
Ocrelizumab in Patients with Active Primary Progressive Multiple Sclerosis: Clinical Outcomes and Immune Markers of Treatment Response

 ,
,  , ,
, ,  , ,
, ,  and
and
Abstract
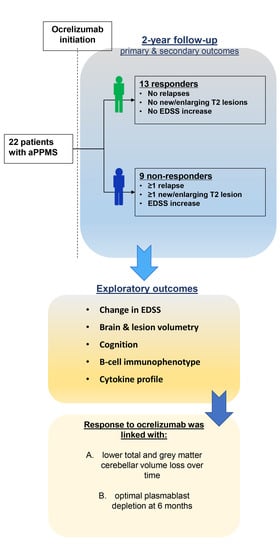
:
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Study Procedures
2.3. B-Cell Immunophenotype
2.4. Serum Cytokine Profile
2.5. Clinical, MRI and Laboratory Outcomes
2.5.1. Primary Outcome
2.5.2. Secondary Outcomes
- The proportion of patients with optimal response at 12 months. Optimal response is defined as absence of relapses, absence of new/enlarged T2 lesions on the brain and cervical MRI and absence of CDA, defined as 1 point of EDSS increase (0.5 point if baseline EDSS ≥ 5.5, confirmed after 6 months from the previous last evaluation) at 12 and at 24 months.
- The mean time to optimal response for patients who exhibited optimal response at 24 months. As per study design, patients were evaluated every 6 months.
- The proportion of patients with CDA defined as 1 point of EDSS increase (0.5 point if baseline EDSS ≥ 5.5), at 12 and at 24 months, relative to the last previous evaluation time point.
- The proportion of patients with MRI activity at 12 and 24 months (defined as the presence of new/enlarged T2 lesions with respect to previous brain MRI.
2.5.3. Exploratory Outcomes
- Mean change in EDSS from baseline at 12- and 24-month point estimates (for all patients and for responders vs. non-responders at 24 months).
- Mean change in total number of new and/or enlarged T2 lesions at 12- and 24-month point estimates (for all patients and for responders vs. non-responders at 24 months).
- Mean change in cognitive performance scales’ scores, namely, BICAMS (including SDMT, GVLT, BVMT-R), for the assessment of processing speed, verbal and visuospatial memory, respectively, from baseline at 12- and 24-month point estimates (for all patients and for responders vs. non-responders at 24 months).
- Mean change in volumetric parameters in cm3 and in percentage of total brain volume from baseline at 12- and 24-month point estimates (for all patients and for responders vs. non-responders at 24 months). Example measurements include, but are not restricted to, overall, cerebrum and cerebellar white and gray matter, brainstem and areas of subcortical gray matter.
- Mean change in lesion (white matter) analysis parameters, namely, lesion count, lesion volume in cm3 and normalized lesion volume, as well as overall lesion burden, from baseline at 12- and 24-month point estimates (for all patients and for responders vs. non-responders at 24 months).
- Possible association between mean EDSS score and/or mean cognitive scores and MRI volumetry and/or lesion analysis at 12- and 24-month point estimates.
- Mean change in B-cell subtypes from baseline at 6-, 12-, 18- and 24-month point estimates (for all patients and for responders vs. non-responders at 24 months).
- Mean change in serum cytokine profile from baseline at 6- and 12-month point estimates (for all patients and for responders vs. non-responders at 24 months).
