
Mechanotransduction in Skin Inflammation


Abstract
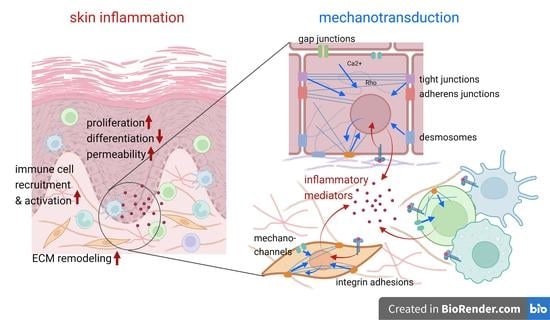
:
1. Introduction
2. Inflammatory Skin Disorders
3. ECM and the Mechanical Environment in the Skin
4. Integrins: Mechanosensitive ECM Receptors
5. Epidermal Cell–Cell Adhesions
6. Actin–Myosin Cytoskeleton and Cell Contractility
7. Intermediate Filaments
8. Mechanosensitive Ion Channels
9. Mechanoactivated Intracellular Signaling
10. Skin Mechanical Stretch and the Koebner Phenomenon
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | atopic dermatitis |
| AJ | adherens junction |
| BM | basement membrane |
| ECM | extracellular matrix |
| FAK | focal adhesion kinase |
| IF | intermediate filaments |
| IFN-γ | interferon-γ |
| IL | interleukin |
| NMII | nonmuscle myosin II |
| TGF | transforming growth factor |
| TJ | tight junction |
| TLR | Toll-like receptor |
References
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, D.M.; Burridge, K. Mechanotransduction and nuclear function. Curr. Opin. Cell Biol. 2016, 40, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehlmann, B.; Bonham, C.A.; Zucal, I.; Prantl, L.; Gurtner, G.C. Mechanotransduction in Wound Healing and Fibrosis. J. Clin. Med. 2020, 9, 1423. [Google Scholar] [CrossRef]
- Moll, I. Duale Reihe Dermatologie; Thieme: Stuttgart, Deutschland, 2016. [Google Scholar]
- Wang, J.; Zhang, Y.; Zhang, N.; Wang, C.; Herrler, T.; Li, Q. An updated review of mechanotransduction in skin disorders: Transcriptional regulators, ion channels, and microRNAs. Cell. Mol. Life Sci. 2015, 72, 2091–2106. [Google Scholar] [CrossRef]
- Wong, V.W.; Akaishi, S.; Longaker, M.T.; Gurtner, G.C. Pushing Back: Wound Mechanotransduction in Repair and Regeneration. J. Investig. Dermatol. 2011, 131, 2186–2196. [Google Scholar] [CrossRef] [Green Version]
- Brembilla, N.C.; Senra, L.; Boehncke, W.-H. The IL-17 Family of Cytokines in Psoriasis: IL-17A and Beyond. Front. Immunol. 2018, 9, 1682. [Google Scholar] [CrossRef] [Green Version]
- Boehncke, W.-H.; Dressel, D.; Zollner, T.M.; Kaufmann, R. Pulling the trigger on psoriasis. Nature 1996, 379, 777. [Google Scholar] [CrossRef] [Green Version]
- Boehncke, W.-H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Boehncke, W.-H. Systemic Inflammation and Cardiovascular Comorbidity in Psoriasis Patients: Causes and Consequences. Front. Immunol. 2018, 9, 579. [Google Scholar] [CrossRef]
- Sagi, L.; Trau, H. The Koebner phenomenon. Clin. Dermatol. 2011, 29, 231–236. [Google Scholar] [CrossRef]
- Bäsler, K.; Brandner, J.M. Tight junctions in skin inflammation. Pflügers Arch. Eur. J. Physiol. 2017, 469, 3–14. [Google Scholar] [CrossRef] [PubMed]
- David Boothe, W.; Tarbox, J.A.; Tarbox, M.B. Atopic Dermatitis: Pathophysiology. Adv. Exp. Med. Biol. 2017, 1027, 21–37. [Google Scholar] [PubMed]
- Jimenez, S.A.; Hitraya, E.; Varga, J. Pathogenesis of scleroderma: Collagen. Rheum. Dis. Clin. N. Am. 1996, 22, 647–674. [Google Scholar] [CrossRef]
- Gabrielli, A.; Avvedimento, E.V.; Krieg, T. Scleroderma. N. Engl. J. Med. 2009, 360, 1989–2003. [Google Scholar] [CrossRef]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.M.; Fujiwara, H. Cell-Extracellular Matrix Interactions in Normal and Diseased Skin. Cold Spring Harb. Perspect. Biol. 2011, 3, a005124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalton, C.J.; Lemmon, C.A. Fibronectin: Molecular Structure, Fibrillar Structure and Mechanochemical Signaling. Cells 2021, 10, 2443. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, K.; Shaw, L.E.; Symmank, D.; Weninger, W. The Extracellular Matrix in Skin Inflammation and Infection. Front. Cell Dev. Biol. 2021, 9, 682414. [Google Scholar] [CrossRef]
- Dufort, C.C.; Paszek, M.J.; Weaver, V.M. Balancing forces: Architectural control of mechanotransduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 308–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egeblad, M.; Rasch, M.G.; Weaver, V.M. Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 2010, 22, 697–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, J.C.; Barnes, J.M.; Desai, S.R.; Sistrunk, C.; Conklin, M.W.; Schedin, P.; Eliceiri, K.W.; Keely, P.J.; Seewaldt, V.L.; Weaver, V.L. Tumor mechanics and metabolic dysfunction. Free. Radic. Biol. Med. 2015, 79, 269–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serup, J.; Northeved, A. Skin elasticity in psoriasis. In vivo measurement of tensile distensibility, hysteresis and resilient distension with a new method. Comparison with skin thickness as measured with high-frequency ultrasound. J. Dermatol. 1985, 12, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, H. In vivo Study of Skin Mechanical Properties in Psoriasis Vulgaris. Acta Derm. Venereol. 2000, 80, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- van Caam, A.; Vonk, M.; van den Hoogen, F.; van Lent, P.; van der Kraan, P. Unraveling SSc Pathophysiology; The Myofibroblast. Front Immunol. 2018, 9, 2452. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Plotnikov, S. Mechanosensitive Regulation of Fibrosis. Cells 2021, 10, 994. [Google Scholar] [CrossRef]
- Ho, Y.Y.; Lagares, D.; Tager, A.M.; Kapoor, M. Fibrosis—A lethal component of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 390–402. [Google Scholar] [CrossRef]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalkwijk, J.; Vlijmen, I.; Oosterling, B.; Perret, C.; Koopman, R.; Born, J.V.D.; Mackie, E.J. Tenascin expression in hyperproliferative skin diseases. Br. J. Dermatol. 1991, 124, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Ito, M.; Takeuchi, K.; Nakada, A.; Heishi, M.; Suto, H.; Mitsuishi, K.; Sugita, Y.; Ogawa, H.; Ra, C. Tenascin-C is upregulated in the skin lesions of patients with atopic dermatitis. J. Dermatol. Sci. 2005, 40, 35–41. [Google Scholar] [CrossRef] [PubMed]
- McFadden, J.; Baker, B.; Powles, A.; Fry, L. Psoriasis and extra domain A fibronectin loops. Br. J. Dermatol. 2010, 163, 5–11. [Google Scholar] [CrossRef]
- Arima, K.; Ohta, S.; Takagi, A.; Shiraishi, H.; Masuoka, M.; Ontsuka, K.; Suto, H.; Suzuki, S.; Yamamoto, K.-I.; Ogawa, M.; et al. Periostin contributes to epidermal hyperplasia in psoriasis common to atopic dermatitis. Allergol. Int. 2014, 64, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Gubán, B.; Vas, K.; Balog, Z.; Manczinger, M.; Bebes, A.; Groma, G.; Széll, M.; Kemény, L.; Bata-Csörgő, Z. Abnormal regulation of fibronectin production by fibroblasts in psoriasis. Br. J. Dermatol. 2015, 174, 533–541. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, O.; Ayyangar, U.; Kurbet, A.S.; Ashok, D.; Raghavan, S. Unraveling the ECM-Immune Cell Crosstalk in Skin Diseases. Front. Cell Dev. Biol. 2019, 7, 68. [Google Scholar] [CrossRef]
- Choi, Y.; Song, M.; Hara, M.; Imanaka-Yoshida, K.; Lee, D.; Chung, J.; Lee, S.-T. Effects of Tenascin C on the Integrity of Extracellular Matrix and Skin Aging. Int. J. Mol. Sci. 2020, 21, 8693. [Google Scholar] [CrossRef]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [Green Version]
- Imanaka-Yoshida, K. Tenascin-C in Heart Diseases—The Role of Inflammation. Int. J. Mol. Sci. 2021, 22, 5828. [Google Scholar] [CrossRef]
