
Population Scale Analysis of Centromeric Satellite DNA Reveals Highly Dynamic Evolutionary Patterns and Genomic Organization in Long-Tailed and Rhesus Macaques



 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimen Collection and DNA Extraction
2.2. Fosmid DNA Library and Isolation of Satellite DNA Sequences
2.3. Cell Culture and Chromosome Preparation
2.4. C-Banding
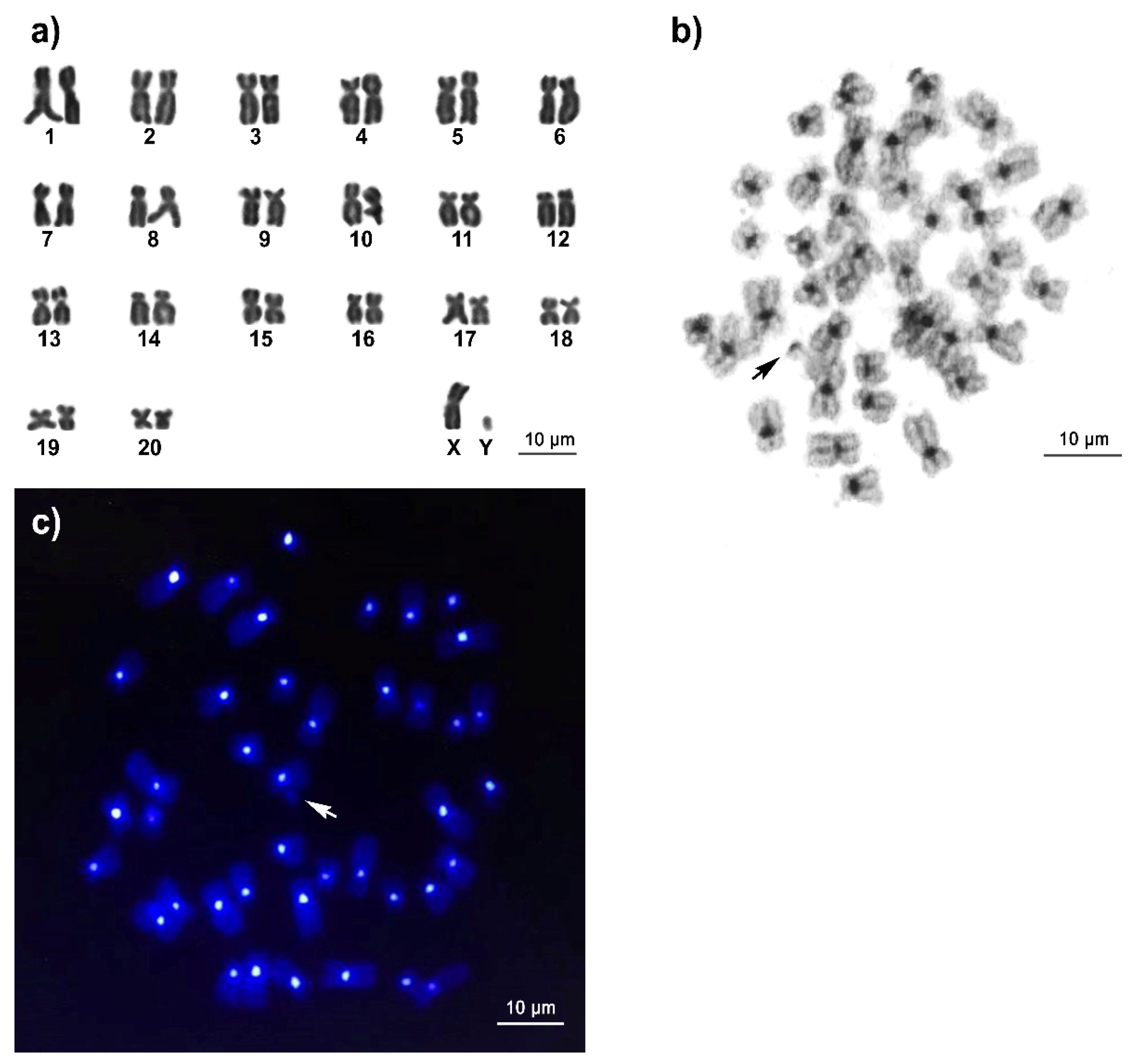
2.5. Fluorescence In Situ Hybridization Mapping of Cen-satDNA
2.6. Centromeric Satellite DNA Sequencing in Macaques
2.7. Identification of Putative Satellite DNA in Long-Tailed and Rhesus Macaque Populations
2.8. Genomic Organization and Comparative Genomics of Satellite DNA Sequences
2.9. Tests of Genetic Diversity within and between Macaque Populations
3. Results
3.1. Karyotype and C-Positive Heterochromatin of Long-Tailed Macaque
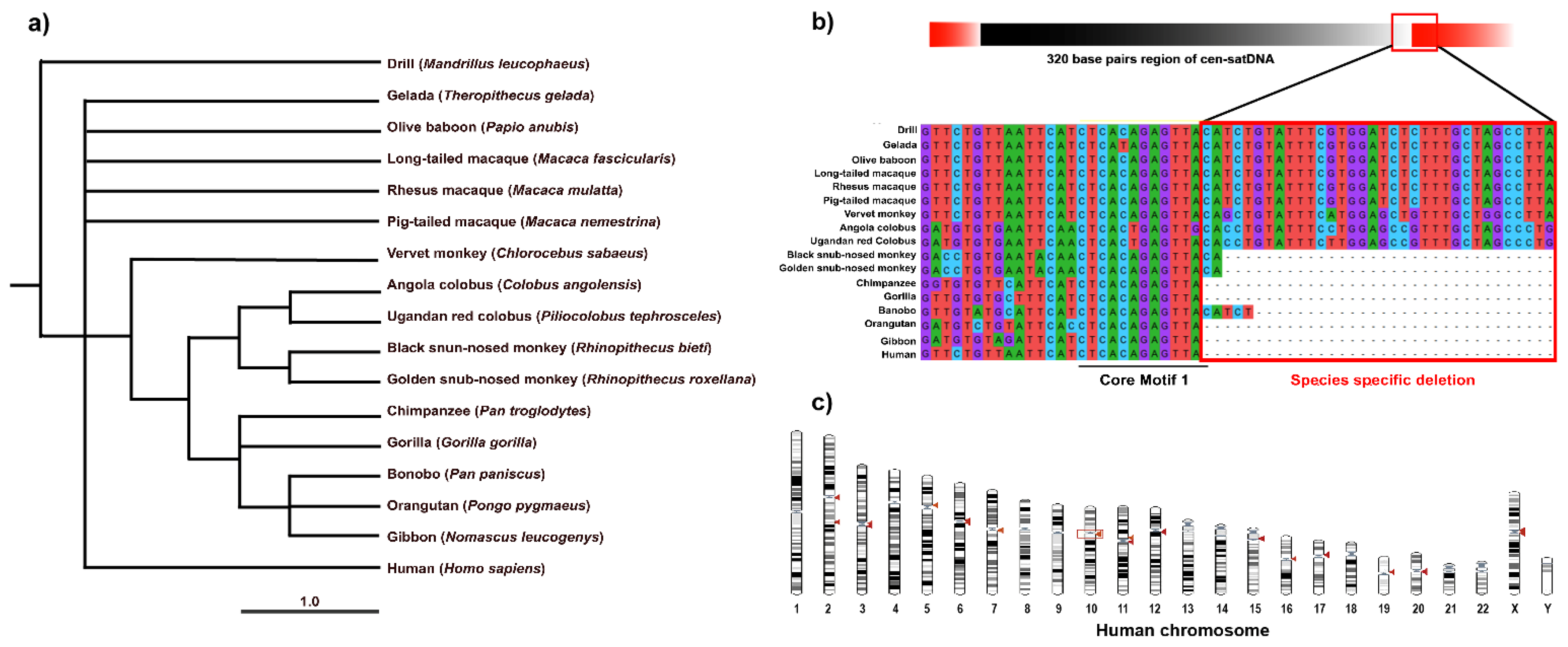
3.2. Isolation of Highly Repetitive DNA Sequences and Their Nucleotide Sequences
3.3. Striking Sequence Variability of Cen-satDNA in Macaque Populations
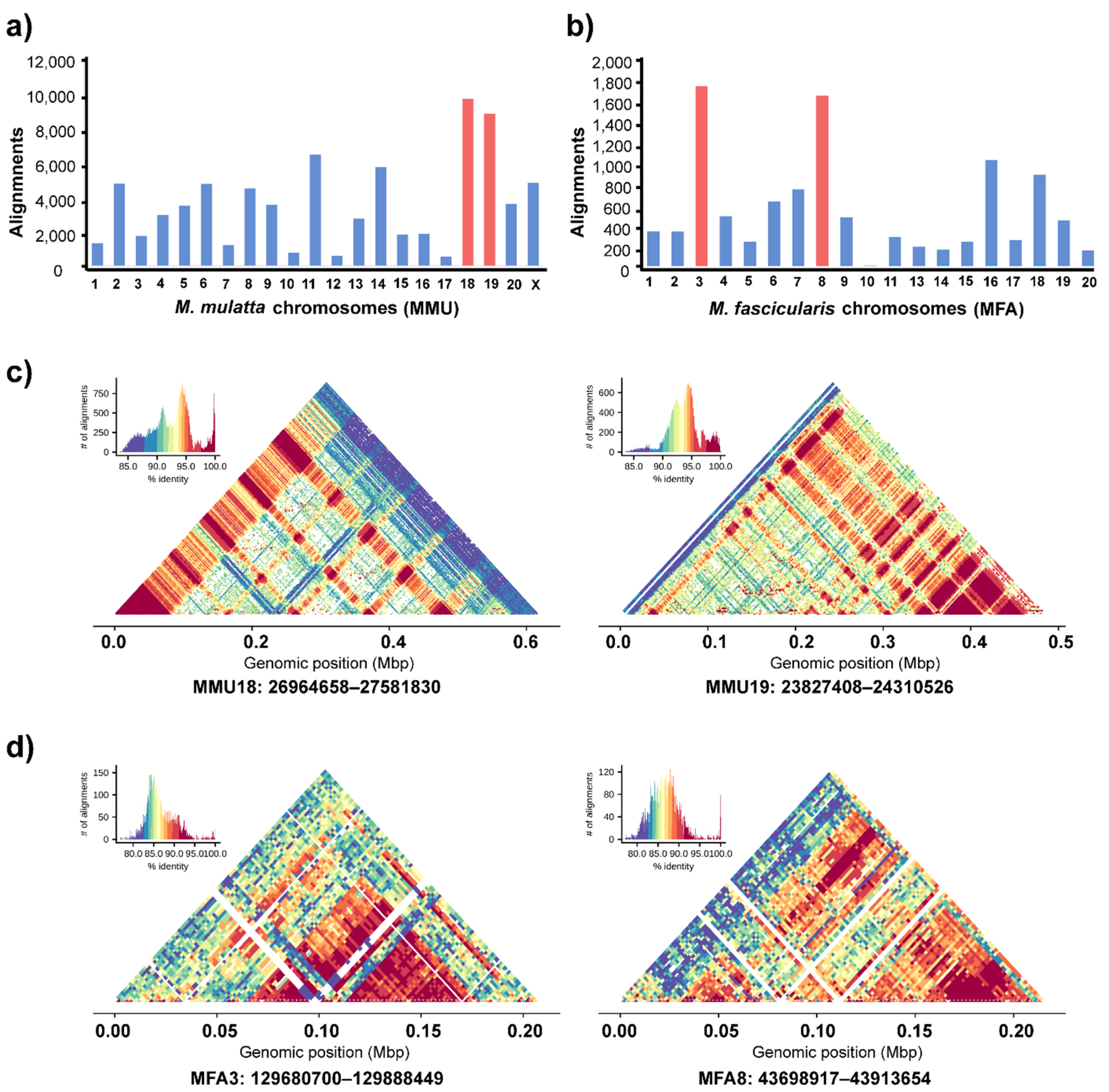
3.4. Genomic Organization of Cen-satDNA Sequences
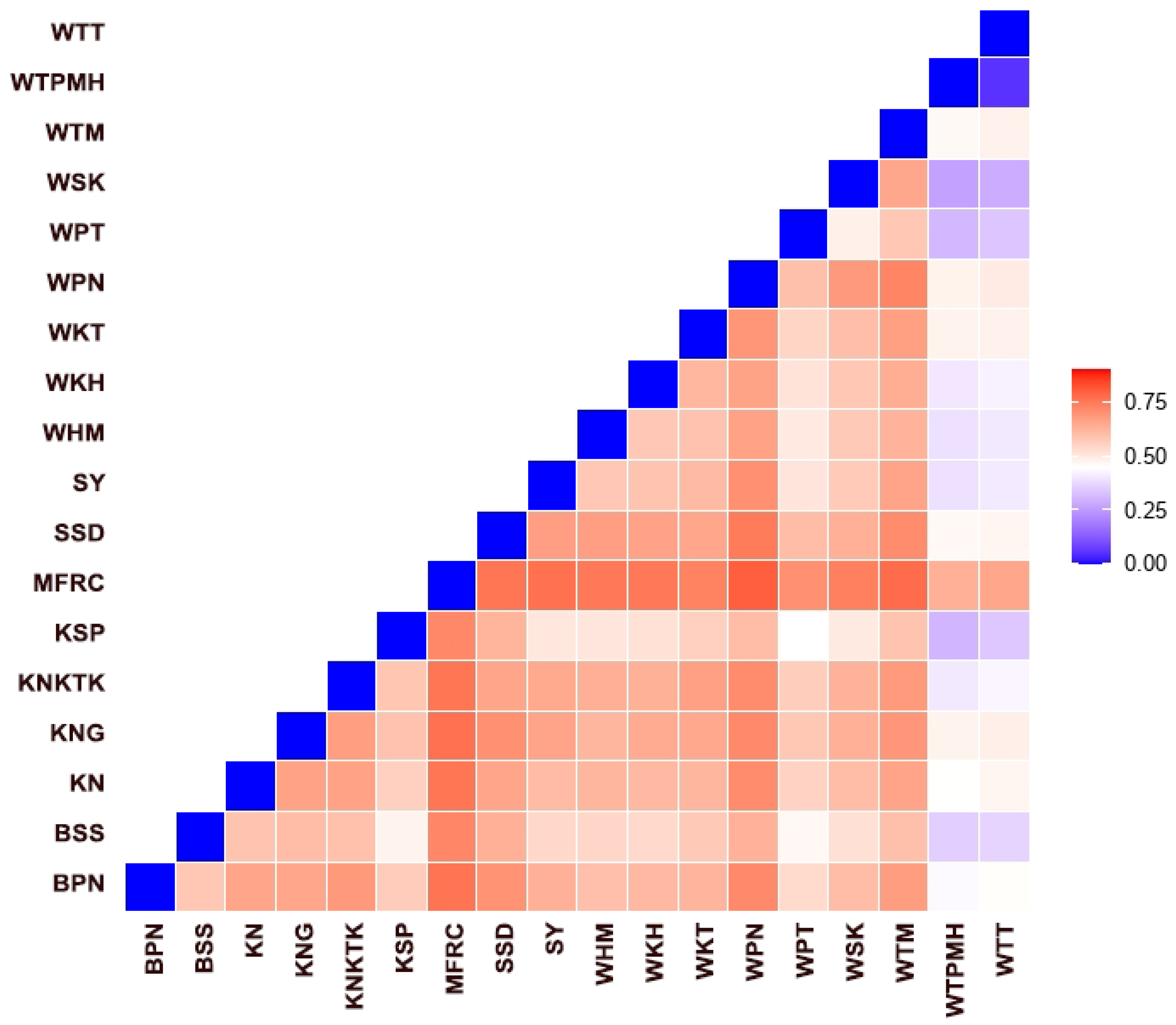
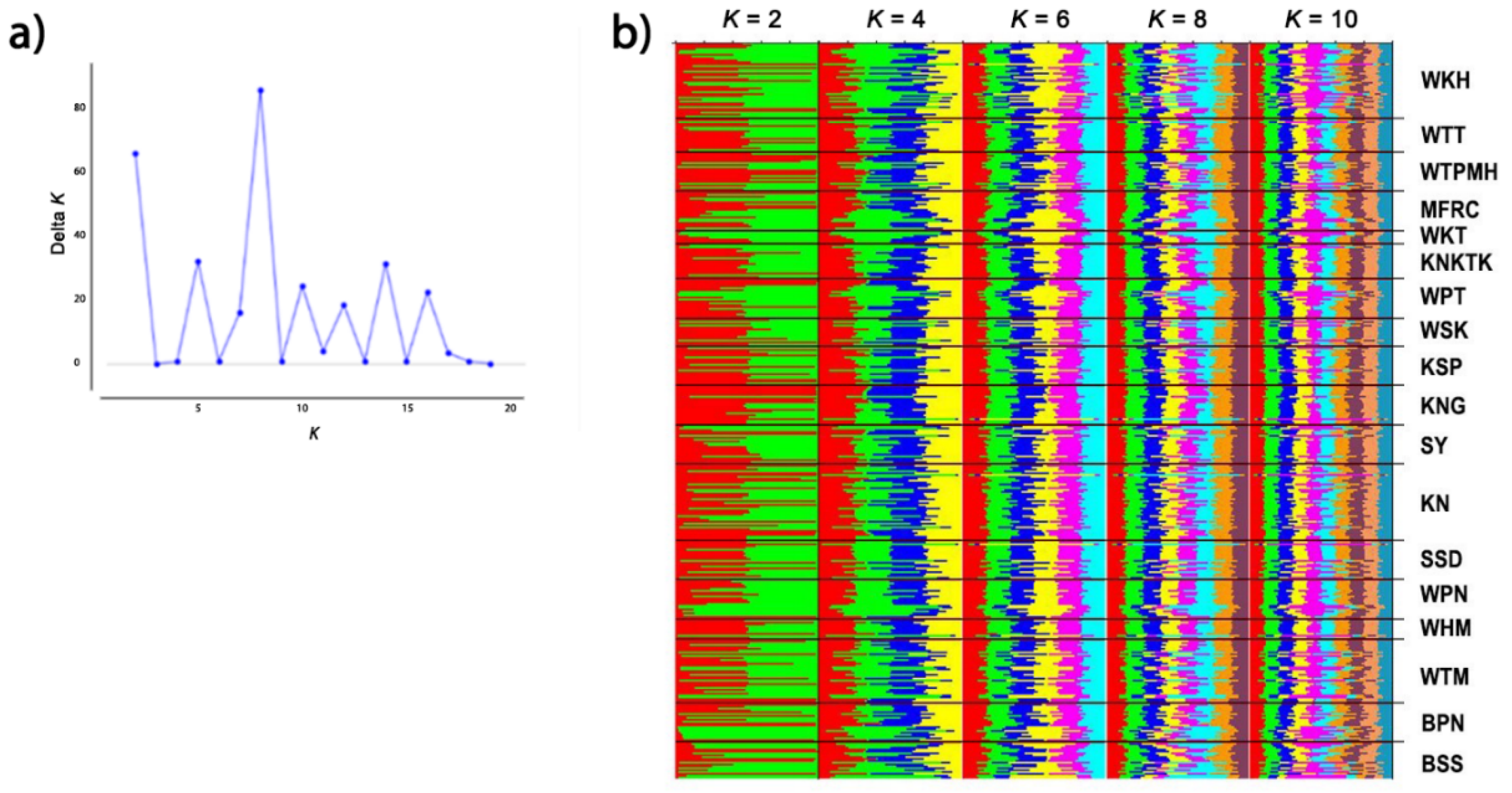
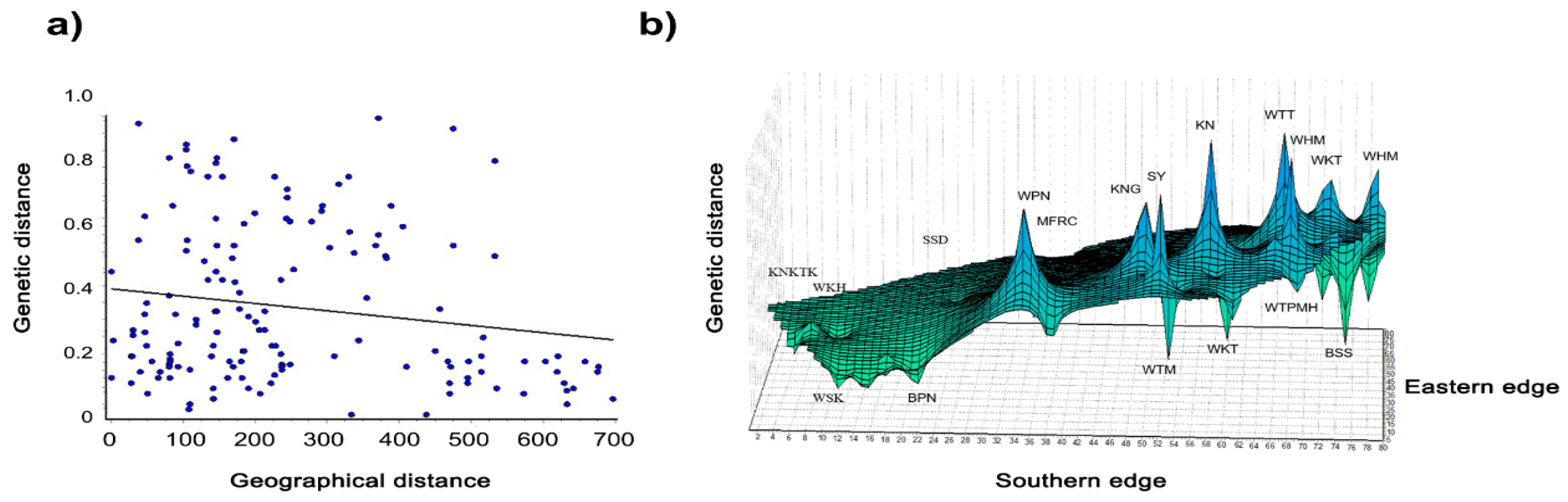
3.5. Genetic Diversity within and between Macaque Populations
4. Discussion
4.1. Turnover of Cen-satDNA Sequences with Multiple Subfamilies in Long-Tailed and Rhesus Macaque Populations
4.2. Nonrandom Cen-satDNA Sequences in Long-Tailed and Rhesus Macaque Chromosomes
4.3. Cen-satDNA in Long-Tailed and Rhesus Macaque Populations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McKinley, K.L.; Cheeseman, I.M. The molecular basis for centromere identity and function. Nat. Rev. Mol. Cell. Biol. 2016, 17, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. What makes a centromere? Exp. Cell Res. 2020, 389, 111895. [Google Scholar] [CrossRef] [PubMed]
- Miga, K.H. Centromeric satellite DNAs: Hidden sequence variation in the human population. Genes 2019, 10, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, G.; O′Neill, R.J. Centromere repeats: Hidden gems of the genome. Genes 2019, 10, 223. [Google Scholar] [CrossRef] [Green Version]
- Henikoff, S.; Ahmad, K.; Malik, H.S. The centromere paradox: Stable inheritance with rapidly evolving DNA. Science 2001, 293, 1098–1102. [Google Scholar] [CrossRef] [Green Version]
- Balzano, E.; Giunta, S. Centromeres under pressure: Evolutionary innovation in conflict with conserved function. Genes 2020, 11, 912. [Google Scholar] [CrossRef]
- Thongchum, R.; Singchat, W.; Laopichienpong, N.; Tawichasri, P.; Kraichak, E.; Prakhongcheep, O.; Sillapaprayoon, S.; Muangmai, N.; Baicharoen, S.; Suntrarachun, S.; et al. Diversity of PBI-DdeI satellite DNA in snakes correlates with rapid independent evolution and different functional roles. Sci. Rep. 2019, 9, 15459. [Google Scholar] [CrossRef] [Green Version]
- Suntronpong, A.; Singchat, W.; Kruasuwan, W.; Prakhongcheep, O.; Sillapaprayoon, S.; Muangmai, N.; Somyong, S.; Indananda, C.; Kraichak, E.; Peyachoknagul, S.; et al. Characterization of centromeric satellite DNAs (MALREP) in the Asian swamp eel (Monopterus albus) suggests the possible origin of repeats from transposable elements. Genomics 2020, 112, 3097–3107. [Google Scholar] [CrossRef]
- Ahmad, S.F.; Singchat, W.; Jehangir, M.; Suntronpong, A.; Panthum, T.; Malaivijitnond, S.; Srikulnath, K. Dark matter of primate genomes: Satellite dna repeats and their evolutionary dynamics. Cells 2020, 9, 2714. [Google Scholar] [CrossRef]
- Feliciello, I.; Picariello, O.; Chinali, G. Intra-specific variability and unusual organization of the repetitive units in a satellite DNA from Rana dalmatina: Molecular evidence of a new mechanism of DNA repair acting on satellite DNA. Gene 2006, 383, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Prakhongcheep, O.; Thapana, W.; Suntronpong, A.; Singchat, W.; Pattanatanang, K.; Phatcharakullawarawat, R.; Muangmai, N.; Peyachoknagul, S.; Matsubara, K.; Ezaz, T.; et al. Lack of satellite DNA species-specific homogenization and relationship to chromosomal rearrangements in monitor lizards (Varanidae, Squamata). BMC Evol. Biol. 2017, 17, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, U.P.; Charlebois, C.; Lawal, R.A.; Dumont, B.L. Population and subspecies diversity at mouse centromere satellites. BMC Genomics 2021, 22, 279. [Google Scholar] [CrossRef] [PubMed]