2.6. Statistical Analysis
3. Results
3.1. Baseline Characteristics, Safety and Treatment Withdrawal
3.2. Primary and Secondary Outcomes
3.3. Exploratory Outcomes
- Cognitive function
- MRI volumetry—association with cognitive performance
- Immune cell phenotype
- Serum cytokine analysis
4. Discussion
Limitations and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ben-Nun, A.; Wekerle, H.; Cohen, I.R. The rapid isolation of clonable antigen-specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur. J. Immunol. 1981, 11, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Probstel, A.K.; Hauser, S.L. Multiple Sclerosis: B Cells Take Center Stage. J. Neuro-Ophthalmol. 2018, 38, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Obermeier, B.; Mentele, R.; Malotka, J.; Kellermann, J.; Kumpfel, T.; Wekerle, H.; Lottspeich, F.; Hohlfeld, R.; Dornmair, K. Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat. Med. 2008, 14, 688–693. [Google Scholar] [CrossRef]
- Stern, J.N.; Yaari, G.; Vander Heiden, J.A.; Church, G.; Donahue, W.F.; Hintzen, R.Q.; Huttner, A.J.; Laman, J.D.; Nagra, R.M.; Nylander, A.; et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci. Transl. Med. 2014, 6, 248ra107. [Google Scholar] [CrossRef] [Green Version]
- Krumbholz, M.; Derfuss, T.; Hohlfeld, R.; Meinl, E. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat. Rev. Neurol. 2012, 8, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Fawaz, L.; Fan, B.; Darlington, P.J.; Rieger, A.; Ghorayeb, C.; Calabresi, P.A.; Waubant, E.; Hauser, S.L.; Zhang, J.; et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann. Neurol. 2010, 67, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007, 130, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Lossius, A.; Johansen, J.N.; Vartdal, F.; Robins, H.; Jurate Saltyte, B.; Holmoy, T.; Olweus, J. High-throughput sequencing of TCR repertoires in multiple sclerosis reveals intrathecal enrichment of EBV-reactive CD8+ T cells. Eur. J. Immunol. 2014, 44, 3439–3452. [Google Scholar] [CrossRef] [PubMed]
- Von Budingen, H.C.; Kuo, T.C.; Sirota, M.; van Belle, C.J.; Apeltsin, L.; Glanville, J.; Cree, B.A.; Gourraud, P.A.; Schwartzburg, A.; Huerta, G.; et al. B cell exchange across the blood-brain barrier in multiple sclerosis. J. Clin. Investig. 2012, 122, 4533–4543. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Polychroniadou, E.; Bakirtzis, C.; Langdon, D.; Lagoudaki, R.; Kesidou, E.; Theotokis, P.; Tsalikakis, D.; Poulatsidou, K.; Kyriazis, O.; Boziki, M.; et al. Validation of the Brief International Cognitive Assessment for Multiple Sclerosis (BICAMS) in Greek population with multiple sclerosis. Mult. Scler. Relat. Disord. 2016, 9, 68–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakirtzis, C.; Nikolaidis, I.; Boziki, M.K.; Artemiadis, A.; Andravizou, A.; Messinis, L.; Ioannidis, P.; Grigoriadis, N. Cognitive Fatigability is Independent of Subjective Cognitive Fatigue and Mood in Multiple Sclerosis. Cogn. Behav. Neurol. 2020, 33, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Benedict, R.H.B.; Amato, M.P.; DeLuca, J.; Geurts, J.J.G. Cognitive impairment in multiple sclerosis: Clinical management, MRI, and therapeutic avenues. Lancet Neurol. 2020, 19, 860–871. [Google Scholar] [CrossRef]
- Battaglia, S.; Serio, G.; Scarpazza, C.; D’Ausilio, A.; Borgomaneri, S. Frozen in (e)motion: How reactive motor inhibition is influenced by the emotional content of stimuli in healthy and psychiatric populations. Behav. Res. Ther. 2021, 146, 103963. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Fabius, J.H.; Moravkova, K.; Fracasso, A.; Borgomaneri, S. The Neurobiological Correlates of Gaze Perception in Healthy Individuals and Neurologic Patients. Biomedicines 2022, 10, 627. [Google Scholar] [CrossRef]