- Chiquet-Ehrismann, R.; Chiquet, M. Tenascins: Regulation and putative functions during pathological stress. J. Pathol. 2003, 200, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Bata-Csorgo, Z.; Cooper, K.; Ting, K.M.; Voorhees, J.J.; Hammerberg, C. Fibronectin and alpha5 integrin regulate keratinocyte cell cycling. A mechanism for increased fibronectin potentiation of T cell lymphokine-driven keratinocyte hyperproliferation in psoriasis. J. Clin. Investig. 1998, 101, 1509–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohan, M.; Muro, A.F.; White, E.S.; Berkman, N. EDA-containing cellular fibronectin induces fibroblast differentiation through binding to alpha4β7 integrin receptor and MAPK/Erk 1/2-dependent signaling. FASEB J. 2010, 24, 4503–4512. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Tamaki, Z.; Wang, W.; Hinchcliff, M.; Hoover, P.; Getsios, S.; White, E.S.; Varga, J. FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci. Transl. Med. 2014, 6, 232ra50. [Google Scholar] [CrossRef] [Green Version]
- Nowicka, D.; Grywalska, E. The Role of Immune Defects and Colonization of Staphylococcus aureus in the Pathogenesis of Atopic Dermatitis. Anal. Cell. Pathol. 2018, 2018, 1956403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suresh, A.; Biswas, A.; Perumal, S.; Khurana, S. Periostin and Integrin Signaling in Stem Cell Regulation. Adv. Exp. Med. Biol. 2019, 1132, 163–176. [Google Scholar] [CrossRef]
- Zhou, H.-M.; Wang, J.; Elliott, C.; Wen, W.; Hamilton, D.W.; Conway, S.J. Spatiotemporal expression of periostin during skin development and incisional wound healing: Lessons for human fibrotic scar formation. J. Cell Commun. Signal. 2010, 4, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, S.K.; Wheeler, J.J.; Pitake, S.; Ding, H.; Jiang, C.; Fukuyama, T.; Paps, J.S.; Ralph, P.; Coyne, J.; Parkington, M.; et al. Periostin Activation of Integrin Receptors on Sensory Neurons Induces Allergic Itch. Cell Rep. 2020, 31, 107472. [Google Scholar] [CrossRef]
- Koivukangas, V.; Kallionen, M.; Karvonen, J.; Autio-Harmainen, H.; Risteli, J.; Risteli, L.; Oikarinen, A. Increased collagen synthesis in psoriasis in vivo. Arch. Dermatol. Res. 1995, 287, 171–175. [Google Scholar] [CrossRef]
- Kounadi, E.; Fountos, G.; Tzaphlidou, M. The influence of inflammation-mediated osteopenia (IMO) on the structure of rabbit bone and skin collagen fibrils. Connect. Tissue Res. 1998, 37, 69–76. [Google Scholar] [CrossRef]
- Cui, S.-J.; Fu, Y.; Liu, Y.; Kou, X.-X.; Zhang, J.-N.; Gan, Y.-H.; Zhou, Y.-H.; Wang, X.-D. Chronic inflammation deteriorates structure and function of collagen fibril in rat temporomandibular joint disc. Int. J. Oral Sci. 2019, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Oyoshi, M.K.; He, R.; Kanaoka, Y.; ElKhal, A.; Kawamoto, S.; Lewis, C.N.; Austen, K.F.; Geha, R.S. Eosinophil-derived leukotriene C4 signals via type 2 cysteinyl leukotriene receptor to promote skin fibrosis in a mouse model of atopic dermatitis. Proc. Natl. Acad. Sci. USA 2012, 109, 4992–4997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, M.S.; Marinkovich, M.P. Role of Dermal-Epidermal Basement Membrane Zone in Skin, Cancer, and Developmental Disorders. Dermatol. Clin. 2010, 28, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Fleischmajer, R.; Kuroda, K.; Hazan, R.; Gordon, R.E.; Lebwohl, M.G.; Sapadin, A.N.; Unda, F.; Iehara, N.; Yamada, Y. Basement Membrane Alterations in Psoriasis are Accompanied by Epidermal Overexpression of MMP-2 and its Inhibitor TIMP-2. J. Investig. Dermatol. 2000, 115, 771–777. [Google Scholar] [CrossRef] [Green Version]
- Suomela, S.; Kariniemi, A.-L.; Snellman, E.; Saarialho-Kere, U. Metalloelastase (MMP-12) and 92-kDa gelatinase (MMP-9) as well as their inhibitors, TIMP-1 and -3, are expressed in psoriatic lesions. Exp. Dermatol. 2001, 10, 175–183. [Google Scholar] [CrossRef]
- Seetharaman, S.; Etienne-Manneville, S. Integrin diversity brings specificity in mechanotransduction. Biol. Cell 2018, 110, 49–64. [Google Scholar] [CrossRef]
- Tamada, M.; Sheetz, M.P.; Sawada, Y. Activation of a Signaling Cascade by Cytoskeleton Stretch. Dev. Cell 2004, 7, 709–718. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Watt, F.M. NEW EMBO MEMBER′S REVIEW: Role of integrins in regulating epidermal adhesion, growth and differentiation. EMBO J. 2002, 21, 3919–3926. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.H.; Harper, S.; Watt, F. Stem cell patterning and fate in human epidermis. Cell 1995, 80, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Molès, J.-P.; Watt, F.M. The Epidermal Stem Cell Compartment: Variation in Expression Levels of E–Cadherin and Catenins Within the Basal Layer of Human Epidermis. J. Histochem. Cytochem. 1997, 45, 867–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peñas, P.F.; Buezo, G.F.; Rios, L.; García-Díez, A.; Gómez, M.; Yáñez-Mo, M.; Cabañas, C.; Sánchez-Madrid, F. Differential Expression of Activation Epitopes of β1 Integrins in Psoriasis and Normal Skin. J. Investig. Dermatol. 1998, 111, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertle, M.D.; Kubler, M.D.; Leigh, I.M.; Watt, F.M. Aberrant integrin expression during epidermal wound healing and in psoriatic epidermis. J. Clin. Investig. 1992, 89, 1892–1901. [Google Scholar] [CrossRef]
- Carroll, J.M.; Romero, M.; Watt, F. Suprabasal integrin expression in the epidermis of transgenic mice results in developmental defects and a phenotype resembling psoriasis. Cell 1995, 83, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Martin, P. Wound Healing--Aiming for Perfect Skin Regeneration. Science 1997, 276, 75–81. [Google Scholar] [CrossRef]
- Grose, R.; Hutter, C.; Bloch, W.; Thorey, I.; Watt, F.M.; Fässler, R.; Brakebusch, C.; Werner, S. A crucial role of β 1 integrins for keratinocyte migration in vitro and during cutaneous wound repair. Development 2002, 129, 2303–2315. [Google Scholar] [CrossRef]
- Staunstrup, N.H.; Stenderup, K.; Mortensen, S.; Primo, M.N.; Rosada, C.; Steiniche, T.; Liu, Y.; Li, R.; Schmidt, M.; Purup, S.; et al. Psoriasiform skin disease in transgenic pigs with high-copy ectopic expression of human integrins α2 and β1. Dis. Model. Mech. 2017, 10, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, G.; De Luca, M.; Orecchia, G.; Balzac, F.; Cremona, O.; Savoia, P.; Cancedda, R.; Marchisio, P.C. Expression, topography, and function of integrin receptors are severely altered in keratinocytes from involved and uninvolved psoriatic skin. J. Clin. Investig. 1992, 89, 1783–1795. [Google Scholar] [CrossRef] [Green Version]
- Conrad, C.; Boyman, O.; Tonel, G.; Tun-Kyi, A.; Laggner, U.; de Fougerolles, A.; Kotelianski, V.; Gardner, H.; Nestle, F.O. α1β1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat. Med. 2007, 13, 836–842. [Google Scholar] [CrossRef]
- Shannon, M.J.; Mace, E.M. Natural Killer Cell Integrins and Their Functions in Tissue Residency. Front. Immunol. 2021, 12, 647358. [Google Scholar] [CrossRef]
- Creamer, D.; Allen, M.; Sousa, A.; Poston, R.; Barker, J. Altered vascular endothelium integrin expression in psoriasis. Am. J. Pathol. 1995, 147, 1661–1667. [Google Scholar]
- Millard, M.; Odde, S.; Neamati, N. Integrin Targeted Therapeutics. Theranostics 2011, 1, 154–188. [Google Scholar] [CrossRef]
- Pandolfi, F.; Franza, L.; Altamura, S.; Mandolini, C.; Cianci, R.; Ansari, A.; Kurnick, J.T. Integrins: Integrating the Biology and Therapy of Cell–cell Interactions. Clin. Ther. 2017, 39, 2420–2436. [Google Scholar] [CrossRef]
- Ho, T.-C.; Yeh, S.-I.; Chen, S.-L.; Tsao, Y.-P. The Psoriasis Therapeutic Potential of a Novel Short Laminin Peptide C16. Int. J. Mol. Sci. 2019, 20, 3144. [Google Scholar] [CrossRef] [Green Version]
- Conroy, K.P.; Kitto, L.J.; Henderson, N.C. αv integrins: Key regulators of tissue fibrosis. Cell Tissue Res. 2016, 365, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Park, E.J.; Myint, P.K.; Ito, A.; Appiah, M.G.; Darkwah, S.; Kawamoto, E.; Shimaoka, M. Integrin-Ligand Interactions in Inflammation, Cancer, and Metabolic Disease: Insights Into the Multifaceted Roles of an Emerging Ligand Irisin. Front. Cell Dev. Biol. 2020, 8, 588066. [Google Scholar] [CrossRef]