- Choo, K.H. Domain organization at the centromere and neocentromere. Dev. Cell 2001, 1, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Nakano, M.; Okamoto, Y.; Ohzeki, J.; Masumoto, H. Epigenetic assembly of centromeric chromatin at ectopic alpha-satellite sites on human chromosomes. J. Cell Sci. 2003, 116, 4021–4034. [Google Scholar] [CrossRef] [Green Version]
- Masumoto, H.; Nakano, M.; Ohzeki, J. The role of CENPB and alpha-satellite DNA: De novo assembly and epigenetic maintenance of human centromeres. Chromosome Res. 2004, 12, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Suntronpong, A.; Kugou, K.; Masumoto, H.; Srikulnath, K.; Ohshima, K.; Hirai, H.; Koga, A. CENP-B box, a nucleotide motif involved in centromere formation, occurs in a New World monkey. Biol. Lett. 2016, 12, 20150817. [Google Scholar] [CrossRef]
- Thongchum, R.; Nishihara, H.; Srikulnath, K.; Hirai, H.; Koga, A. The CENP-B box, a nucleotide motif involved in centromere formation, has multiple origins in New World monkeys. Genes. Genet. Syst. 2020, 94, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Dover, G. Molecular drive: A cohesive mode of species evolution. Nature 1982, 299, 111–117. [Google Scholar] [CrossRef]
- Plohl, M.; Mestrovic, N.; Mravinac, B. Satellite DNA evolution. Genome Dyn. 2012, 7, 126–152. [Google Scholar]
- Lower, S.S.; McGurk, M.P.; Clark, A.G.; Barbash, D.A. Satellite DNA evolution: Old ideas, new approaches. Curr. Opin. Genet. Dev. 2018, 49, 70–78. [Google Scholar] [CrossRef]
- Charlesworth, B.; Sniegowski, P.; Stephan, W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 1994, 371, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, I.; Kazakov, A.; Tumeneva, I.; Shepelev, V.; Yurov, Y. α-Satellite DNA of primates: Old and new families. Chromosoma 2001, 110, 253–266. [Google Scholar] [CrossRef]
- Chaiprasertsri, N.; Uno, Y.; Peyachoknagul, S.; Prakhongcheep, O.; Baicharoen, S.; Charernsuk, S.; Nishida, C.; Matsuda, Y.; Koga, A.; Srikulnath, K. Highly species-specific centromeric repetitive DNA sequences in lizards: Molecular cytogenetic characterization of a novel family of satellite DNA sequences isolated from the water monitor lizard (Varanus salvator macromaculatus, Platynota). J. Hered. 2013, 104, 798–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meštrović, N.; Plohl, M.; Mravinac, B.; Ugarković, Ð. Evolution of satellite DNAs from the genus Palorus—experimental evidence for the library hypothesis. Mol. Biol. Evol. 1998, 15, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Nijman, I.J.; Lenstra, J.A. Mutation and recombination in cattle satellite DNA: A feedback model for the evolution of satellite repeats. J. Mol. Evol. 2001, 52, 361–371. [Google Scholar] [CrossRef]
- Prakhongcheep, O.; Chaiprasertsri, N.; Terada, S.; Hirai, Y.; Srikulnath, K.; Hirai, H.; Koga, A. Heterochromatin blocks constituting the entire short arms of acrocentric chromosomes of Azara′s owl monkey: Formation processes inferred from chromosomal locations. DNA Res. 2013, 20, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Prakhongcheep, O.; Hirai, Y.; Hara, T.; Srikulnath, K.; Hirai, H.; Koga, A. Two types of alpha satellite DNA in distinct chromosomal locations in Azara′s owl monkey. DNA Res. 2013, 20, 235–240. [Google Scholar] [CrossRef] [Green Version]
- Koga, A.; Hirai, Y.; Terada, S.; Jahan, I.; Baicharoen, S.; Arsaithamkul, V.; Hirai, H. Evolutionary origin of higher-order repeat structure in alpha-satellite DNA of primate centromeres. DNA Res. 2014, 21, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Sujiwattanarat, P.; Thapana, W.; Srikulnath, K.; Hirai, Y.; Hirai, H.; Koga, A. Higher-order repeat structure in alpha satellite DNA occurs in New World monkeys and is not confined to hominoids. Sci. Rep. 2015, 5, 10315. [Google Scholar] [CrossRef] [Green Version]