- Battaglia, S.; Thayer, J.F. Functional interplay between central and autonomic nervous systems in human fear conditioning. Trends Neurosci. 2022. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Orsolini, S.; Borgomaneri, S.; Barbieri, R.; Diciotti, S.; di Pellegrino, G. Characterizing cardiac autonomic dynamics of fear learning in humans. Psychophysiology 2022, e14122. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Watanabe, E.; Fukuchi, M. Psychiatric Neural Networks and Precision Therapeutics by Machine Learning. Biomedicines 2021, 9, 403. [Google Scholar] [CrossRef]
- Eshaghi, A.; Young, A.L.; Wijeratne, P.A.; Prados, F.; Arnold, D.L.; Narayanan, S.; Guttmann, C.R.G.; Barkhof, F.; Alexander, D.C.; Thompson, A.J.; et al. Author Correction: Identifying multiple sclerosis subtypes using unsupervised machine learning and MRI data. Nat. Commun. 2021, 12, 3169. [Google Scholar] [CrossRef]
- Zarzuelo-Romero, M.J.; Perez-Ramirez, C.; Cura, Y.; Carrasco-Campos, M.I.; Marangoni-Iglecias, L.M.; Ramirez-Tortosa, M.C.; Jimenez-Morales, A. Influence of Genetic Polymorphisms on Clinical Outcomes of Glatiramer Acetate in Multiple Sclerosis Patients. J. Pers. Med. 2021, 11, 1032. [Google Scholar] [CrossRef] [PubMed]
- Bivona, G.; Gambino, C.M.; Lo Sasso, B.; Scazzone, C.; Giglio, R.V.; Agnello, L.; Ciaccio, M. Serum Vitamin D as a Biomarker in Autoimmune, Psychiatric and Neurodegenerative Diseases. Diagnostics 2022, 12, 130. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Vecsei, L. Monitoring the Redox Status in Multiple Sclerosis. Biomedicines 2020, 8, 406. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Vecsei, L. Monitoring the kynurenine system: Concentrations, ratios or what else? Adv. Clin. Exp. Med. Off. Organ Wroc. Med. Univ. 2021, 30, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Toldi, J.; Vecsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Avan, R.; Sahebnasagh, A.; Hashemi, J.; Monajati, M.; Faramarzi, F.; Henney, N.C.; Montecucco, F.; Jamialahmadi, T.; Sahebkar, A. Update on Statin Treatment in Patients with Neuropsychiatric Disorders. Life 2021, 11, 1365. [Google Scholar] [CrossRef] [PubMed]
- Torok, N.; Tanaka, M.; Vecsei, L. Searching for Peripheral Biomarkers in Neurodegenerative Diseases: The Tryptophan-Kynurenine Metabolic Pathway. Int. J. Mol. Sci. 2020, 21, 9338. [Google Scholar] [CrossRef] [PubMed]
- Chiaravalloti, N.D.; Amato, M.P.; Brichetto, G.; Chataway, J.; Dalgas, U.; DeLuca, J.; Meza, C.; Moore, N.B.; Feys, P.; Filippi, M.; et al. The emotional impact of the COVID-19 pandemic on individuals with progressive multiple sclerosis. J. Neurol 2021, 268, 1598–1607. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palanichamy, A.; Jahn, S.; Nickles, D.; Derstine, M.; Abounasr, A.; Hauser, S.L.; Baranzini, S.E.; Leppert, D.; von Budingen, H.C. Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J. Immunol. 2014, 193, 580–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Velasco, J.I.; Kuhle, J.; Monreal, E.; Meca-Lallana, V.; Meca-Lallana, J.; Izquierdo, G.; Gascon-Gimenez, F.; Sainz de la Maza, S.; Walo-Delgado, P.E.; Maceski, A.; et al. Effect of Ocrelizumab in Blood Leukocytes of Patients With Primary Progressive MS. Neurol. (R) Neuroimmunol. Neuroinflamm. 2021, 8, e940. [Google Scholar] [CrossRef]