- Häkkinen, L.; Koivisto, L.; Gardner, H.; Saarialho-Kere, U.; Carroll, J.M.; Lakso, M.; Rauvala, H.; Laato, M.; Heino, J.; Larjava, H. Increased expression of β6-integrin in skin leads to spontaneous development of chronic wounds. Am. J. Pathol. 2004, 164, 229–242. [Google Scholar] [CrossRef]
- Henderson, N.C.; Sheppard, D. Integrin-mediated regulation of TGFβ in fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 891–896. [Google Scholar] [CrossRef] [Green Version]
- Thannickal, V.J.; Lee, D.Y.; White, E.S.; Cui, Z.; Larios, J.M.; Chacon, R.; Horowitz, J.C.; Day, R.M.; Thomas, P.E. Myofibroblast differentiation by transforming growth factor-β1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J. Biol. Chem. 2003, 278, 12384–12389. [Google Scholar] [CrossRef] [Green Version]
- Gerber, E.E.; Gallo, E.M.; Fontana, S.C.; Davis, E.C.; Wigley, F.M.; Huso, D.L.; Dietz, H.C. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature 2013, 503, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Angulo-Urarte, A.; van der Wal, T.; Huveneers, S. Cell-cell junctions as sensors and transducers of mechanical forces. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183316. [Google Scholar] [CrossRef]
- Borghi, N.; Sorokina, M.; Shcherbakova, O.G.; Weis, W.I.; Pruitt, B.L.; Nelson, W.J.; Dunn, A.R. E-cadherin is under constitutive actomyosin-generated tension that is increased at cell–cell contacts upon externally applied stretch. Proc. Natl. Acad. Sci. USA 2012, 109, 12568–12573. [Google Scholar] [CrossRef] [Green Version]
- Yap, A.S.; Duszyc, K.; Viasnoff, V. Mechanosensing and Mechanotransduction at Cell–Cell Junctions. Cold Spring Harb. Perspect. Biol. 2017, 10, a028761. [Google Scholar] [CrossRef]
- Yonemura, S.; Wada, Y.; Watanabe, T.; Nagafuchi, A.; Shibata, M. α-Catenin as a tension transducer that induces adherens junction development. Nat. Cell Biol. 2010, 12, 533–542. [Google Scholar] [CrossRef]
- Ray, S.; Foote, H.P.; Lechler, T. β-Catenin protects the epidermis from mechanical stresses. J. Cell Biol. 2013, 202, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Karayiannakis, A.J.; Syrigos, K.N.; Efstathiou, J.; Valizadeh, A.; Noda, M.; Playford, R.J.; Kmiot, W.; Pignatelli, M. Expression of catenins and E-cadherin during epithelial restitution in inflammatory bowel disease. J. Pathol. 1998, 185, 413–418. [Google Scholar] [CrossRef]
- Bruewer, M.; Samarin, S.; Nusrat, A. Inflammatory Bowel Disease and the Apical Junctional Complex. Ann. N. Y. Acad. Sci. 2006, 1072, 242–252. [Google Scholar] [CrossRef]
- Gavard, J.; Gutkind, J.S. VEGF controls endothelial-cell permeability by promoting the β-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef]
- Ivanov, A.I.; Parkos, C.A.; Nusrat, A. Cytoskeletal Regulation of Epithelial Barrier Function During Inflammation. Am. J. Pathol. 2010, 177, 512–524. [Google Scholar] [CrossRef]
- Naydenov, N.G.; Baranwal, S.; Khan, S.; Feygin, A.; Gupta, P.; Ivanov, A.I. Novel mechanism of cytokine-induced disruption of epithelial barriers. Tissue Barriers 2013, 1, e25231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Blikslager, A.T. The Regulation of Intestinal Mucosal Barrier by Myosin Light Chain Kinase/Rho Kinases. Int. J. Mol. Sci. 2020, 21, 3550. [Google Scholar] [CrossRef] [PubMed]
- Ghadially, R.; Reed, J.T.; Elias, P.M. Stratum Corneum Structure and Function Correlates with Phenotype in Psoriasis. J. Investig. Dermatol. 1996, 107, 558–564. [Google Scholar] [CrossRef] [Green Version]
- Wolf, R.; Orion, E.; Ruocco, E.; Ruocco, V. Abnormal epidermal barrier in the pathogenesis of psoriasis. Clin. Dermatol. 2012, 30, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Montero-Vilchez, T.; Segura-Fernández-Nogueras, M.-V.; Pérez-Rodríguez, I.; Soler-Gongora, M.; Martinez-Lopez, A.; Fernández-González, A.; Molina-Leyva, A.; Arias-Santiago, S. Skin Barrier Function in Psoriasis and Atopic Dermatitis: Transepidermal Water Loss and Temperature as Useful Tools to Assess Disease Severity. J. Clin. Med. 2021, 10, 359. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, A.; Altznauer, F.; Akdis, M.; Simon, H.-U.; Blaser, K.; Akdis, C.A.; Disch, R.; Bröcker, E.-B. The Differential Fate of Cadherins during T-Cell-Induced Keratinocyte Apoptosis Leads to Spongiosis in Eczematous Dermatitis. J. Investig. Dermatol. 2001, 117, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Matsuyoshi, N.; Takeuchi, T.; Ohtsuki, Y.; Miyachi, Y. Reciprocal altered expression of T-cadherin and P-cadherin in psoriasis vulgaris. Br. J. Dermatol. 2003, 149, 268–273. [Google Scholar] [CrossRef]
- Li, Z.; Peng, Z.; Wang, Y.; Geng, S.; Ji, F. Decreased expression of E-cadherin and β-catenin in the lesional skin of patients with active psoriasis. Int. J. Dermatol. 2008, 47, 207–209. [Google Scholar] [CrossRef]
- El Rebey, H.S.; Maree, A.H.; El Tahmody, M.; Abdel Naby, H. Role of E-cadherin in psoriasis. Egypt. J. Pathol. 2014, 34, 148–154. [Google Scholar] [CrossRef]
- Xie, G.; Ao, X.; Lin, T.; Zhou, G.; Wang, M.; Wang, H.; Chen, Y.; Li, X.; Xu, B.; He, W.; et al. E-Cadherin–Mediated Cell Contact Controls the Epidermal Damage Response in Radiation Dermatitis. J. Investig. Dermatol. 2017, 137, 1731–1739. [Google Scholar] [CrossRef]
- Anderson, E.D.; Alishahedani, M.E.; Myles, I.A. Epithelial-Mesenchymal Transition in Atopy: A Mini-Review. Front. Allergy 2020, 1, 628381. [Google Scholar] [CrossRef]
- Hampton, P.J.; Ross, O.K.; Reynolds, N.J. Increased nuclear β-catenin in suprabasal involved psoriatic epidermis. Br. J. Dermatol. 2007, 157, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- El-Wahed Gaber, M.A.; El-Halim Kandil, M.A.; El-Farargy, S.M.; Galbet, D.A.E. Beta-catenin expression in psoriasis. Indian Derm. Online J. 2015, 6, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Nowell, C.S.; Odermatt, P.D.; Azzolin, L.; Hoehnel, S.; Wagner, E.F.; Fantner, G.E.; Lutolf, M.P.; Barrandon, Y.; Piccolo, S.; Radtke, F. Chronic inflammation imposes aberrant cell fate in regenerating epithelia through mechanotransduction. Nat. Cell Biol. 2015, 18, 168–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yu, X.; Wu, C.; Jin, H. IL-36γ inhibits differentiation and induces inflammation of keratinocyte via Wnt signaling pathway in psoriasis. Int. J. Med Sci. 2017, 14, 1002–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef] [Green Version]
- Beyer, C.; Schramm, A.; Akhmetshina, A.; Dees, C.; Kireva, T.; Gelse, K.; Sonnylal, S.; De Crombrugghe, B.; Taketo, M.M.; Distler, O.; et al. β-catenin is a central mediator of pro-fibrotic Wnt signaling in systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 761–767. [Google Scholar] [CrossRef] [Green Version]
- Astudillo, P. Extracellular matrix stiffness and Wnt/β-catenin signaling in physiology and disease. Biochem. Soc. Trans. 2020, 48, 1187–1198. [Google Scholar] [CrossRef]
- Perez-Moreno, M.; Davis, M.A.; Wong, E.; Pasolli, H.A.; Reynolds, A.B.; Fuchs, E. p120-Catenin Mediates Inflammatory Responses in the Skin. Cell 2006, 124, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Brandner, J.M. Importance of Tight Junctions in Relation to Skin Barrier Function. Ski. Barrier Funct. 2016, 49, 27–37. [Google Scholar] [CrossRef]
- Rübsam, M.; Mertz, A.F.; Kubo, A.; Marg, S.; Jüngst, C.; Goranci-Buzhala, G.; Schauss, A.C.; Horsley, V.; Dufresne, E.R.; Moser, M.; et al. E-cadherin integrates mechanotransduction and EGFR signaling to control junctional tissue polarization and tight junction positioning. Nat. Commun. 2017, 8, 1250. [Google Scholar] [CrossRef]