- Cacheux, L.; Ponger, L.; Gerbault-Seureau, M.; Loll, F.; Gey, D.; Richard, F.A.; Escudé, C. The targeted sequencing of alpha satellite DNA in Cercopithecus pogonias provides new insight into the diversity and dynamics of centromeric repeats in old world monkeys. Genome Biol. Evol. 2018, 10, 1837–1851. [Google Scholar] [CrossRef]
- Sullivan, L.L.; Chew, K.; Sullivan, B.A. α satellite DNA variation and function of the human centromere. Nucleus 2017, 8, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNulty, S.M.; Sullivan, B.A. Alpha satellite DNA biology: Finding function in the recesses of the genome. Chromosome Res. 2018, 26, 115–138. [Google Scholar] [CrossRef]
- Willard, H.F.; Waye, J.S. Chromosome-specific subsets of human α satellite DNA: Analysis of sequence divergence within and between chromosomal subsets and evidence for an ancestral pentameric repeat. J. Mol. Evol. 1987, 25, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Wevrick, R.; Fisher, R.B.; Ferguson-Smith, M.A.; Lin, C.C. Human centromeric DNAs. Hum. Genet. 1997, 100, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Blumenbach. The Integrated Taxonomic Information System. The Paleobiology Database. Homotypic Synonym of Simia Troglodytes. De Generis Humani Varietate Native. 1997, p. 37. Available online: https://www.gbif.org/dataset/cbb6498e-8927-405a-916b-576d00a6289b (accessed on 1 December 2021).
- Savage, T.S.; Wyman, J. Notice of the external characters and habits of Troglodytes gorilla, a new species of orang from the Gaboon River, osteology of the same. Boston J. Nat. Hist. 1847, 5, 417–443. [Google Scholar]
- Lesson, R.P. Manuel de Mammalogie ou Histoire Naturelle des Mammifères; Nabu Press: Dublin, Ireland; London, UK, 1827; p. 32. (In French) [Google Scholar]
- Durfy, S.J.; Willard, H.F. Concerted evolution of primate alpha satellite DNA. Evidence for an ancestral sequence shared by gorilla and human X chromosome alpha satellite. J. Mol. Biol. 1990, 216, 555–566. [Google Scholar] [CrossRef]
- Rudd, M.K.; Wray, G.A.; Willard, H.F. The evolutionary dynamics of alpha-satellite. Genome Res. 2006, 16, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Cechova, M.; Harris, R.S.; Tomaszkiewicz, M.; Arbeithuber, B.; Chiaromonte, F.; Makova, K.D. High satellite repeat turnover in great apes studied with short- and long-read technologies. Mol. Biol. Evol. 2019, 36, 2415–2431. [Google Scholar] [CrossRef] [Green Version]
- Chmátal, L.; Gabriel, S.I.; Mitsainas, G.P.; Martínez-Vargas, J.; Ventura, J.; Searle, J.B.; Schultz, R.M.; Lampson, M.A. Centromere strength provides the cell biological basis for meiotic drive and karyotype evolution in mice. Curr. Biol. 2014, 24, 2295–2300. [Google Scholar] [CrossRef] [Green Version]
- Iwata-Otsubo, A.; Dawicki-McKenna, J.M.; Akera, T.; Falk, S.J.; Chmátal, L.; Yang, K.; Sullivan, B.A.; Schultz, R.M.; Lampson, M.A.; Black, B.E. Expanded satellite repeats amplify a discrete CENP-A nucleosome assembly site on chromosomes that drive in female meiosis. Curr. Biol. 2017, 27, 2365–2373. [Google Scholar] [CrossRef]
- Groves, C.P. Species Macaca mulatta. In Mammal Species of the World: A Taxonomic and Geographic Reference, 3rd ed.; Wilson, D.E., Reeder, D.M., Eds.; Johns Hopkins University Press: Baltimore, MD, USA, 2005; p. 163. [Google Scholar]
- Raffles, T.S. Descriptive catalogue of a zoological collection, made on account of the honourable east india company, in the island of sumatra and its vicinity, under the direction of sir thomas stamford raffles, lieutenant-governor of fort marlborough. Trans. Linn. Soc. Lond. 1821, 13, 246–247. [Google Scholar] [CrossRef] [Green Version]
- Roos, C.; Zinner, D. Chapter 1—Diversity and Evolutionary History of Macaques with Special Focus on Macaca mulatta and Macaca fascicularis. In The Nonhuman Primate in Nonclinical Drug Development and Safety Assessment; Bluemel, J., Korte, S., Schenck, E., Weinbauer, G., Eds.; Academic Press: Cambridge, MA, USA, 2015; pp. 3–16. [Google Scholar]