- Sabatino, J.J., Jr.; Wilson, M.R.; Calabresi, P.A.; Hauser, S.L.; Schneck, J.P.; Zamvil, S.S. Anti-CD20 therapy depletes activated myelin-specific CD8(+) T cells in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 25800–25807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstock-Guttman, B.; Bermel, R.; Cutter, G.; Freedman, M.S.; Leist, T.P.; Ma, X.; Kile, D.; Musch, B.; Reder, A.T.; Wolinsky, J.S. Ocrelizumab treatment for relapsing-remitting multiple sclerosis after a suboptimal response to previous disease-modifying therapy: A nonrandomized controlled trial. Mult. Scler. J. 2022, 28, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Pellkofer, H.L.; Krumbholz, M.; Berthele, A.; Hemmer, B.; Gerdes, L.A.; Havla, J.; Bittner, R.; Canis, M.; Meinl, E.; Hohlfeld, R.; et al. Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology 2011, 76, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Huh, S.Y.; Lee, S.J.; Joung, A.; Kim, H.J. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol 2013, 70, 1110–1117. [Google Scholar] [CrossRef] [Green Version]
- Stuve, O.; Leussink, V.I.; Frohlich, R.; Hemmer, B.; Hartung, H.P.; Menge, T.; Kieseier, B.C. Long-term B-lymphocyte depletion with rituximab in patients with relapsing-remitting multiple sclerosis. Arch. Neurol. 2009, 66, 259–261. [Google Scholar] [CrossRef] [Green Version]
- Ellrichmann, G.; Bolz, J.; Peschke, M.; Duscha, A.; Hellwig, K.; Lee, D.H.; Linker, R.A.; Gold, R.; Haghikia, A. Peripheral CD19(+) B-cell counts and infusion intervals as a surrogate for long-term B-cell depleting therapy in multiple sclerosis and neuromyelitis optica/neuromyelitis optica spectrum disorders. J. Neurol. 2019, 266, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Evertsson, B.; Hoyt, T.; Christensen, A.; Nimer, F.A.; Foley, J.; Piehl, F. A comparative study of tolerability and effects on immunoglobulin levels and CD19 cell counts with ocrelizumab vs low dose of rituximab in multiple sclerosis. Mult. Scler. J. Exp. Transl. Clin. 2020, 6, 2055217320964505. [Google Scholar] [CrossRef]
- Nissimov, N.; Hajiyeva, Z.; Torke, S.; Grondey, K.; Bruck, W.; Hausser-Kinzel, S.; Weber, M.S. B cells reappear less mature and more activated after their anti-CD20-mediated depletion in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2020, 117, 25690–25699. [Google Scholar] [CrossRef] [PubMed]
- Kurtzke, J.F. Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS). Neurology 1983, 33, 1444–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz Soldan, M.M.; Novotna, M.; Abou Zeid, N.; Kale, N.; Tutuncu, M.; Crusan, D.J.; Atkinson, E.J.; Siva, A.; Keegan, B.M.; Pirko, I.; et al. Relapses and disability accumulation in progressive multiple sclerosis. Neurology 2015, 84, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedict, R.H.; Amato, M.P.; Boringa, J.; Brochet, B.; Foley, F.; Fredrikson, S.; Hamalainen, P.; Hartung, H.; Krupp, L.; Penner, I.; et al. Brief International Cognitive Assessment for MS (BICAMS): International standards for validation. BMC Neurol. 2012, 12, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, C.; Rigueiro-Neves, M.; Miranda, T.; Alegria, P.; Vale, J.; Passos, A.M.; Langdon, D.; Sa, M.J. Validation of the brief international cognitive assessment for multiple sclerosis (BICAMS) in the Portuguese population with multiple sclerosis. BMC Neurol. 2018, 18, 172. [Google Scholar] [CrossRef] [PubMed]
- Smith, A. Symbol Digit Modalities Test (SDMT). In Manual (Revised); Western Psychological Services: Los Angeles, CA, USA, 1982. [Google Scholar]