- Al-Sadi, R.; Boivin, M.; Ma, T. Mechanism of cytokine modulation of epithelial tight junction barrier. Front. Biosci. 2009, 14, 2765–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuki, T.; Tobiishi, M.; Kusaka-Kikushima, A.; Ota, Y.; Tokura, Y. Impaired Tight Junctions in Atopic Dermatitis Skin and in a Skin-Equivalent Model Treated with Interleukin-17. PLoS ONE 2016, 11, e0161759. [Google Scholar] [CrossRef]
- De Benedetto, A.; Rafaels, N.M.; McGirt, L.Y.; Ivanov, A.I.; Georas, S.N.; Cheadle, C.; Berger, A.E.; Zhang, K.; Vidyasagar, S.; Yoshida, T.; et al. Tight junction defects in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2011, 127, 773–786.e1-7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschner, N.; Poetzl, C.; Driesch, P.V.D.; Wladykowski, E.; Moll, I.; Behne, M.J.; Brandner, J.M. Alteration of Tight Junction Proteins Is an Early Event in Psoriasis: Putative Involvement of Proinflammatory Cytokines. Am. J. Pathol. 2009, 175, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Tokumasu, R.; Yamaga, K.; Yamazaki, Y.; Murota, H.; Suzuki, K.; Tamura, A.; Bando, K.; Furuta, Y.; Katayama, I.; Tsukita, S. Dose-dependent role of claudin-1 in vivo in orchestrating features of atopic dermatitis. Proc. Natl. Acad. Sci. USA 2016, 113, E4061–E4068. [Google Scholar] [CrossRef] [Green Version]
- Uttagomol, J.; Ahmad, U.S.; Rehman, A.; Huang, Y.; Laly, A.C.; Kang, A.; Soetaert, J.; Chance, R.; Teh, M.-T.; Connelly, J.T.; et al. Evidence for the Desmosomal Cadherin Desmoglein-3 in Regulating YAP and Phospho-YAP in Keratinocyte Responses to Mechanical Forces. Int. J. Mol. Sci. 2019, 20, 6221. [Google Scholar] [CrossRef] [Green Version]
- Kobielak, A.; Boddupally, K. Junctions and Inflammation in the Skin. Cell Commun. Adhes. 2014, 21, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Samuelov, L.; Sarig, O.; Harmon, R.M.; Rapaport, D.; Ishida-Yamamoto, A.; Isakov, O.; Koetsier, J.L.; Gat, A.; Goldberg, I.; Bergman, R.; et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat. Genet. 2013, 45, 1244–1248. [Google Scholar] [CrossRef] [Green Version]
- Willebrords, J.; Yanguas, S.C.; Maes, M.; Decrock, E.; Wang, N.; Leybaert, L.; Kwak, B.R.; Green, C.R.; Cogliati, B.; Vinken, M. Connexins and their channels in inflammation. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 413–439. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Sachs, F.; Dahl, G. Connexins are mechanosensitive. Am. J. Physiol. Cell Physiol. 2004, 287, C1389-95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svenningsen, P.; Burford, J.; Peti-Peterdi, J. ATP Releasing Connexin 30 Hemichannels Mediate Flow-Induced Calcium Signaling in the Collecting Duct. Front. Physiol. 2013, 4, 292. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Riquelme, M.A.; Li, Z.; Li, Y.; Tong, Y.; Quan, Y.; Pei, C.; Gu, S.; Jiang, J.X. Mechanosensitive collaboration between integrins and connexins allows nutrient and antioxidant transport into the lens. J. Cell Biol. 2020, 219, e202002154. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, M.A.; Gu, S.; Hua, R.; Jiang, J.X. Mechanotransduction via the coordinated actions of integrins, PI3K signaling and Connexin hemichannels. Bone Res. 2021, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- van Steensel, M.A.M. Gap junction diseases of the skin. In American Journal of Medical Genetics Part C: Seminars in Medical Genetics; Wiley Subscription Services, Inc.: Hoboken, NJ, USA, 2004; Volume 131, pp. 12–19. [Google Scholar]
- Salameh, A.; Dhein, S. Effects of mechanical forces and stretch on intercellular gap junction coupling. Biochim. Biophys. Acta Biomembr. 2013, 1828, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.A.; Tattersall, D.; O’Toole, E.; Kelsell, D. Connexins in epidermal homeostasis and skin disease. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Krendel, M.; Mooseker, M.S. Myosins: Tails (and Heads) of Functional Diversity. Physiology 2005, 20, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Newell-Litwa, K.A.; Horwitz, R.; Lamers, M. Non-muscle myosin II in disease: Mechanisms and therapeutic opportunities. Dis. Model. Mech. 2015, 8, 1495–1515. [Google Scholar] [CrossRef] [Green Version]
- Walko, G.; Woodhouse, S.; Pisco, A.O.; Rognoni, E.; Liakath-Ali, K.; Lichtenberger, B.M.; Mishra, A.; Telerman, S.B.; Viswanathan, P.; Logtenberg, M.; et al. A genome-wide screen identifies YAP/WBP2 interplay conferring growth advantage on human epidermal stem cells. Nat. Commun. 2017, 8, 14744. [Google Scholar] [CrossRef] [Green Version]
- Discher, D.E.; Janmey, P.; Wang, Y.-L. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Gao, X.P.; Ramchandran, R.; Zhao, Y.Y.; Vogel, S.M.; Malik, A.B. Nonmuscle myosin light-chain kinase mediates neutrophil transmigration in sepsis-induced lung inflammation by activating β2 integrins. Nat. Immunol. 2008, 9, 880–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraning-Rush, C.M.; Carey, S.P.; Califano, J.P.; Smith, B.N.; Reinhart-King, C.A. The role of the cytoskeleton in cellular force generation in 2D and 3D environments. Phys. Biol. 2011, 8, 015009. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Kim, J.J.; Shen, J.; Dai, N. Crosstalk between Inflammation and ROCK/MLCK Signaling Pathways in Gastrointestinal Disorders with Intestinal Hyperpermeability. Gastroenterol. Res. Pract. 2016, 2016, 7374197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Srinivasan, S.; Srivastava, A.; Kumar, S.; Das, G.; Das, S.; Dwivedi, A.; Karulkar, A.; Makkad, K.; Bilala, R.; et al. Differential Influence of IL-9 and IL-17 on Actin Cytoskeleton Regulates the Migration Potential of Human Keratinocytes. J. Immunol. 2019, 202, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Borowczyk, J.; Buerger, C.; Tadjrischi, N.; Drukala, J.; Wolnicki, M.; Wnuk, D.; Modarressi, A.; Boehncke, W.-H.; Brembilla, N.C. IL-17E (IL-25) and IL-17A Differentially Affect the Functions of Human Keratinocytes. J. Investig. Dermatol. 2020, 140, 1379–1389.e2. [Google Scholar] [CrossRef]
- Burger, D.; Fickentscher, C.; De Moerloose, P.; Brandt, K.J. F-actin dampens NLRP3 inflammasome activity via Flightless-I and LRRFIP2. Sci. Rep. 2016, 6, 29834. [Google Scholar] [CrossRef] [Green Version]
- Joshi, H.; Morley, S.C. Cells under stress: The mechanical environment shapes inflammasome responses to danger signals. J. Leukoc. Biol. 2019, 106, 119–125. [Google Scholar] [CrossRef]
- Hall, A. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 2005, 33, 891–895. [Google Scholar] [CrossRef]
- Sahai, E.; Marshall, C.J. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat. Cell Biol. 2002, 4, 408–415. [Google Scholar] [CrossRef]
- Thumkeo, D.; Watanabe, S.; Narumiya, S. Physiological roles of Rho and Rho effectors in mammals. Eur. J. Cell Biol. 2013, 92, 303–315. [Google Scholar] [CrossRef] [PubMed]
- McMullan, R.; Lax, S.; Robertson, V.H.; Radford, D.J.; Broad, S.; Watt, F.M.; Rowles, A.; Croft, D.R.; Olson, M.F.; Hotchi, N.A. Keratinocyte Differentiation Is Regulated by the Rho and ROCK Signaling Pathway. Curr. Biol. 2003, 13, 2185–2189. [Google Scholar] [CrossRef] [PubMed]
- Lock, F.E.; Hotchin, N.A. Distinct Roles for ROCK1 and ROCK2 in the Regulation of Keratinocyte Differentiation. PLoS ONE 2009, 4, e8190. [Google Scholar] [CrossRef] [Green Version]
- Βpudi, V.; Rai, V.; Beach, J.R.; Egelhoff, T. Novel regulation and dynamics of myosin II activation during epidermal wound responses. Exp. Cell Res. 2010, 316, 980–991. [Google Scholar] [CrossRef] [Green Version]