- Srikulnath, K.; Ahmad, S.F.; Panthum, T.; Malaivijitnond, S. Importance of Thai macaque bioresources for biological research and human health. J. Med. Primatol. 2022, 51, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Thierry, B. The macaques: A double-layered social organization. In Primates in Perspective, 2nd ed.; Campbell, C.J., Fuentes, A., MacKinnon, K.C., Bearder, S.K., Stumpf, R.M., Eds.; Oxford University Press: Oxford, UK, 2011; pp. 229–241. [Google Scholar]
- Phillips, K.A.; Bales, K.L.; Capitanio, J.P.; Conley, A.; Czoty, P.W.; Hart, B.A.; Hopkins, W.D.; Hu, S.L.; Miller, L.A.; Nader, M.A. Why primate models matter. Am. J. Primatol. 2014, 76, 801–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fooden, J. Tail-length variation in Macaca fascicularis and M. mulatta. Primates 1997, 38, 221–231. [Google Scholar] [CrossRef]
- Hamada, Y.; Watanabe, T.; Chatani, K.; Hayakawa, S.; Iwamoto, M. Morphometrical comparison between Indian- and Chinese-derived rhesus macaques (Macaca mulatta). Anthropol. Sci. 2005, 113, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Bunlungsup, S.; Imai, H.; Hamada, Y.; Matsudaira, K.; Malaivijitnond, S. Mitochondrial DNA and two Y chromosome genes of common long-tailed macaques (Macaca fascicularis fascicularis) throughout Thailand and vicinity. Am. J. Primatol. 2017, 79, e22596. [Google Scholar] [CrossRef]
- Sunyakumthorn, P.; Somponpun, S.J.; Im-Erbsin, R.; Anantatat, T.; Jenjaroen, K.; Dunachie, S.J.; Lombardini, E.D.; Burke, R.L.; Blacksell, S.D.; Jones, J.W.; et al. Characterization of the rhesus macaque (Macaca mulatta) scrub typhus model: Susceptibility to intradermal challenge with the human pathogen Orientia tsutsugamushi Karp. PLoS Negl. Trop. Dis. 2018, 12, e0006305. [Google Scholar] [CrossRef] [Green Version]
- Balasubramaniam, K.N.; Malaivijitnond, S.; Kemthong, T.; Meesawat, S.; Hamada, Y.; Jeamsripong, S.; Srisamran, J.; Kuldee, M.; Thaotumpitak, V.; McCowan, B.; et al. Prevalence of enterobacteriaceae in wild long-tailed macaques (Macaca fascicularis) in Thailand. Int. J. Primatol. 2021, 42, 337–341. [Google Scholar] [CrossRef]
- Phadphon, P.; Kanthaswamy, S.; Oldt, R.F.; Hamada, Y.; Malaivijitnond, S. Population structure of Macaca fascicularis aurea, and their genetic relationships with M. f. fascicularis and M. mulatta Determined by 868 RADseq-Derived Autosomal SNPs–A consideration for biomedical research. J. Med. Primatol. 2022, 51, 33–44. [Google Scholar] [CrossRef]
- Gupta, A.; Galinski, M.R.; Voit, E.O. Dynamic control balancing cell proliferation and inflammation is crucial for an effective immune response to malaria. Front. Mol. Biosci. 2022, 8, 800721. [Google Scholar] [CrossRef]
- Suchkova, I.O.; Baranova, T.V.; Klustova, M.E.; Kisljakova, T.V.; Vassiliev, V.B.; Slominskaja, N.O.; Alenina, N.V.; Patkin, E.L. Bovine satellite DNA induces heterochromatinization of host chromosomal DNA in cells of transsatellite mouse embryonal carcinoma. Tsitologiia 2004, 46, 53–61. [Google Scholar] [PubMed]
- Piras, F.M.; Nergadze, S.G.; Magnani, E.; Bertoni, L.; Attolini, C.; Khoriauli, L.; Raimondi, E.; Giulotto, E. Uncoupling of satellite DNA and centromeric function in the genus Equus. PLoS Genet. 2010, 6, e1000845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapana, W.; Sujiwattanarat, P.; Srikulnath, K.; Hirai, H.; Koga, A. Reduction in the structural instability of cloned eukaryotic tandem-repeat DNA by low-temperature culturing of host bacteria. Genet. Res. 2014, 96, e13. [Google Scholar] [CrossRef] [PubMed]
- Cacheux, L.; Ponger, L.; Gerbault-Seureau, M.; Richard, F.A.; Escudé, C. Diversity and distribution of alpha satellite DNA in the genome of an Old World monkey: Cercopithecus solatus. BMC Genomics 2016, 17, 916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunlungsup, S.; Kanthaswamy, S.; Oldt, R.F.; Smith, D.G.; Houghton, P.; Hamada, Y.; Malaivijitnond, S. Genetic analysis of samples from wild populations opens new perspectives on hybridization between long-tailed (Macaca fascicularis) and rhesus macaques (M. mulatta). Am. J. Primatol. 2017, 79, e22726. [Google Scholar] [CrossRef]