- Vlahou, C.H.; Kosmidis, M.H.; Dardagani, A.; Tsotsi, S.; Giannakou, M.; Giazkoulidou, A.; Zervoudakis, E.; Pontikakis, N. Development of the Greek Verbal Learning Test: Reliability, construct validity, and normative standards. Arch. Clin. Neuropsychol. 2013, 28, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Strober, L.; DeLuca, J.; Benedict, R.H.; Jacobs, A.; Cohen, J.A.; Chiaravalloti, N.; Hudson, L.D.; Rudick, R.A.; LaRocca, N.G.; Multiple Sclerosis Outcome Assessments, C. Symbol Digit Modalities Test: A valid clinical trial endpoint for measuring cognition in multiple sclerosis. Mult. Scler. 2019, 25, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Benedict, R.H.B. Brief. Visuospatial Memory Test–Revised: Professional Manual; Psychological Assessment Resources, Inc.: Odessa, FL, USA, 1997. [Google Scholar]
- Bakalidou, D.; Voumvourakis, K.; Tsourti, Z.; Papageorgiou, E.; Poulios, A.; Giannopoulos, S. Validity and reliability of the Greek version of the Modified Fatigue Impact Scale in multiple sclerosis patients. Int. J. Rehabil. Res. 2014, 37, 271–276. [Google Scholar] [CrossRef]
- Beckerman, H.; Eijssen, I.C.; van Meeteren, J.; Verhulsdonck, M.C.; de Groot, V. Fatigue Profiles in Patients with Multiple Sclerosis are Based on Severity of Fatigue and not on Dimensions of Fatigue. Sci. Rep. 2020, 10, 4167. [Google Scholar] [CrossRef] [PubMed]
- Wattjes, M.P.; Ciccarelli, O.; Reich, D.S.; Banwell, B.; de Stefano, N.; Enzinger, C.; Fazekas, F.; Filippi, M.; Frederiksen, J.; Gasperini, C.; et al. 2021 MAGNIMS-CMSC-NAIMS consensus recommendations on the use of MRI in patients with multiple sclerosis. Lancet Neurol. 2021, 20, 653–670. [Google Scholar] [CrossRef]
- Manjon, J.V.; Coupe, P. volBrain: An Online MRI Brain Volumetry System. Front. Neuroinform. 2016, 10, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tustison, N.J.; Cook, P.A.; Klein, A.; Song, G.; Das, S.R.; Duda, J.T.; Kandel, B.M.; van Strien, N.; Stone, J.R.; Gee, J.C.; et al. Large-scale evaluation of ANTs and FreeSurfer cortical thickness measurements. NeuroImage 2014, 99, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Manjon, J.V.; Tohka, J.; Robles, M. Improved estimates of partial volume coefficients from noisy brain MRI using spatial context. NeuroImage 2010, 53, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Burggraaff, J.; Liu, Y.; Prieto, J.C.; Simoes, J.; de Sitter, A.; Ruggieri, S.; Brouwer, I.; Lissenberg-Witte, B.I.; Rocca, M.A.; Valsasina, P.; et al. Manual and automated tissue segmentation confirm the impact of thalamus atrophy on cognition in multiple sclerosis: A multicenter study. NeuroImage Clin. 2021, 29, 102549. [Google Scholar] [CrossRef] [PubMed]
- Dass, S.; Rawstron, A.C.; Vital, E.M.; Henshaw, K.; McGonagle, D.; Emery, P. Highly sensitive B cell analysis predicts response to rituximab therapy in rheumatoid arthritis. Arthritis Rheum. 2008, 58, 2993–2999. [Google Scholar] [CrossRef] [PubMed]
- Warnatz, K.; Schlesier, M. Flowcytometric phenotyping of common variable immunodeficiency. Cytom. Part B Clin. Cytom. 2008, 74, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Malekzadeh, A.; Twaalfhoven, H.; Wijnstok, N.J.; Killestein, J.; Blankenstein, M.A.; Teunissen, C.E. Comparison of multiplex platforms for cytokine assessments and their potential use for biomarker profiling in multiple sclerosis. Cytokine 2017, 91, 145–152. [Google Scholar] [CrossRef]
- Lang, J.D.; Olmes, D.G.; Proske, M.; Hagge, M.; Dogan Onugoren, M.; Rothhammer, V.; Schwab, S.; Hamer, H.M. Pre- and Postictal Changes in the Innate Immune System: Cause or Effect? Eur. Neurol. 2021, 84, 380–388. [Google Scholar] [CrossRef]