- Samuel Michael, S.; Lopez Jose, I.; McGhee Ewan, J.; Croft Daniel, R.; Strachan, D.; Timpson, P.; Munro, J.; Schröder, E.; Zhou, J.; Brunton, V.G.; et al. Actomyosin-Mediated Cellular Tension Drives Increased Tissue Stiffness and β-Catenin Activation to Induce Epidermal Hyperplasia and Tumor Growth. Cancer Cell. 2011, 19, 776–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, S.; Wickström, S.A. Mechanical regulation of chromatin and transcription. Nat. Rev. Genet. 2022, 2022, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kassianidou, E.; Hughes, J.; Kumar, S. Activation of ROCK and MLCK tunes regional stress fiber formation and mechanics via preferential myosin light chain phosphorylation. Mol. Biol. Cell 2017, 28, 3832–3843. [Google Scholar] [CrossRef]
- Kazakova, O.A.; Khapchaev, A.Y.; Shirinsky, V.P. MLCK and ROCK mutualism in endothelial barrier dysfunction. Biochimie 2019, 168, 83–91. [Google Scholar] [CrossRef]
- Xiong, Y.; Wang, C.; Shi, L.; Wang, L.; Zhou, Z.; Chen, D.; Wang, J.; Guo, H. Myosin Light Chain Kinase: A Potential Target for Treatment of Inflammatory Diseases. Front. Pharmacol. 2017, 8, 292. [Google Scholar] [CrossRef] [Green Version]
- Shutova, M.S.; Svitkina, T.M. Mammalian nonmuscle myosin II comes in three flavors. Biochem. Biophys. Res. Commun. 2018, 506, 394–402. [Google Scholar] [CrossRef]
- Heuzé, M.L.; Narayana, G.H.N.S.; D’Alessandro, J.; Cellerin, V.; Dang, T.; Williams, D.S.; Van Hest, J.C.; Marcq, P.; Mège, R.-M.; Ladoux, B. Myosin II isoforms play distinct roles in adherens junction biogenesis. eLife 2019, 8, 46599. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Yuan, X.; Liu, Y.; Cai, X.; Guo, S.; Wang, A. Non-muscle Myosin II: Role in Microbial Infection and Its Potential as a Therapeutic Target. Front. Microbiol. 2019, 10, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cane, S.; Ponnappan, S.; Ponnappan, U. Impairment of non-muscle myosin IIA in human CD4+ T cells contributes to functional deficits in the elderly. Cell. Mol. Immunol. 2011, 9, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Hindman, B.; Goeckeler, Z.; Sierros, K.; Wysolmerski, R. Non-Muscle Myosin II Isoforms Have Different Functions in Matrix Rearrangement by MDA-MB-231 Cells. PLoS ONE 2015, 10, e0131920. [Google Scholar] [CrossRef]
- Bond, J.E.; Ho, T.Q.; Selim, M.A.; Hunter, C.L.; Bowers, E.V.; Levinson, H. Temporal spatial expression and function of non-muscle myosin II isoforms IIA and IIB in scar remodeling. Lab. Investig. 2010, 91, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-T.; Yin, W.; Jin, Y.-J.; Panza, P.; Gunawan, F.; Grohmann, B.; Buettner, C.; Sokol, A.M.; Preussner, J.; Guenther, S.; et al. Myh10 deficiency leads to defective extracellular matrix remodeling and pulmonary disease. Nat. Commun. 2018, 9, 4600. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-G.; Niu, J.-T.; Wu, H.-W.; Si, X.-L.; Zhang, S.-J.; Li, D.-H.; Bian, T.-T.; Li, Y.-F.; Yan, X.-K. Actin-Binding Proteins as Potential Biomarkers for Chronic Inflammation-Induced Cancer Diagnosis and Therapy. Anal. Cell. Pathol. 2021, 2021, 6692811. [Google Scholar] [CrossRef]
- Jayo, A.; Malboubi, M.; Antoku, S.; Chang, W.; Ortiz-Zapater, E.; Groen, C.; Pfisterer, K.; Tootle, T.; Charras, G.; Gundersen, G.G.; et al. Fascin Regulates Nuclear Movement and Deformation in Migrating Cells. Dev. Cell 2016, 38, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Lamb, M.C.; Tootle, T.L. Fascin in Cell Migration: More Than an Actin Bundling Protein. Biology 2020, 9, 403. [Google Scholar] [CrossRef]
- Mo, Y.-K.; Kankavi, O.; Masci, P.P.; Mellick, G.; Whitehouse, M.W.; Boyle, G.; Parsons, P.; Roberts, M.; Cross, S.E. Surfactant Protein Expression in Human Skin: Evidence and Implications. J. Investig. Dermatol. 2007, 127, 381–386. [Google Scholar] [CrossRef]
- Hsu, R.M.; Tsai, M.H.; Hsieh, Y.J.; Lyu, P.C.; Yu, J.S. Identification of MYO18A as a novel interacting partner of the PAK2/βPIX/GIT1 complex and its potential function in modulating epithelial cell migration. Mol. Biol. Cell 2010, 21, 287–301. [Google Scholar] [CrossRef] [Green Version]
- De Masson, A.; Giustiniani, J.; Marie-Cardine, A.; Bouaziz, J.D.; Dulphy, N.; Gossot, D.; Validire, P.; Tazi, A.; Garbar, C.; Bagot, M.; et al. Identification of CD245 as myosin 18A, a receptor for surfactant A: A novel pathway for activating human NK lymphocytes. Oncoimmunology 2016, 5, e1127493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Naydenov, N.G.; Feygin, A.; Baranwal, S.; Kuemmerle, J.F.; Ivanov, A.I. Actin-Depolymerizing Factor and Cofilin-1 Have Unique and Overlapping Functions in Regulating Intestinal Epithelial Junctions and Mucosal Inflammation. Am. J. Pathol. 2016, 186, 844–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, M.; Calvo, F. Regulation of mechanotransduction: Emerging roles for septins. Cytoskeleton 2018, 76, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, A.I.; Le, H.T.; Naydenov, N.G.; Rieder, F. Novel Functions of the Septin Cytoskeleton: Shaping Up Tissue Inflammation and Fibrosis. Am. J. Pathol. 2021, 191, 40–51. [Google Scholar] [CrossRef]
- Weber, G.F.; Bjerke, M.A.; DeSimone, D.W. A Mechanoresponsive Cadherin-Keratin Complex Directs Polarized Protrusive Behavior and Collective Cell Migration. Dev. Cell 2012, 22, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Ramms, L.; Fabris, G.; Windoffer, R.; Schwarz, N.; Springer, R.; Zhou, C.; Lazar, J.; Stiefel, S.; Hersch, N.; Schnakenberg, U.; et al. Keratins as the main component for the mechanical integrity of keratinocytes. Proc. Natl. Acad. Sci. USA 2013, 110, 18513–18518. [Google Scholar] [CrossRef] [Green Version]
- Sanghvi-Shah, R.; Weber, G.F. Intermediate Filaments at the Junction of Mechanotransduction, Migration, and Development. Front. Cell Dev. Biol. 2017, 5, 81. [Google Scholar] [CrossRef]
- Sapra, K.T.; Medalia, O. Bend, Push, Stretch: Remarkable Structure and Mechanics of Single Intermediate Filaments and Meshworks. Cells 2021, 10, 1960. [Google Scholar] [CrossRef]
- Zuidema, A.; Wang, W.; Sonnenberg, A. Crosstalk between Cell Adhesion Complexes in Regulation of Mechanotransduction. BioEssays 2020, 42, 2000119. [Google Scholar] [CrossRef]
- Zhang, L.-J. Keratins in skin epidermal development and diseases. In Keratin; IntechOpen: London, UK, 2018. [Google Scholar]
- Laly, A.C.; Sliogeryte, K.; Pundel, O.J.; Ross, R.; Keeling, M.C.; Avisetti, D.; Waseem, A.; Gavara, N.; Connelly, J.T. The keratin network of intermediate filaments regulates keratinocyte rigidity sensing and nuclear mechanotransduction. Sci. Adv. 2021, 7, eabd6187. [Google Scholar] [CrossRef] [PubMed]
- Haines, R.L.; Lane, E.B. Keratins and disease at a glance. J. Cell Sci. 2012, 125, 3923–3928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komine, M. Regulation of Expression of Keratins and their Pathogenic Roles in Keratinopathies. In Keratin; Blumenberg, M., Ed.; IntechOpen: London, UK, 2018. [Google Scholar]
- Leyva-Castillo, J.M.; Sun, L.; Wu, S.-Y.; Rockowitz, S.; Sliz, P.; Geha, R. Single cell transcriptome profile of mouse skin undergoing antigen driven allergic inflammation recapitulates findings in atopic dermatitis skin lesions. J. Allergy Clin. Immunol. 2022; in press. [Google Scholar] [CrossRef]
- Hobbs, R.P.; Lessard, J.C.; Coulombe, P.A. Keratin intermediate filament proteins—novel regulators of inflammation and immunity in skin. J. Cell Sci. 2012, 125, 5257–5258. [Google Scholar] [CrossRef] [Green Version]