- Fooden, J. Comparative review of fascicularis-group species of Macaques (primates: Macaca). Fieldiana Zool. 2006, 2006, 1–43. [Google Scholar]
- Malaivijitnond, S.; Arsaithamkul, V.; Tanaka, H.; Pomchote, P.; Jaroenporn, S.; Suryobroto, B.; Hamada, Y. Boundary zone between northern and southern pig-tailed macaques and their morphological differences. Primates 2012, 53, 377–389. [Google Scholar] [CrossRef]
- Hamada, Y.; San, A.M.; Malaivijitnond, S. Assessment of the hybridization between rhesus (Macaca mulatta) and long-tailed macaques (M. fascicularis) based on morphological characters. Am. J. Phys. Anthropol. 2016, 159, 189–198. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Purification of nucleic acids by extraction with phenol: Chloroform. CSH Protoc. 2006, 2006, pdbprot4455. [Google Scholar]
- Singchat, W.; O′Connor, R.E.; Tawichasri, P.; Suntronpong, A.; Sillapaprayoon, S.; Suntrarachun, S.; Muangmai, N.; Baicharoen, S.; Peyachoknagul, S.; Chanhome, L.; et al. Chromosome map of the Siamese cobra: Did partial synteny of sex chromosomes in the amniote represent “a hypothetical ancestral super-sex chromosome” or random distribution? BMC Genomics 2018, 19, 939. [Google Scholar] [CrossRef]
- Sumner, A.T. A simple technique for demonstrating centromeric heterochromatin. Exp. Cell Res. 1972, 75, 304–306. [Google Scholar] [CrossRef]
- Matsuda, Y.; Chapman, V.M. Application of fluorescence in situ hybridization in genome analysis of the mouse. Electrophoresis 1995, 16, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Srikulnath, K.; Matsubara, K.; Uno, Y.; Thongpan, A.; Suputtitada, S.; Apisitwanich, S.; Matsuda, Y.; Nishida, C. Karyological characterization of the butterfly lizard (Leiolepis reevesii rubritaeniata, Agamidae, Squamata) by molecular cytogenetic approach. Cytogenet Genome Res. 2009, 125, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Novák, P.; Ávila Robledillo, L.; Koblížková, A.; Vrbová, I.; Neumann, P.; Macas, J. TAREAN: A computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 2017, 45, e111. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Macas, J. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat. Protoc. 2020, 15, 3745–3776. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P.; RepeatMasker Open-4.0. 2013–2015. Available online: http://www.repeatmasker.org (accessed on 1 December 2021).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollger, M.R.; Kerpedjiev, P.; Phillippy, A.M.; Eichler, E.E. StainedGlass: Interactive visualization of massive tandem repeat structures with identity heatmaps. Bioinformatics 2022, 38, 2049–2051. [Google Scholar] [CrossRef] [PubMed]
- Köster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2018, 34, 3600. [Google Scholar] [CrossRef] [Green Version]
- Mölder, F.; Jablonski, K.P.; Letcher, B.; Hall, M.B.; Tomkins-Tinch, C.H.; Sochat, V.; Forster, J.; Lee, S.; Twardziok, S.O.; Kanitz, A.; et al. Sustainable data analysis with Snakemake. F1000research 2021, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT-the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, 884–891. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 1 December 2021).
- R Core Team. R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria. Available online: https://www.R-project.org/ (accessed on 1 December 2021).
- Geoffroy, I.; de Blainville. Mammal Species of the World: A Taxonomic and Geographic Reference; Johns Hopkins University Press: Baltimore, MD, USA, 1834; Available online: https://eol.org/pages/323910#:~:text=Geoffroy%20Saint%2DHilaire%20%26%20de%20Blainville%201834)&text=Saimiri%20boliviensis%20is%20a%20species,the%20family%20New%20World%20monkeys (accessed on 1 December 2021).
- Linnaeus, C. Systema Naturae per Regna Tria Naturae: Secundum Classes, Ordines, Genera, Species, cum Characteribus, Differentiis, Synonymis, Locis; Ed. 12. 1., Regnum Animale. 1 & 2. Holmiae [Stockholm], Laurentii Salvii. 1766, pp. 1–532. Available online: https://www.marinespecies.org/aphia.php?p=sourcedetails&id=671 (accessed on 1 December 2021).