- Kappos, L.; Wolinsky, J.S.; Giovannoni, G.; Arnold, D.L.; Wang, Q.; Bernasconi, C.; Model, F.; Koendgen, H.; Manfrini, M.; Belachew, S.; et al. Contribution of Relapse-Independent Progression vs Relapse-Associated Worsening to Overall Confirmed Disability Accumulation in Typical Relapsing Multiple Sclerosis in a Pooled Analysis of 2 Randomized Clinical Trials. JAMA Neurol. 2020, 77, 1132–1140. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohlfeld, R.; Meinl, E. Ocrelizumab in multiple sclerosis: Markers and mechanisms. Lancet Neurol. 2017, 16, 259–261. [Google Scholar] [CrossRef]
- Li, R.; Rezk, A.; Miyazaki, Y.; Hilgenberg, E.; Touil, H.; Shen, P.; Moore, C.S.; Michel, L.; Althekair, F.; Rajasekharan, S.; et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci. Transl. Med. 2015, 7, 310ra166. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.; Assemlal, H.; Traboulsee, A.; Kolind, S.; Sprenger, T.; Bar-Or, A.; Wolinsky, J.; Kappos, L.; Inglese, M.; Schneble, H.; et al. Effect of Ocrelizumab on Cerebellar Atrophy in RMS and PPMS: Results from OPERA I/OPERA II and ORATORIO (OPR-085). Eur. J. Neurol. 2021, 28, 120. [Google Scholar]
- Valentino, P.; Cerasa, A.; Chiriaco, C.; Nistico, R.; Pirritano, D.; Gioia, M.; Lanza, P.; Canino, M.; Del Giudice, F.; Gallo, O.; et al. Cognitive deficits in multiple sclerosis patients with cerebellar symptoms. Mult. Scler. 2009, 15, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Damasceno, A.; Damasceno, B.P.; Cendes, F. The clinical impact of cerebellar grey matter pathology in multiple sclerosis. PLoS ONE 2014, 9, e96193. [Google Scholar] [CrossRef] [Green Version]
- Cerasa, A.; Valentino, P.; Chiriaco, C.; Pirritano, D.; Nistico, R.; Gioia, C.M.; Trotta, M.; Del Giudice, F.; Tallarico, T.; Rocca, F.; et al. MR imaging and cognitive correlates of relapsing-remitting multiple sclerosis patients with cerebellar symptoms. J. Neurol 2013, 260, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Romascano, D.; Meskaldji, D.E.; Bonnier, G.; Simioni, S.; Rotzinger, D.; Lin, Y.C.; Menegaz, G.; Roche, A.; Schluep, M.; Pasquier, R.D.; et al. Multicontrast connectometry: A new tool to assess cerebellum alterations in early relapsing-remitting multiple sclerosis. Hum. Brain. Mapp. 2015, 36, 1609–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoodley, C.J.; Schmahmann, J.D. Evidence for topographic organization in the cerebellum of motor control versus cognitive and affective processing. Cortex 2010, 46, 831–844. [Google Scholar] [CrossRef] [Green Version]
- Schmahmann, J.D. An emerging concept. The cerebellar contribution to higher function. Arch. Neurol 1991, 48, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Weier, K.; Penner, I.K.; Magon, S.; Amann, M.; Naegelin, Y.; Andelova, M.; Derfuss, T.; Stippich, C.; Radue, E.W.; Kappos, L.; et al. Cerebellar abnormalities contribute to disability including cognitive impairment in multiple sclerosis. PLoS ONE 2014, 9, e86916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarica, A.; Cerasa, A.; Quattrone, A. The neurocognitive profile of the cerebellum in multiple sclerosis. Int. J. Mol. Sci. 2015, 16, 12185–12198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margoni, M.; Preziosa, P.; Tortorella, P.; Filippi, M.; Rocca, M.A. Does Ocrelizumab Limit Multiple Sclerosis Progression? Current Evidence from Clinical, MRI, and Fluid Biomarkers. Neurother. J. Am. Soc. Exp. Neurother. 2022, 1–13, online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Owczarczyk, K.; Lal, P.; Abbas, A.R.; Wolslegel, K.; Holweg, C.T.; Dummer, W.; Kelman, A.; Brunetta, P.; Lewin-Koh, N.; Sorani, M.; et al. A plasmablast biomarker for nonresponse to antibody therapy to CD20 in rheumatoid arthritis. Sci. Transl. Med. 2011, 3, 101ra192. [Google Scholar] [CrossRef] [PubMed]