- Roth, W.; Kumar, V.; Beer, H.-D.; Richter, M.; Wohlenberg, C.; Reuter, U.; Thiering, S.; Staratschek-Jox, A.; Hofmann, A.; Kreusch, F.; et al. Keratin 1 maintains skin integrity and participates in an inflammatory network in skin via interleukin-18. J Cell Sci. 2012, 125 Pt 22, 5269–5279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, S.; Deguchi, S.; Magin, T.M. Disease-associated keratin mutations reduce traction forces and compromise adhesion and collective migration. J. Cell Sci. 2020, 133, 243956. [Google Scholar] [CrossRef]
- Barberis, L.; Pasquali, C.; Bertschy-Meier, D.; Cuccurullo, A.; Costa, C.; Ambrogio, C.; Vilbois, F.; Chiarle, R.; Wymann, M.; Altruda, F.; et al. Leukocyte transmigration is modulated by chemokine-mediated PI3Kgamma-dependent phosphorylation of vimentin. Eur. J. Immunol. 2009, 39, 1136–1146. [Google Scholar] [CrossRef]
- Wang, L.; Mohanasundaram, P.; Lindström, M.; Asghar, M.N.; Sultana, G.; Misiorek, J.O.; Jiu, Y.; Chen, H.; Chen, Z.; Toivola, D.M.; et al. Vimentin Suppresses Inflammation and Tumorigenesis in the Mouse Intestine. Front. Cell Dev. Biol. 2022, 10, 862237. [Google Scholar] [CrossRef]
- dos Santos, G.; Rogel, M.R.; Baker, M.A.; Troken, J.R.; Urich, D.; Morales-Nebreda, L.; Sennello, J.A.; Kutuzov, M.A.; Sitikov, A.; Davis, J.M.; et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat. Commun. 2015, 6, 6574. [Google Scholar] [CrossRef] [Green Version]
- Isermann, P.; Lammerding, J. Nuclear Mechanics and Mechanotransduction in Health and Disease. Curr. Biol. 2013, 23, R1113–R1121. [Google Scholar] [CrossRef] [Green Version]
- Alam, S.G.; Zhang, Q.; Prasad, N.; Li, Y.; Chamala, S.; Kuchibhotla, R.; Kc, B.; Aggarwal, V.; Shrestha, S.; Jones, A.L.; et al. The mammalian LINC complex regulates genome transcriptional responses to substrate rigidity. Sci. Rep. 2016, 6, 38063. [Google Scholar] [CrossRef]
- Cho, S.; Irianto, J.; Discher, D.E. Mechanosensing by the nucleus: From pathways to scaling relationships. J. Cell Biol. 2017, 216, 305–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, T.J.; Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 2018, 20, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the nucleus and cytoplasm: Role of the LINC complex. J. Cell Biol. 2005, 172, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristiani, L.; Kim, M.; Kim, Y. Role of the Nuclear Lamina in Age-Associated Nuclear Reorganization and Inflammation. Cells 2020, 9, 718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.; Kuebler, W.M. Mechanotransduction by TRP Channels: General Concepts and Specific Role in the Vasculature. Cell Biophys. 2009, 56, 1. [Google Scholar] [CrossRef]
- Tóth, B.I.; Oláh, A.; Szöllősi, A.G.; Bíró, T. TRP channels in the skin. Br. J. Pharmacol. 2014, 171, 2568–2581. [Google Scholar] [CrossRef] [Green Version]
- Martinac, B.; Cox, C.D. Mechanosensory Transduction: Focus on Ion Channels. In Reference Module in Life Sciences; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Koizumi, S.; Fujishita, K.; Inoue, K.; Shigemoto-Mogami, Y.; Tsuda, M.; Inoue, K. Ca2+ waves in keratinocytes are transmitted to sensory neurons: The involvement of extracellular ATP and P2Y2 receptor activation. Biochem. J. 2004, 380, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Elias, P.M.; Ahn, S.K.; Denda, M.; Brown, B.E.; Crumrine, D.; Kimutai, L.K.; Kömüves, L.; Lee, S.H.; Feingold, K.R. Modulations in Epidermal Calcium Regulate the Expression of Differentiation-Specific Markers. J. Investig. Dermatol. 2002, 119, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Bautista, D.M.; Jordt, S.-E.; Nikai, T.; Tsuruda, P.R.; Read, A.J.; Poblete, J.; Yamoah, E.N.; Basbaum, A.I.; Julius, D. TRPA1 Mediates the Inflammatory Actions of Environmental Irritants and Proalgesic Agents. Cell 2006, 124, 1269–1282. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Sun, Y.; Li, L.; Wu, P.; Dkw, O.; Shi, H. Calcium Channels: Noteworthy Regulators and Therapeutic Targets in Dermatological Diseases. Front. Pharmacol. 2021, 12, 702264. [Google Scholar] [CrossRef]
- Leuner, K.; Kraus, M.; Wölfle, U.; Beschmann, H.; Harteneck, C.; Boehncke, W.-H.; Schempp, C.M.; Müller, W.E. Reduced TRPC Channel Expression in Psoriatic Keratinocytes Is Associated with Impaired Differentiation and Enhanced Proliferation. PLoS ONE 2011, 6, e14716. [Google Scholar] [CrossRef] [PubMed]
- Lakk, M.; Križaj, D. Mechanically induced cytoskeletal remodeling in trabecular meshwork cells requires TRPV4-Rho signaling interactions. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cambria, E.; Arlt, M.; Wandel, S.; Krupkova, O.; Hitzl, W.; Passini, F.; Hausmann, O.; Snedeker, J.; Ferguson, S.; Wuertz-Kozak, K. TRPV4 Inhibition and CRISPR-Cas9 Knockout Reduce Inflammation Induced by Hyperphysiological Stretching in Human Annulus Fibrosus Cells. Cells 2020, 9, 1736. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, J.; Zheng, Q.; Feng, X.; Zhan, F.; Wang, X.; Xu, G.; Hua, F. Piezo1 Channels as Force Sensors in Mechanical Force-Related Chronic Inflammation. Front. Immunol. 2022, 13, 816149. [Google Scholar] [CrossRef]
- Solis, A.G.; Bielecki, P.; Steach, H.R.; Sharma, L.; Harman, C.C.D.; Yun, S.; De Zoete, M.R.; Warnock, J.N.; To, S.D.F.; York, A.G.; et al. Mechanosensation of cyclical force by PIEZO1 is essential for innate immunity. Nature 2019, 573, 69–74. [Google Scholar] [CrossRef]
- Orsini, E.M.; Perelas, A.; Southern, B.D.; Grove, L.M.; Olman, M.A.; Scheraga, R.G. Stretching the Function of Innate Immune Cells. Front. Immunol. 2021, 12, 767319. [Google Scholar] [CrossRef]
- Ellefsen, K.L.; Holt, J.R.; Chang, A.C.; Nourse, J.L.; Arulmoli, J.; Mekhdjian, A.H. Myosin-II mediated traction forces evoke localized Piezo1-dependent Ca(2+) flickers. Commun. Biol. 2019, 2, 298. [Google Scholar] [CrossRef]
- Aglialoro, F.; Hofsink, N.; Hofman, M.; Brandhorst, N.; Akker, E.V.D. Inside Out Integrin Activation Mediated by PIEZO1 Signaling in Erythroblasts. Front. Physiol. 2020, 11, 958. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, J.; Yang, X.; Zhou, G.; Wang, L.; Xiao, B. Tethering Piezo channels to the actin cytoskeleton for mechanogating via the cadherin-β-catenin mechanotransduction complex. Cell Rep. 2022, 38, 110342. [Google Scholar] [CrossRef]
- Holt, J.R.; Zeng, W.Z.; Evans, E.L.; Woo, S.H.; Ma, S.; Abuwarda, H.; Loud, M.; Patapoutian, A.; Pathak, M.M. Spatiotemporal dynamics of PIEZO1 localization controls keratinocyte migration during wound healing. eLife 2021, 10, e65415. [Google Scholar] [CrossRef]
- Kim, T.-H.; Jeon, W.-Y.; Ji, Y.; Park, E.J.; Yoon, D.S.; Lee, N.-H.; Park, S.-M.; Mandakhbayar, N.; Lee, J.-H.; Lee, H.-H.; et al. Electricity auto-generating skin patch promotes wound healing process by activation of mechanosensitive ion channels. Biomaterials 2021, 275, 120948. [Google Scholar] [CrossRef] [PubMed]
- Dubin Adrienne, E.; Schmidt, M.; Mathur, J.; Petrus Matthew, J.; Xiao, B.; Coste, B.; Patapoutian, A. Inflammatory Signals Enhance Piezo2-Mediated Mechanosensitive Currents. Cell Rep. 2012, 2, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Luo, J.; Yang, P.; Du, J.; Kim, B.S.; Hu, H. Piezo2 channel–Merkel cell signaling modulates the conversion of touch to itch. Science 2018, 360, 530–533. [Google Scholar] [CrossRef] [Green Version]
- Kanoldt, V.; Fischer, L.; Grashoff, C. Unforgettable force—crosstalk and memory of mechanosensitive structures. Biol. Chem. 2018, 400, 687–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, C.-L.; Chang, W.; Bikle, D.D. The Calcium-Sensing Receptor-Dependent Regulation of Cell–Cell Adhesion and Keratinocyte Differentiation Requires Rho and Filamin, A. J. Investig. Dermatol. 2011, 131, 1119–1128. [Google Scholar] [CrossRef] [Green Version]
