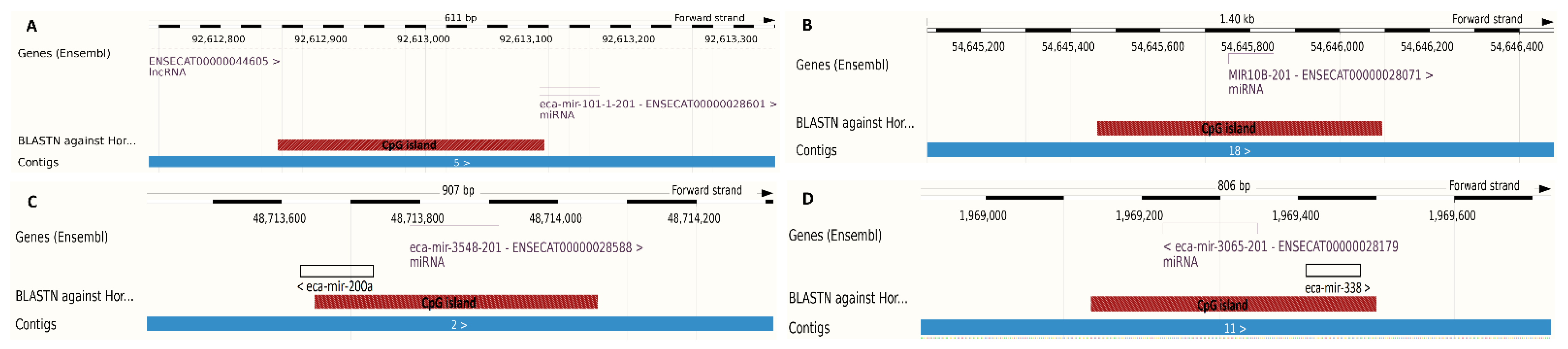
| Average CpG methylation level in the sarcoid samples | 85.4% | 67.8% | 79.9% | 73.9% |
| Average CpG methylation level in the control samples | 82.5% | 58.8% | 78.3% | 77.3% |
| Methylation difference | 2.9% (p value = 0.009) | 9.0% (p value = 0.011) | 1.6% (p value = 1.49 × 10−5) | −3.4% (p value = 0.623) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawlina-Tyszko, K.; Semik-Gurgul, E.; Ząbek, T.; Witkowski, M. Methylation Status of Gene Bodies of Selected microRNA Genes Associated with Neoplastic Transformation in Equine Sarcoids. Cells 2022, 11, 1917. https://doi.org/10.3390/cells11121917
Pawlina-Tyszko K, Semik-Gurgul E, Ząbek T, Witkowski M. Methylation Status of Gene Bodies of Selected microRNA Genes Associated with Neoplastic Transformation in Equine Sarcoids. Cells. 2022; 11(12):1917. https://doi.org/10.3390/cells11121917
Chicago/Turabian StylePawlina-Tyszko, Klaudia, Ewelina Semik-Gurgul, Tomasz Ząbek, and Maciej Witkowski. 2022. "Methylation Status of Gene Bodies of Selected microRNA Genes Associated with Neoplastic Transformation in Equine Sarcoids" Cells 11, no. 12: 1917. https://doi.org/10.3390/cells11121917
APA StylePawlina-Tyszko, K., Semik-Gurgul, E., Ząbek, T., & Witkowski, M. (2022). Methylation Status of Gene Bodies of Selected microRNA Genes Associated with Neoplastic Transformation in Equine Sarcoids. Cells, 11(12), 1917. https://doi.org/10.3390/cells11121917