- Md Yusof, M.Y.; Shaw, D.; El-Sherbiny, Y.M.; Dunn, E.; Rawstron, A.C.; Emery, P.; Vital, E.M. Predicting and managing primary and secondary non-response to rituximab using B-cell biomarkers in systemic lupus erythematosus. Ann. Rheum. Dis. 2017, 76, 1829–1836. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Jeong, I.H.; Hyun, J.W.; Joung, A.; Jo, H.J.; Hwang, S.H.; Yun, S.; Joo, J.; Kim, H.J. Treatment Outcomes with Rituximab in 100 Patients with Neuromyelitis Optica: Influence of FCGR3A Polymorphisms on the Therapeutic Response to Rituximab. JAMA Neurol. 2015, 72, 989–995. [Google Scholar] [CrossRef] [Green Version]
- Choy, E.H.; De Benedetti, F.; Takeuchi, T.; Hashizume, M.; John, M.R.; Kishimoto, T. Translating IL-6 biology into effective treatments. Nat. Rev. Rheumatol. 2020, 16, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Gobel, K.; Ruck, T.; Meuth, S.G. Cytokine signaling in multiple sclerosis: Lost in translation. Mult. Scler. 2018, 24, 432–439. [Google Scholar] [CrossRef]
- Comabella, M. Tocilizumab and multiple sclerosis: A causal relationship? Clinical Commentary on the case report entitled--MS arising during Tocilizumab therapy for rheumatoid arthritis. Mult. Scler. 2016, 22, 257–258. [Google Scholar] [CrossRef]
- Beauchemin, P.; Carruthers, R. MS arising during Tocilizumab therapy for rheumatoid arthritis. Mult. Scler. 2016, 22, 254–256. [Google Scholar] [CrossRef]
- Cruz-Velásquez, G.; Artal, J.; Sebastián, B.; Seral, M.; Pérez-Moreiras, J.-V.; Martín, J.; Ara, J.-R. Multiple sclerosis triggered duringtocilizumab therapy. Report of a new case. Mult. Scler. J. 2016, 22, 375. [Google Scholar] [CrossRef]
- Garcia, A.; Morille, J.; Shah, S.; Le Frère, F.; Nicot, A.; Berthelot, L.; Wiertlewski, S.; Gourraud, P.A.; Raposo, C.; Bakdache, F.; et al. A broad effect of ocrelizumab on the peripheral immune component in patients with early relapsing-remitting multiple sclerosis. Mult. Scler. J. 2021, 27, 593. [Google Scholar] [CrossRef]
- Miscioscia, A.; Puthenparampil, M.; Miante, S.; Pengo, M.; Rinaldi, F.; Perini, P.; Gallo, P. Retinal inner nuclear layer thinning is decreased and associates with the clinical outcome in ocrelizumab-treated primary progressive multiple sclerosis. J. Neurol. 2022, 1–7, online ahead of print. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All (N = 22) | Non-Responders (N = 9) | Responders (N = 13) | p * | |
|---|---|---|---|---|
| Gender (male/female) Age Education (years) | 12/10 | 5/4 | 7/6 | 0.937 |
| 48.5 ± 1.69 | 49.56 ± 3.45 | 47.77 ± 1.69 | 0.948 | |
| 14.05 ± 0.7 | 15.44 ± 1.16 | 13 ± 0.78 | 0.058 | |
| Disease duration (years) Time from diagnosis (years) | 9.05 ± 0.92 | 8.89 ± 1.79 | 9.15 ± 1.02 | 0.556 |
| 5.59 ± 0.84 | 6.44 ± 1.44 | 6.69 ± 1.06 | 0.512 | |
| Smoking (yes/no) Cardiovascular comorbidities ** Other (non-cardiovascular) comorbidities *** EDSS | 10/12 | 2/7 | 8/5 | 0.069 |
| 16/2/4 | 8/0/1 | 8/2/3 | 0.309 | |
| 16/6/1/1/1 | 7/2/0/0/0 | 9/4/1/1/1 | 0.556 | |
| 4.91 ± 0.3 | 4.56 ± 0.56 | 5.15 ± 0.34 | 0.324 | |
| MRI new/enlarged T2 lesions (brain and cervical) MRI Gd+ lesions (brain and cervical) | 1.05 ± 0.26 | 0.89 ± 0.39 | 1.15 ± 0.36 | 0.647 |
| 0.36 ± 0.12 | 0.44 ± 0.18 | 0.31 ± 0.17 | 0.512 |