- Citi, S.; Guerrera, D.; Spadaro, D.; Shah, J. Epithelial junctions and Rho family GTPases: The zonular signalosome. Small GTPases 2014, 5, e973760. [Google Scholar] [CrossRef] [Green Version]
- Malakou, L.S.; Gargalionis, A.N.; Piperi, C.; Papadavid, E.; Papavassiliou, A.G.; Basdra, E.K. Molecular mechanisms of mechanotransduction in psoriasis. Ann. Transl. Med. 2018, 6, 245. [Google Scholar] [CrossRef]
- Heim, J.B.; McDonald, C.A.; Wyles, S.P.; Sominidi-Damodaran, S.; Squirewell, E.J.; Li, M.; Motsonelidze, C.; Böttcher, R.T.; Van Deursen, J.; Meves, A. FAK auto-phosphorylation site tyrosine 397 is required for development but dispensable for normal skin homeostasis. PLoS ONE 2018, 13, e0200558. [Google Scholar] [CrossRef]
- Duperret, E.K.; Dahal, A.; Ridky, T.W. Focal adhesion-independent integrin αv regulation of FAK and c-myc is necessary for 3D skin formation and tumor invasion. J. Cell Sci. 2015, 128, 3997–4013. [Google Scholar] [CrossRef] [Green Version]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gómez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK Controls Chemokine Transcription, Tregs, and Evasion of Anti-tumor Immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef] [Green Version]
- Chung, I.-C.; OuYang, C.-N.; Yuan, S.-N.; Li, H.-P.; Chen, J.-T.; Shieh, H.-R.; Chen, Y.-J.; Ojcius, D.M.; Chu, C.-L.; Yu, J.-S.; et al. Pyk2 activates the NLRP3 inflammasome by directly phosphorylating ASC and contributes to inflammasome-dependent peritonitis. Sci. Rep. 2016, 6, 36214. [Google Scholar] [CrossRef] [PubMed]
- Mimura, Y.; Ihn, H.; Jinnin, M.; Asano, Y.; Yamane, K.; Tamaki, K. Constitutive Phosphorylation of Focal Adhesion Kinase Is Involved in the Myofibroblast Differentiation of Scleroderma Fibroblasts. J. Investig. Dermatol. 2005, 124, 886–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, V.W.; Rustad, K.C.; Akaishi, S.; Sorkin, M.; Glotzbach, J.P.; Januszyk, M.; Nelson, E.R.; Levi, K.; Paterno, J.; Vial, I.N.; et al. Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat. Med. 2011, 18, 148–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.-K.; Cheng, Y.; Cheng, M.L.; Yu, L.; Mu, M.; Li, H.; Liu, Y.; Zhang, B.; Yao, Y.; Guo, H.; et al. Focal Adhesion Kinase Regulates Fibroblast Migration via Integrin β-1 and Plays a Central Role in Fibrosis. Sci. Rep. 2016, 6, 19276. [Google Scholar] [CrossRef] [Green Version]
- Baeuerle, P.A.; Henkel, T. Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef]
- Pasparakis, M. Role of NF-κB in epithelial biology. Immunol. Rev. 2012, 246, 346–358. [Google Scholar] [CrossRef]
- Khachigian, L.M.; Resnick, N.; Gimbrone, M.A.; Collins, T. Nuclear factor-kappa B interacts functionally with the platelet-derived growth factor B-chain shear-stress response element in vascular endothelial cells exposed to fluid shear stress. J. Clin. Investig. 1995, 96, 1169–1175. [Google Scholar] [CrossRef]
- Ishihara, S.; Yasuda, M.; Harada, I.; Mizutani, T.; Kawabata, K.; Haga, H. Substrate stiffness regulates temporary NF-κB activation via actomyosin contractions. Exp. Cell Res. 2013, 319, 2916–2927. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.; Aguda, B.D.; Rath, B.; Agarwal, S. Biomechanical thresholds regulate inflammation through the NF-kappaB pathway: Experiments and modeling. PLoS ONE 2009, 4, e5262. [Google Scholar] [CrossRef] [Green Version]
- Tong, L.; Tergaonkar, V. Rho protein GTPases and their interactions with NFκB: Crossroads of inflammation and matrix biology. Biosci. Rep. 2014, 34, e00115. [Google Scholar] [CrossRef]
- Németh, Z.H.; Deitch, E.A.; Davidson, M.T.; Szabó, C.; Vizi, E.S.; Haskó, G. Disruption of the actin cytoskeleton results in nuclear factor-kappaB activation and inflammatory mediator production in cultured human intestinal epithelial cells. J. Cell Physiol. 2004, 200, 71–81. [Google Scholar] [CrossRef]
- Cowell, C.F.; Yan, I.K.; Eiseler, T.; Leightner, A.C.; Döppler, H.; Storz, P. Loss of cell-cell contacts induces NF-kappaB via RhoA-mediated activation of protein kinase D1. J. Cell. Biochem. 2009, 106, 714–728. [Google Scholar] [CrossRef] [Green Version]
- Sharili Amir, S.; Connelly John, T. Nucleocytoplasmic shuttling: A common theme in mechanotransduction. Biochem. Soc. Trans. 2014, 42, 645–649. [Google Scholar] [CrossRef]
- Connelly, J.T.; Gautrot, J.E.; Trappmann, B.; Tan, D.W.-M.; Donati, G.; Huck, W.T.; Watt, F.M. Actin and serum response factor transduce physical cues from the microenvironment to regulate epidermal stem cell fate decisions. Nat. Cell Biol. 2010, 12, 711–718. [Google Scholar] [CrossRef]
- Busche, S.; Descot, A.; Julien, S.; Genth, H.; Posern, G. Epithelial cell-cell contacts regulate SRF-mediated transcription via Rac-actin-MAL signalling. J. Cell Sci. 2008, 121, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Tang, W.; Bruscia, E.M.; Zhang, P.-X.; Lin, A.; Gaines, P.; Wu, D.; Halene, S. SRF is required for neutrophil migration in response to inflammation. Blood 2014, 123, 3027–3036. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Hindes, A.; Burns, C.J.; Koppel, A.C.; Kiss, A.; Yin, Y.; Ma, L.; Blumenberg, M.; Khnykin, D.; Jahnsen, F.L.; et al. Serum Response Factor Controls Transcriptional Network Regulating Epidermal Function and Hair Follicle Morphogenesis. J. Investig. Dermatol. 2013, 133, 608–617. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [Green Version]
- Schlegelmilch, K.; Mohseni, M.; Kirak, O.; Pruszak, J.; Rodriguez, J.R.; Zhou, D.; Kreger, B.T.; Vasioukhin, V.; Avruch, J.; Brummelkamp, T.R.; et al. Yap1 Acts Downstream of α-Catenin to Control Epidermal Proliferation. Cell 2011, 144, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Rognoni, E.; Walko, G. The Roles of YAP/TAZ and the Hippo Pathway in Healthy and Diseased Skin. Cells 2019, 8, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, J.; Li, C.; Yang, J.; Wang, X.; Li, R.; Luo, S.; Li, Z.; Liu, J.; Liu, Z.; Zheng, Y. Yes-associated protein promotes the abnormal proliferation of psoriatic keratinocytes via an amphiregulin dependent pathway. Sci. Rep. 2018, 8, 14513. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Yu, Q.; Xu, H.; Dai, X.; Yu, Y.; Cui, L.; Chen, Y.; Gu, J.; Zhang, X.; Guo, C.; et al. IL-17A Promotes Psoriasis-Associated Keratinocyte Proliferation through ACT1-Dependent Activation of YAP-AREG Axis. J. Investig. Dermatol. 2022. [Google Scholar] [CrossRef]
- Toyama, T.; Looney, A.P.; Baker, B.; Stawski, L.; Haines, P.; Simms, R.; Szymaniak, A.D.; Varelas, X.; Trojanowska, M. Therapeutic Targeting of TAZ and YAP by Dimethyl Fumarate in Systemic Sclerosis Fibrosis. J. Investig. Dermatol. 2017, 138, 78–88. [Google Scholar] [CrossRef]
- Shi-Wen, X.; Racanelli, M.; Ali, A.; Simon, A.; Quesnel, K.; Stratton, R.J.; Leask, A. Verteporfin inhibits the persistent fibrotic phenotype of lesional scleroderma dermal fibroblasts. J. Cell Commun. Signal. 2021, 15, 71–80. [Google Scholar] [CrossRef]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ Incorporation in the β-Catenin Destruction Complex Orchestrates the Wnt Response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhou, L.; Ling, L.; Meng, X.; Chu, F.; Zhang, S.; Zhou, F. The Crosstalk Between Hippo-YAP Pathway and Innate Immunity. Front. Immunol. 2020, 11, 323. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar]
- Novoszel, P.; Holcmann, M.; Stulnig, G.; De Sa Fernandes, C.; Zyulina, V.; Borek, I.; Linder, M.; Bogusch, A.; Drobits, B.; Bauer, T.; et al. Psoriatic skin inflammation is promoted by c-Jun/AP-1-dependent CCL2 and IL-23 expression in dendritic cells. EMBO Mol. Med. 2021, 13, e12409. [Google Scholar] [CrossRef]