- Guzen, A. Studbook of Pan paniscus Schwarz, 1929. Acta. Zool. Pathol. Antverp. 1975, 61, 119–164. [Google Scholar]
- Elliot. The Integrated Taxonomic Information Systemc. Ann. Mag. Nat. Hist. 1907, 20, 195. [Google Scholar]
- Milne-Edwards, A. The Integrated Taxonomic Information Systemc. Bull. Mus. Hist. Nat. Paris 1897, 3, 157. [Google Scholar]
- Sclater. The Integrated Taxonomic Information System. Proc. Zool. Soc. Lond. 1860, 1860, 245. [Google Scholar]
- Cuvier, F. The Integrated Taxonomic Information Systemc. Ann. Mus. Hist. Nat. Paris 1807, 9, 477. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Lischer, H.E.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2012, 28, 298–299. [Google Scholar] [CrossRef] [Green Version]
- Gruber, B.; Unmack, P.J.; Berry, O.F.; Georges, A. dartr: An R package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol. Ecol. Resour. 2018, 18, 691–699. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Wright, S. Evolution and the Genetics of Populations. In Variability within and among Natural Populations; University of Chicago Press: Chicago, IL, USA, 1978; Volume 4. [Google Scholar]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Fu, Y.X.; Li, W.H. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, B.; Wittelsbürger, U.; Ramos-Onsins, S.E.; Lercher, M.J. PopGenome: An efficient Swiss army knife for population genomic analyses in R. Mol. Biol Evol. 2014, 31, 1929–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinaro, A.M.; Simon, R.; Pfeiffer, R.M. Prediction error estimation: A comparison of resampling methods. Bioinformatics 2005, 21, 3301–3307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diniz-Filho, J.A.F.; Soares, T.N.; Lima, J.S.; Dobrovolski, R.; Landeiro, V.L.; Telles, M.P.D.C.; Rangel, T.F.; Bini, L.M. Mantel test in population genetics. Genet. Mol. Biol. 2013, 36, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.P. Alleles In Space (AIS): Computer software for the joint analysis of interindividual spatial and genetic information. J. Hered. 2005, 96, 722–724. [Google Scholar] [CrossRef]
- Scott, L.M.; Janikas, M.V. Spatial Statistics in ArcGIS. In Handbook of Applied Spatial Analysis; Fischer, M., Getis, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Cawthon, L.K.A. Primate Factsheets: Pigtail macaque (Macaca nemestrina) Taxonomy, Morphology, & Ecology. 2005. Available online: https://primate.wisc.edu/primate-info-net/pin-factsheets/pin-factsheet-pig-tailed-macaque/ (accessed on 1 December 2021).
- Geoffroy, E. The Integrated Taxonomic Information System. Ann. Mus. Hist. Nat. Paris 1812, 19, 98. [Google Scholar]
- Masumoto, H.; Masukata, H.; Muro, Y.; Nozaki, N.; Okazaki, T. A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J. Cell Biol. 1989, 109, 1963–1973. [Google Scholar] [CrossRef]
- Ogilby. The Integrated Taxonomic Information Systemc. Proc. Zool. Soc. Lond. 1840, 1840, 20. [Google Scholar]
- Du, K.; Stöck, M.; Kneitz, S.; Klopp, C.; Woltering, J.M.; Adolfi, M.C.; Feron, R.; Prokopov, D.; Makunin, A.; Kichigin, I.; et al. The sterlet sturgeon genome sequence and the mechanisms of segmental rediploidization. Nat. Ecol. Evol. 2020, 4, 841–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnaeus, C. Systema Naturæ per Regna tria Naturæ, Secundum Classes, Ordines, Genera, Species, cum Characteribus, Differentiis, Syn-onymis, Locis; Tomus I. Editio decima, reformata. pp. [1–4], 1–824. Holmiæ. (Salvius). 1758; pp. 1–824. Available online: https://www.marinespecies.org/aphia.php?p=sourcedetails&id=8 (accessed on 1 December 2021).
- The Integrated Taxonomic Information System. Cat. Monkeys Lemurs Fruit Eat. Bats Brit. Mus. 1870, 133. Available online: https://www.gbif.org/species/4266938 (accessed on 1 December 2021).
- Nguyen, N.; Vincens, P.; Roest Crollius, H.; Louis, A. Genomicus 2018: Karyotype evolutionary trees and on-the-fly synteny computing. Nucleic Acids Res. 2018, 46, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Barra, V.; Fachinetti, D. The dark side of centromeres: Types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 2018, 9, 4340. [Google Scholar] [CrossRef] [Green Version]
- Deakin, J.E.; Potter, S.; O′Neill, R.; Ruiz-Herrera, A.; Cioffi, M.B.; Eldridge, M.D.; Fukui, K.; Marshall Graves, J.A.; Griffin, D.; Grutzner, F.; et al. Chromosomics: Bridging the gap between genomes and chromosomes. Genes 2019, 10, 627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valeri, M.P.; Dias, G.B.; do Espírito Santo, A.A.; Moreira, C.N.; Yonenaga-Yassuda, Y.; Sommer, I.B.; Kuhn, G.; Svartman, M. First description of a satellite DNA in Manatees′ centromeric regions. Front. Genet. 2021, 12, 694866. [Google Scholar] [CrossRef] [PubMed]
- Willard, H.F. Evolution of alpha satellite. Curr. Opin. Genet. Dev. 1991, 1, 509–514. [Google Scholar] [CrossRef]
- Hommelsheim, C.; Frantzeskakis, L.; Huang, M.; Ülker, B. PCR amplification of repetitive DNA: A limitation to genome editing technologies and many other applications. Sci. Rep. 2014, 4, 5052. [Google Scholar] [CrossRef] [Green Version]
- Rovie-Ryan, J.J.; Khan, F.A.A.; Abdullah, M.T. Evolutionary pattern of Macaca fascicularis in Southeast Asia inferred using Y-chromosomal gene. BMC Ecol. Evol. 2021, 21, 26. [Google Scholar] [CrossRef]
- Hall, S.E.; Kettler, G.; Preuss, D. Centromere satellites from Arabidopsis populations: Maintenance of conserved and variable domains. Genome Res. 2003, 13, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Schueler, M.G.; Higgins, A.W.; Rudd, M.K.; Gustashaw, K.; Willard, H.F. Genomic and genetic definition of a functional human centromere. Science 2001, 294, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Schueler, M.G.; Dunn, J.M.; Bird, C.P.; Ross, M.T.; Viggiano, L.; Rocchi, M.; Willard, H.F.; Green, E.D.; NISC Comparative Sequencing Program. Progressive proximal expansion of the primate X chromosome centromere. Proc. Natl. Acad. Sci. USA 2005, 102, 10563–10568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pike, L.M.; Carlisle, A.; Newell, C.; Hong, S.B.; Musich, P.R. Sequence and evolution of rhesus monkey alphoid DNA. J. Mol. Evol. 1986, 23, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Alves, G.; Seuánez, H.N.; Fanning, T. Alpha satellite DNA in neotropical primates (Platyrrhini). Chromosoma 1994, 103, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Terada, S.; Hirai, Y.; Hirai, H.; Koga, A. Higher-order repeat structure in alpha satellite DNA is an attribute of hominoids rather than hominids. J. Hum. Genet. 2013, 58, 752–754. [Google Scholar] [CrossRef] [Green Version]
- Choo, K.H.; Earle, E.; McQuillan, C. A homologous subfamily of satellite III DNA on human chromosomes 14 and 22. Nucleic Acids Res. 1990, 18, 5641–5648. [Google Scholar] [CrossRef] [Green Version]
- Waye, J.S.; Willard, H.F. Human beta satellite DNA: Genomic organization and sequence definition of a class of highly repetitive tandem DNA. Proc. Natl. Acad. Sci. USA 1989, 86, 6250–6254. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.K.C.; Biddle, F.G.; Rattner, J.B. The chromosomal distribution of the major and minor satellite is not conserved in the genus Mus. Chromosoma 1990, 99, 190–195. [Google Scholar] [CrossRef]
- Komissarov, A.S.; Gavrilova, E.V.; Demin, S.J.; Ishov, A.M.; Podgornaya, O.I. Tandemly repeated DNA families in the mouse genome. BMC Genomics 2011, 12, 531. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.R.; Hayden, K.E.; Willard, H.F. Organization and molecular evolution of CENP-A-associated satellite DNA families in a basal primate genome. Genome Biol. Evol. 2011, 3, 1136–1149. [Google Scholar] [CrossRef] [Green Version]
- Smurova, K.; De Wulf, P. Centromere and pericentromere transcription: Roles and regulation in sickness and in health. Front. Genet. 2018, 9, 674. [Google Scholar] [CrossRef] [Green Version]
- Ewens, W.J. Mathematical Population Genetics, 2nd ed.; Springer: New York, NY, USA, 2004. [Google Scholar]
- Malaspinas, A.S.; Malaspinas, O.; Evans, S.N.; Slatkin, M. Estimating allele age and selection coefficient from time-serial data. Genetics 2012, 192, 599–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomiento, M.; Jiang, Z.; D′Addabbo, P.; Eichler, E.E.; Rocchi, M. Evolutionary-new centromeres preferentially emerge within gene deserts. Genome Biol. 2018, 9, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rop, V.; Padeganeh, A.; Maddox, P.S. CENP-A: The key player behind centromere identity, propagation, and kinetochore assembly. Chromosoma 2012, 121, 527–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, H.S.; Henikoff, S. Adaptive evolution of Cid, a centromere-specific histone in Drosophila. Genetics 2001, 157, 1293–1298. [Google Scholar] [CrossRef]
- Talbert, P.B.; Bryson, T.D.; Henikoff, S. Adaptive evolution of centromere proteins in plants and animals. J. Biol. 2004, 3, 18. [Google Scholar] [CrossRef] [Green Version]
- Catacchio, C.; Ragone, R.; Chiatante, G.; Ventura, M. Organization and evolution of Gorilla centromeric DNA from old strategies to new approaches. Sci. Rep. 2015, 5, 14189. [Google Scholar] [CrossRef] [Green Version]
- Blumenbach, J.F. 2. Africanus. In Handbuch der Naturgeschichte [Handbook of Natural History], 5th ed.; Johann Christian Dieterich: Göttingen, Germany, 1797; p. 125. [Google Scholar]
- Alkan, C.; Cardone, M.F.; Catacchio, C.R.; Antonacci, F.; O′Brien, S.J.; Ryder, O.A.; Purgato, S.; Zoli, M.; Della Valle, G.; Eichler, E.E.; et al. Genome-wide characterization of centromeric satellites from multiple mammalian genomes. Genome Res. 2011, 21, 137–145. [Google Scholar] [CrossRef] [Green Version]
- The Integrated Taxonomic Information System. Prodr. Syst. Mamm. Avium. 1811, p. 108. Available online: https://www.gbif.org/fr/species/2436362 (accessed on 1 December 2021).