| Proportion of Patients, n/N (%) 95% CI | 12 Months (N = 22) | 24 Months (N = 22) |
|---|---|---|
| Responders | 14/22 (63.6%) 0.41–0.83 | 13/22 (59.1%) 0.36–0.79 |
| No CDA | 14/22 (63.6%) 0.41–0.83 | 13/22 (59.1%) 0.36–0.79 |
| No new/enlarged T2 lesions (brain and cervical MRI) | 20/22 (90.9%) 0.71–0.99 | 20/22 (90.9%) 0.71–0.99 |
| All (N = 22) | Non-Responders (N = 9) | Responders (N = 13) | p * | |
|---|---|---|---|---|
| SDMT | ||||
| baseline | 42.09 ± 2.4 | 47.11 ± 3.21 | 38.62 ± 3.14 | 0.096 |
| 12 months | 39.91 ± 2.21 | 45 ± 3.37 | 36.38 ± 2.6 | 0.06 |
| 24 months | 41.59 ± 2.25 | 46.67 ± 3.18 | 38.08 ± 2.79 | 0.071 |
| change from baseline (12 months) | −2.18 ± 0.89 | −2.11 ± 1.45 | −2.23 ± 1.17 | 0.061 |
| change from baseline (24 months) | −5 ± 1.28 | −0.44 ± 1.45 | −0.54 ± 1.96 | 0.253 |
| GVLT | ||||
| baseline | 52.82 ± 2.33 | 53.56 ± 3.2 | 52.31 ± 3.37 | 0.556 |
| 12 months | 54.91 ± 2.52 | 58.11 ± 3.73 | 52.69 ± 3.37 | 0.324 |
| 24 months | 55.73 ± 2.43 | 60.33 ± 3.34 | 52.54 ± 3.2 | 0.096 |
| change from baseline (12 months) | 2.09 ± 2.09 | 4.56 ± 3.04 | 0.38 ± 2.83 | 0.058 |
| change from baseline (24 months) | 2.91 ± 2.43 | 6.78 ± 2.81 | 0.23 ± 3.53 | 0.288 |
| BVMT-R | ||||
| baseline | 19.14 ± 1.7 | 20.89 ± 1.94 | 17.92 ± 2.56 | 0.647 |
| 12 months | 20.41 ± 1.95 | 22.56 ± 2.87 | 18.92 ± 2.64 | 0.357 |
| 24 months | 18.45 ± 1.78 | 21.89 ± 2.55 | 16.08 ± 2.29 | 0.126 |
| change from baseline (12 months) | 1.27 ± 1.57 | 1.67 ± 2.67 | 1 ± 2 | 0.338 |
| change from baseline (24 months) | −0.68 ± 1.64 | 1 ± 2.44 | −1.85 ± 2.22 | 0.165 |
| MFIS | ||||
| baseline | 36.32 ± 3.2 | 30.22 ± 3.94 | 40.54 ± 4.42 | 0.082 |
| 12 months | 37.95 ± 3.86 | 33.89 ± 7.15 | 40.77 ± 4.34 | 0.512 |
| 24 months | 41.55 ± 3.64 | 36.11 ± 6.09 | 45.31 ± 4.4 | 0.235 |
| change from baseline (12 months) | 1.64 ± 3.56 | 3.67 ± 7.17 | 0.23 ± 3.66 | 0.173 |
| change from baseline (24 months) | 5.23 ± 3.07 | 5.89 ± 5.96 | 4.77 ± 3.38 | 0.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boziki, M.; Bakirtzis, C.; Sintila, S.-A.; Kesidou, E.; Gounari, E.; Ioakimidou, A.; Tsavdaridou, V.; Skoura, L.; Fylaktou, A.; Nikolaidou, V.; et al. Ocrelizumab in Patients with Active Primary Progressive Multiple Sclerosis: Clinical Outcomes and Immune Markers of Treatment Response. Cells 2022, 11, 1959. https://doi.org/10.3390/cells11121959
Boziki M, Bakirtzis C, Sintila S-A, Kesidou E, Gounari E, Ioakimidou A, Tsavdaridou V, Skoura L, Fylaktou A, Nikolaidou V, et al. Ocrelizumab in Patients with Active Primary Progressive Multiple Sclerosis: Clinical Outcomes and Immune Markers of Treatment Response. Cells. 2022; 11(12):1959. https://doi.org/10.3390/cells11121959
Chicago/Turabian StyleBoziki, Marina, Christos Bakirtzis, Styliani-Aggeliki Sintila, Evangelia Kesidou, Evdoxia Gounari, Aliki Ioakimidou, Vasiliki Tsavdaridou, Lemonia Skoura, Asimina Fylaktou, Vasiliki Nikolaidou, and et al. 2022. "Ocrelizumab in Patients with Active Primary Progressive Multiple Sclerosis: Clinical Outcomes and Immune Markers of Treatment Response" Cells 11, no. 12: 1959. https://doi.org/10.3390/cells11121959
APA StyleBoziki, M., Bakirtzis, C., Sintila, S. -A., Kesidou, E., Gounari, E., Ioakimidou, A., Tsavdaridou, V., Skoura, L., Fylaktou, A., Nikolaidou, V., Stangou, M., Nikolaidis, I., Giantzi, V., Karafoulidou, E., Theotokis, P., & Grigoriadis, N. (2022). Ocrelizumab in Patients with Active Primary Progressive Multiple Sclerosis: Clinical Outcomes and Immune Markers of Treatment Response. Cells, 11(12), 1959. https://doi.org/10.3390/cells11121959