- Zenz, R.; Eferl, R.; Kenner, L.; Florin, L.; Hummerich, L.; Mehic, D.; Scheuch, H.; Angel, P.; Tschachler, E.; Wagner, E.F. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature 2005, 437, 369–375. [Google Scholar] [CrossRef]
- Park, C.C.; Kim, K.J.; Woo, S.-Y.; Chun, J.H.; Lee, K.H. Comparison of the Expression Profile of JunB, c-Jun, and S100A8 (Calgranulin A) in Psoriasis Vulgaris and Guttate Psoriasis. Ann. Dermatol. 2009, 21, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Timis, T.L.; Orasan, R.I. Understanding psoriasis: Role of miRNAs (Review). Biomed. Rep. 2018, 9, 367–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro, A.; Driscoll, T.P.; Boraas, L.C.; Armero, W.; Kasper, D.M.; Baeyens, N.; Jouy, C.; Mallikarjun, V.; Swift, J.; Ahn, S.J.; et al. MicroRNA-dependent regulation of biomechanical genes establishes tissue stiffness homeostasis. Nat. Cell Biol. 2019, 21, 348–358. [Google Scholar] [CrossRef]
- Sanchez, D.P.; Sonthalia, S. Koebner Phenomenon; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Camargo, C.M.d.S.; Brotas, A.M.; Ramos-e-Silva, M.; Carneiro, S. Isomorphic phenomenon of Koebner: Facts and controversies. Clin. Dermatol. 2013, 31, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Trepat, X.; Deng, L.; An, S.; Navajas, D.; Tschumperlin, D.J.; Gerthoffer, W.T.; Butler, J.P.; Fredberg, J.J. Universal physical responses to stretch in the living cell. Nature 2007, 447, 592–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, S.; Komine, M.; Fujimoto, M.; Okochi, H.; Tamaki, K. Mechanical Stretching In Vitro Regulates Signal Transduction Pathways and Cellular Proliferation in Human Epidermal Keratinocytes. J. Investig. Dermatol. 2004, 122, 783–790. [Google Scholar] [CrossRef] [Green Version]
- Yano, S.; Komine, M.; Fujimoto, M.; Okochi, H.; Tamaki, K. Activation of Akt by mechanical stretching in human epidermal keratinocytes. Exp. Dermatol. 2006, 15, 356–361. [Google Scholar] [CrossRef]
- Zhang, H.; Landmann, F.; Zahreddine, H.; Rodriguez, D.; Koch, M.; Labouesse, M. A tension-induced mechanotransduction pathway promotes epithelial morphogenesis. Nature 2011, 471, 99–103. [Google Scholar] [CrossRef]
- Skutek, M.; van Griensven, M.; Zeichen, J.; Brauer, N.; Bosch, U. Cyclic mechanical stretching enhances secretion of Interleukin 6 in human tendon fibroblasts. Knee Surg. Sports Traumatol. Arthrosc. 2001, 9, 322–326. [Google Scholar] [CrossRef]
- Yang, G.; Im, H.-J.; Wang, J.H.C. Repetitive mechanical stretching modulates IL-1β induced COX-2, MMP-1 expression, and PGE2 production in human patellar tendon fibroblasts. Gene 2005, 363, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Matsumori, A.; Ono, K.; Furukawa, Y.; Shioi, T.; Iwasaki, A.; Matsushima, K.; Sasayama, S. Cyclic Stretch Upregulates Production of Interleukin-8 and Monocyte Chemotactic and Activating Factor/Monocyte Chemoattractant Protein-1 in Human Endothelial Cells. Arter. Thromb. Vasc. Biol. 1998, 18, 894–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, R.; Hsu, C.-K. Mechanobiological dysregulation of the epidermis and dermis in skin disorders and in degeneration. J. Cell. Mol. Med. 2013, 17, 817–822. [Google Scholar] [CrossRef] [PubMed]




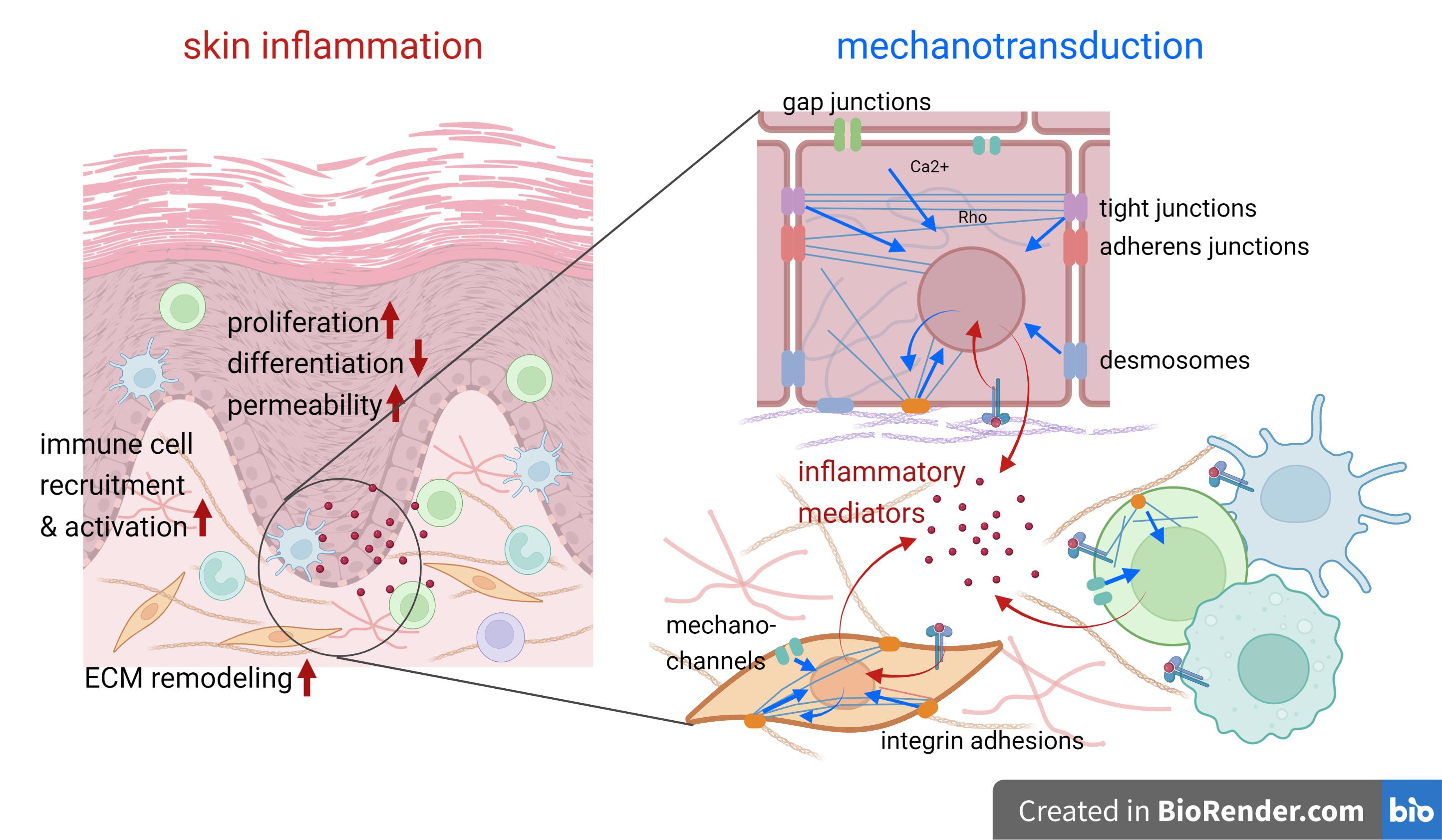{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ECM Protein | Location | Inflammation-Associated Changes | Significance for Pathology | References |
|---|---|---|---|---|
| Collagen I and III | Dermis | Increased in chronic inflammation and fibrosis | Increased skin stiffness and integrin signaling in fibroblasts | [21,30,50,51,52,53] |
| Collagen IV and VII | BM | Disorganization of the fibrils; decreased expression in psoriasis | Increased dermal–epidermal permeability; affects integrin signaling in basal keratinocytes | [38,51,52] |
| Fibronectin (cellular) | Dermis, BM, epidermis | Increased in dermis during fibrosis; found in epidermis in AD | Increased skin stiffness and integrin signaling in fibroblasts; can promote bacterial colonisation in AD epidermis | [37,46] |
| Fibronectin (EDA+ cellular) | Dermis | Increased in inflammation (psoriasis) and fibrosis | Angiogenesis; tissue remodelling; fibroblast differentiation | [35,37,44,45] |
| Fibronectin (plasma) | Epidermis | Accumulates during inflammation (psoriasis) | Signaling and proliferation in basal keratinocytes | [43] |
| Laminin | BM | Overexpression; disorganized structure (psoriasis) | Increased immune cell adhesion; keratinocyte proliferation | [21,38] |
| Periostin | Dermis, BM | Increased expression during inflammation (AD) but not in psoriasis | Modulates dermal cell adhesion and signaling; stimulates cell proliferation; contributes to itch | [21,36,47,48,49] |
| Tenascin | Dermis | Increased expression during inflammation (AD, psoriasis) | Modulates dermal cell adhesion and signalling; stimulates cell proliferation; induces pro-inflammatory pathways | [21,33,34,40,41,42] |
| Integrin Complex | Ligand | Expressing Cells | Inflammation-Associated Changes | Significance for Pathology | References |
|---|---|---|---|---|---|
| α1β1 | Collagen | Epidermal T-cells, fibroblasts, endothelial cells | Increased T cell and macrophage recruitment to inflammation sites | Pro-inflammatory effects | [71,72] |
| α2β1 | Collagen | Keratinocytes, fibroblasts, endothelial cells | Upregulated in all epidermal layers in wound healing and psoriasis | Increased keratinocyte proliferation and altered differentiation | [65,70] |
| α3β1 | Laminin | Keratinocytes | Upregulated in all epidermal layers in wound healing and psoriasis | Increased keratinocyte proliferation and altered differentiation; possible regulation of fibrosis | [65,70,80] |
| α5β1 | Fibronectin | Keratinocytes (low in homeostasis), dermal fibroblasts, endothelial cells | Upregulated in basal and suprabasal keratinocytes during wound healing and in psoriasis; upregulated in fibroblasts under TGF-β | Fibroblast activation; increased keratinocyte proliferation and altered differentiation | [64,65,66,67,68,70,81] |
| α9β1 | Tenascin | Keratinocytes (low in homeostasis), endothelial cells | Upregulated during wound healing | Increased keratinocyte proliferation and altered differentiation | [61] |
| α6β4 (hemidesmosomal) | Laminin | Basal keratinocytes, endothelial cells | Upregulated in psoriatic epidermis; decreased in psoriatic vasculature | Increased keratinocyte proliferation and altered differentiation | [65,70,73] |
| αvβ3 | Vitronectin, fibronectin | Endothelial cells, fibroblasts | Increased in psoriatic skin vasculature | Pro-inflammatory effects | [73] |
| αvβ5 | Vitronectin | Fibroblasts, keratinocytes (low expression) | Upregulated in fibrosis | TGF-β activation; pro-fibrotic effects | [61,80] |
| αvβ6 | Fibronectin, tenascin | Keratinocytes (low in homeostasis) | Upregulated during wound healing | TGF-β activation; pro-fibrotic effects | [61,79] |
| αvβ8 | Vitronectin | Suprabasal keratinocytes, dermal fibroblasts | Upregulated in fibrosis | TGF-β activation; pro-fibrotic effects | [61,80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shutova, M.S.; Boehncke, W.-H. Mechanotransduction in Skin Inflammation. Cells 2022, 11, 2026. https://doi.org/10.3390/cells11132026
Shutova MS, Boehncke W-H. Mechanotransduction in Skin Inflammation. Cells. 2022; 11(13):2026. https://doi.org/10.3390/cells11132026
Chicago/Turabian StyleShutova, Maria S., and Wolf-Henning Boehncke. 2022. "Mechanotransduction in Skin Inflammation" Cells 11, no. 13: 2026. https://doi.org/10.3390/cells11132026
APA StyleShutova, M. S., & Boehncke, W. -H. (2022). Mechanotransduction in Skin Inflammation. Cells, 11(13), 2026. https://doi.org/10.3390/cells11132026