- Sena, R.S.; Heringer, P.; Valeri, M.P.; Pereira, V.S.; Kuhn, G.; Svartman, M. Identification and characterization of satellite DNAs in two-toed sloths of the genus Choloepus (Megalonychidae, Xenarthra). Sci. Rep. 2020, 10, 19202. [Google Scholar] [CrossRef]
- Dernburg, A.F.; Sedat, J.W.; Hawley, R.S. Direct evidence of a role for heterochromatin in meiotic chromosome segregation. Cell 1996, 86, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Karpen, G.H.; Le, M.H.; Le, H. Centric heterochromatin and the efficiency of achiasmate disjunction in Drosophila female meiosis. Science 1996, 273, 118–122. [Google Scholar] [CrossRef]
- Peng, J.C.; Karpen, G.H. Epigenetic regulation of heterochromatic DNA stability. Curr. Opin. Genet. Dev. 2008, 18, 204–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earle, E.; Saxena, A.; MacDonald, A.; Hudson, D.F.; Shaffer, L.G.; Saffery, R.; Cancilla, M.R.; Cutts, S.M.; Howman, E.; Choo, K.A. Poly (ADP-ribose) polymerase at active centromeres and neocentromeres at metaphase. Hum. Mol. Genet. 2000, 9, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Ohzeki, J.I.; Nakano, M.; Yoda, K.; Brinkley, W.R.; Larionov, V.; Masumoto, H. CENP-B controls centromere formation depending on the chromatin context. Cell 2007, 131, 1287–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappelletti, E.; Piras, F.M.; Badiale, C.; Bambi, M.; Santagostino, M.; Vara, C.; Masterson, T.A.; Sullivan, K.F.; Nergadze, S.G.; Ruiz-Herrera, A.; et al. CENP-A binding domains and recombination patterns in horse spermatocytes. Sci. Rep. 2019, 9, 15800. [Google Scholar] [CrossRef]
- Harrison, M.J.S. A new species of guenon (genus Cercopithecus) from Gabon. J. Zool. 1988, 215, 561–575. [Google Scholar] [CrossRef]
- Wei, K.H.; Grenier, J.K.; Barbash, D.A.; Clark, A.G. Correlated variation and population differentiation in satellite DNA abundance among lines of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2014, 111, 18793–18798. [Google Scholar] [CrossRef] [Green Version]
- Malik, H.S.; Henikoff, S. Major evolutionary transitions in centromere complexity. Cell 2009, 138, 1067–1082. [Google Scholar] [CrossRef] [Green Version]
- Charlesworth, D. Effects of inbreeding on the genetic diversity of populations. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 1051–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlesworth, B.; Charlesworth, D. The degeneration of Y chromosomes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 1563–1572. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Fuster, A.; Juan, C.; Petitpierre, E. Genome size in Tribolium flour-beetles: Inter- and intraspecific variation. Genet. Res. 1991, 58, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Bosco, G.; Campbell, P.; Leiva-Neto, J.T.; Markow, T.A. Analysis of Drosophila species genome size and satellite DNA content reveals significant differences among strains as well as between species. Genetics 2007, 177, 1277–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarmuz, M.; Glotzbach, C.D.; Bailey, K.A.; Bandyopadhyay, R.; Shaffer, L.G. The Evolution of satellite III DNA subfamilies among primates. Am. J. Hum. Genet. 2007, 80, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukvic, N.; Susca, F.; Gentile, M.; Tangari Ianniruberto, A.; Guanti, G. An unusual dicentric Y chromosome with a functional centromere with no detectable α-satellite. Hum. Genet. 1996, 97, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Rivera, H.; Vassquez, A.I.; Ayala-Madrigal, M.L.; Ramirez-Duenas, M.L.; Davalos, I.P. Alphoidless centromere of a familial unstable inverted Y chromosome. Ann. Genet. 1996, 39, 236–239. [Google Scholar]
- Tyler-Smith, C.; Gimelli, G.; Giglio, S.; Floridia, G.; Pandya, A.; Terzoli, G.; Warburton, P.E.; Earnshaw, W.C.; Zuffardi, O. Transmission of a fully functional human neocentromere through three generations. Am. J. Hum. Genet. 1999, 64, 1440–1444. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Larkin, D.M.; Cai, Q.; Asan-Zhang, Y.; Ge, R.L.; Auvil, L.; Capitanu, B.; Zhang, G.; Lewin, H.A.; Ma, J. Reference-assisted chromosome assembly. Proc. Natl. Acad. Sci. USA 2013, 110, 1785–1790. [Google Scholar] [CrossRef] [Green Version]
- Nergadze, S.G.; Piras, F.M.; Gamba, R.; Corbo, M.; Cerutti, F.; McCarter, J.G.; Cappelletti, E.; Gozzo, F.; Harman, R.M.; Antczak, D.F. Birth, evolution, and transmission of satellite-free mammalian centromeric domains. Genome Res. 2018, 28, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Ventura, M.; Antonacci, F.; Cardone, M.F.; Stanyon, R.; D′Addabbo, P.; Cellamare, A.; Sprague, L.J.; Eichler, E.E.; Archidiacono, N.; Rocchi, M. Evolutionary formation of new centromeres in macaque. Science 2007, 316, 243–246. [Google Scholar] [CrossRef] [Green Version]
- Cellamare, A.; Catacchio, C.R.; Alkan, C.; Giannuzzi, G.; Antonacci, F.; Cardone, M.F.; Della Valle, G.; Malig, M.; Rocchi, M.; Eichler, E.E.; et al. New insights into centromere organization and evolution from the white-cheeked gibbon and marmoset. Mol. Biol. Evol. 2009, 26, 1889–1900. [Google Scholar] [CrossRef] [Green Version]
- Malaivijitnond, S.; Hamada, Y. Current situation and status of long-tailed macaques (Macaca fascicularis) in Thailand. Nat. Hist. J. Chulalongkorn. Univ. 2008, 8, 185–204. [Google Scholar]
- Zhao, D.; He, H.S.; Wang, W.J.; Wang, L.; Du, H.; Liu, K.; Zong, S. Predicting wetland distribution changes under climate change and human activities in a mid- and high-latitude region. Sustainability 2018, 10, 863. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.M.; Caldas, I.; Cristescu, M.E.; Clark, A.G. Selection constrains high rates of tandem repetitive DNA mutation in Daphnia pulex. Genetics 2017, 207, 697–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Lengths (bp) | GC Content | Motif Sequences |
|---|---|---|
| 170–174 | 36.8–41.5%, (average of 39.5%) | Motif 1: CTCACAGAGTTAC Motif 2: CTTTCTGAGAAACT |
| Population | Tajima’s D Statistic | Fu and Li’s D Test | Fu and Li’s F Test |
|---|---|---|---|
| BPN | 1.34 ns | 0.343 ns | 0.872 ns |
| BSS | 2.69 ns | 0.450 ns | 1.547 ns |
| KN | 1.17 ns | −0.603 ns | 0.200 ns |
| KNG | 3.52 ns | −0.842 ns | 0.918 ns |
| KNKTK | 2.72 ns | 0.085 ns | 1.218 ns |
| KSP | 1.15 ns | −0.397 ns | 0.284 ns |
| MFRC | −1.01 ns | 0.398 ns | 0.519 ns |
| SSD | 3.87 ns | -0.549 ns | 1.221 ns |
| SY | 2.36 ns | 0.885 ns | 1.580 ns |
| WHM | 1.57 ns | 0.235 ns | 0.948 ns |
| WKH | 0.82 ns | −0.289 ns | 0.228 ns |
| WKT | 4.04 ns | 0.214 ns | 1.468 ns |
| WPN | N/A | N/A | N/A |
| WPT | 1.98 ns | 0.113 ns | 0.942 ns |
| WSK | 0.88 ns | −0.285 ns | 0.284 ns |
| WTM | 1.05 ns | −0.153 ns | 0.494 ns |
| WTPMH | 3.03 ns | 0.350 ns | 1.629 ns |
| WTT | 1.45 ns | −0.024 ns | 0.672 ns |
| Population | N | Na | Ne | I | Ho | He | uHe | F | |
|---|---|---|---|---|---|---|---|---|---|
| BPN | Mean | 17 | 0.836 | 0.778 | 0.128 | 0.149 | 0.088 | 0.111 | −0.597 |
| S.E. | 0.067 | 0.061 | 0.022 | 0.028 | 0.016 | 0.021 | 0.031 | ||
| BSS | Mean | 40 | 1.008 | 0.921 | 0.197 | 0.198 | 0.136 | 0.170 | −0.383 |
| S.E. | 0.072 | 0.065 | 0.026 | 0.031 | 0.018 | 0.024 | 0.058 | ||
| KN | Mean | 18 | 0.820 | 0.714 | 0.134 | 0.139 | 0.089 | 0.105 | −0.445 |
| S.E. | 0.072 | 0.061 | 0.021 | 0.025 | 0.015 | 0.018 | 0.032 | ||
| KNG | Mean | 19 | 0.758 | 0.691 | 0.120 | 0.133 | 0.081 | 0.099 | −0.535 |
| S.E. | 0.069 | 0.061 | 0.021 | 0.026 | 0.015 | 0.019 | 0.031 | ||
| KNKTK | Mean | 20 | 0.875 | 0.847 | 0.162 | 0.213 | 0.115 | 0.162 | −0.801 |
| S.E. | 0.069 | 0.067 | 0.025 | 0.034 | 0.018 | 0.027 | 0.026 | ||
| KSP | Mean | 21 | 1.063 | 0.915 | 0.188 | 0.190 | 0.125 | 0.152 | −0.417 |
| S.E. | 0.072 | 0.059 | 0.024 | 0.027 | 0.016 | 0.021 | 0.028 | ||
| MFRC | Mean | 10 | 0.523 | 0.512 | 0.126 | 0.172 | 0.090 | 0.122 | −0.891 |
| S.E. | 0.070 | 0.068 | 0.023 | 0.033 | 0.017 | 0.023 | 0.021 | ||
| SSD | Mean | 20 | 0.852 | 0.788 | 0.174 | 0.217 | 0.121 | 0.189 | −0.693 |
| S.E. | 0.074 | 0.068 | 0.025 | 0.034 | 0.018 | 0.030 | 0.033 | ||
| SY | Mean | 14 | 0.883 | 0.804 | 0.123 | 0.137 | 0.083 | 0.106 | −0.521 |
| S.E. | 0.066 | 0.058 | 0.021 | 0.027 | 0.015 | 0.020 | 0.033 | ||
| WHM | Mean | 20 | 0.914 | 0.824 | 0.150 | 0.138 | 0.102 | 0.130 | −0.293 |
| S.E. | 0.070 | 0.062 | 0.023 | 0.027 | 0.016 | 0.022 | 0.057 | ||
| WKH | Mean | 19 | 0.875 | 0.779 | 0.132 | 0.139 | 0.088 | 0.109 | −0.459 |
| S.E. | 0.069 | 0.059 | 0.021 | 0.026 | 0.015 | 0.020 | 0.030 | ||
| WKT | Mean | 21 | 0.875 | 0.780 | 0.177 | 0.197 | 0.121 | 0.157 | −0.505 |
| S.E. | 0.076 | 0.067 | 0.024 | 0.031 | 0.017 | 0.024 | 0.045 | ||
| WPN | Mean | 6 | 0.727 | 0.715 | 0.110 | 0.140 | 0.078 | 0.125 | −0.767 |
| S.E. | 0.064 | 0.063 | 0.022 | 0.030 | 0.016 | 0.026 | 0.042 | ||
| WPT | Mean | 40 | 1.094 | 0.978 | 0.214 | 0.216 | 0.146 | 0.202 | −0.377 |
| S.E. | 0.072 | 0.064 | 0.026 | 0.032 | 0.018 | 0.028 | 0.056 | ||
| WSK | Mean | 20 | 1.070 | 1.024 | 0.183 | 0.228 | 0.128 | 0.188 | −0.720 |
| S.E. | 0.063 | 0.059 | 0.026 | 0.034 | 0.018 | 0.029 | 0.028 | ||
| WTM | Mean | 20 | 0.766 | 0.729 | 0.125 | 0.160 | 0.088 | 0.110 | −0.732 |
| S.E. | 0.068 | 0.064 | 0.023 | 0.031 | 0.016 | 0.022 | 0.031 | ||
| WTPMH | Mean | 33 | 1.695 | 1.428 | 0.359 | 0.382 | 0.241 | 0.278 | −0.420 |
| S.E. | 0.041 | 0.037 | 0.025 | 0.035 | 0.018 | 0.023 | 0.045 | ||
| WTT | Mean | 20 | 1.648 | 1.345 | 0.325 | 0.323 | 0.214 | 0.240 | −0.373 |
| S.E. | 0.045 | 0.036 | 0.024 | 0.030 | 0.017 | 0.020 | 0.031 | ||
| Total | Mean | 377 | 0.960 | 0.865 | 0.174 | 0.193 | 0.119 | 0.153 | −0.510 |
| S.E. | 0.017 | 0.015 | 0.006 | 0.007 | 0.004 | 0.006 | 0.010 |
| Source | df | Sum of Squares | % Variation | F-Statistics |
|---|---|---|---|---|
| among populations | 17 | 821.495 | 4% | FST = 0.038 * |
| among individuals | 359 | 8864.168 | 67% | FIS = 0.701 * |
| within individuals | 377 | 1634.500 | 29% | FIT = 0.713 * |
| total | 753 | 11320.163 | 100% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singchat, W.; Ahmad, S.F.; Jaisamut, K.; Panthum, T.; Ariyaraphong, N.; Kraichak, E.; Muangmai, N.; Duengkae, P.; Payungporn, S.; Malaivijitnond, S.; et al. Population Scale Analysis of Centromeric Satellite DNA Reveals Highly Dynamic Evolutionary Patterns and Genomic Organization in Long-Tailed and Rhesus Macaques. Cells 2022, 11, 1953. https://doi.org/10.3390/cells11121953
Singchat W, Ahmad SF, Jaisamut K, Panthum T, Ariyaraphong N, Kraichak E, Muangmai N, Duengkae P, Payungporn S, Malaivijitnond S, et al. Population Scale Analysis of Centromeric Satellite DNA Reveals Highly Dynamic Evolutionary Patterns and Genomic Organization in Long-Tailed and Rhesus Macaques. Cells. 2022; 11(12):1953. https://doi.org/10.3390/cells11121953
Chicago/Turabian StyleSingchat, Worapong, Syed Farhan Ahmad, Kitipong Jaisamut, Thitipong Panthum, Nattakan Ariyaraphong, Ekaphan Kraichak, Narongrit Muangmai, Prateep Duengkae, Sunchai Payungporn, Suchinda Malaivijitnond, and et al. 2022. "Population Scale Analysis of Centromeric Satellite DNA Reveals Highly Dynamic Evolutionary Patterns and Genomic Organization in Long-Tailed and Rhesus Macaques" Cells 11, no. 12: 1953. https://doi.org/10.3390/cells11121953
APA StyleSingchat, W., Ahmad, S. F., Jaisamut, K., Panthum, T., Ariyaraphong, N., Kraichak, E., Muangmai, N., Duengkae, P., Payungporn, S., Malaivijitnond, S., & Srikulnath, K. (2022). Population Scale Analysis of Centromeric Satellite DNA Reveals Highly Dynamic Evolutionary Patterns and Genomic Organization in Long-Tailed and Rhesus Macaques. Cells, 11(12), 1953. https://doi.org/10.3390/cells11121953