
Skeletal Muscle Pathogenesis in Polyglutamine Diseases

 , ,
, , 
Abstract
:1. Introduction
2. Polyglutamine Diseases: Clinical Presentation and Disease Pathogenesis
3. Muscle Pathology in Polyglutamine Disease Patients
4. Skeletal Muscle Pathology Is Recapitulated in Mouse Models of Polyglutamine Diseases
5. Pathological Processes in the Skeletal Muscle in Polyglutamine Diseases
6. Modeling Skeletal Muscle in a Dish
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Stoyas, C.A.; La Spada, A.R. The CAG-polyglutamine repeat diseases: A clinical, molecular, genetic, and pathophysiologic nosology. Handb. Clin. Neurol. 2018, 147, 143–170. [Google Scholar] [CrossRef] [PubMed]
- Bunting, E.L.; Hamilton, J.; Tabrizi, S.J. Polyglutamine diseases. Curr. Opin. Neurobiol. 2022, 72, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Tanzi, R.E. The genetic epidemiology of neurodegenerative disease. J. Clin. Investig. 2005, 115, 1449–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- GBD 2017 Risk Factor Collaborators. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1923–1994. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, R.; Tabrizi, S.J. Clinical Features of Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 1–28. [Google Scholar] [CrossRef]
- Mao, R.; Aylsworth, A.S.; Potter, N.; Wilson, W.G.; Breningstall, G.; Wick, M.J.; Babovic-Vuksanovic, D.; Nance, M.; Patterson, M.C.; Gomez, C.M.; et al. Childhood-onset ataxia: Testing for large CAG-repeats in SCA2 and SCA7. Am. J. Med. Genet. 2002, 110, 338–345. [Google Scholar] [CrossRef]
- Roselli, F.; Caroni, P. From intrinsic firing properties to selective neuronal vulnerability in neurodegenerative diseases. Neuron 2015, 85, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Huang, S.; Yin, P.; Yang, S.; Zhang, J.; Jing, L.; Cheng, S.; Tang, B.; Li, X.J.; Pan, Y.; et al. Cerebellum-enriched protein INPP5A contributes to selective neuropathology in mouse model of spinocerebellar ataxias type 17. Nat. Commun. 2020, 11, 1101. [Google Scholar] [CrossRef]
- Pearce, M.M.P.; Kopito, R.R. Prion-Like Characteristics of Polyglutamine-Containing Proteins. Cold Spring Harb. Perspect Med. 2018, 8, a024257. [Google Scholar] [CrossRef] [Green Version]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennuto, M.; Rinaldi, C. From gene to therapy in spinal and bulbar muscular atrophy: Are we there yet? Mol. Cell Endocrinol. 2018, 465, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, S. Dentatorubral-pallidoluysian atrophy. Handb. Clin. Neurol. 2012, 103, 587–594. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Orr, H.T. Spinocerebellar Ataxia Type 1: Molecular Mechanisms of Neurodegeneration and Preclinical Studies. Adv. Exp. Med. Biol. 2018, 1049, 135–145. [Google Scholar] [CrossRef]
- Egorova, P.A.; Bezprozvanny, I.B. Molecular Mechanisms and Therapeutics for Spinocerebellar Ataxia Type 2. Neurotherapeutics 2019, 16, 1050–1073. [Google Scholar] [CrossRef]
- McLoughlin, H.S.; Moore, L.R.; Paulson, H.L. Pathogenesis of SCA3 and implications for other polyglutamine diseases. Neurobiol. Dis. 2020, 134, 104635. [Google Scholar] [CrossRef]
- Casey, H.L.; Gomez, C.M. Spinocerebellar Ataxia Type 6. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; Genetic and Rare Diseases Information Center: Seattle, DC, USA, 1993. [Google Scholar]
- La Spada, A.R. Spinocerebellar Ataxia Type 7. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; Genetic and Rare Diseases Information Center: Seattle, DC, USA, 1993. [Google Scholar]
- Liu, Q.; Pan, Y.; Li, X.J.; Li, S. Molecular Mechanisms and Therapeutics for SCA17. Neurotherapeutics 2019, 16, 1097–1105. [Google Scholar] [CrossRef]
- Hashizume, A.; Fischbeck, K.H.; Pennuto, M.; Fratta, P.; Katsuno, M. Disease mechanism, biomarker and therapeutics for spinal and bulbar muscular atrophy (SBMA). J. Neurol. Neurosurg. Psychiatry 2020, 91, 1085–1091. [Google Scholar] [CrossRef]
- Klickovic, U.; Zampedri, L.; Sinclair, C.D.J.; Wastling, S.J.; Trimmel, K.; Howard, R.S.; Malaspina, A.; Sharma, N.; Sidle, K.; Emira, A.; et al. Skeletal muscle MRI differentiates SBMA and ALS and correlates with disease severity. Neurology 2019, 93, e895–e907. [Google Scholar] [CrossRef] [Green Version]
- Palazzolo, I.; Stack, C.; Kong, L.; Musaro, A.; Adachi, H.; Katsuno, M.; Sobue, G.; Taylor, J.P.; Sumner, C.J.; Fischbeck, K.H.; et al. Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron 2009, 63, 316–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, F.J.; Merry, D.E. Molecular Mechanisms and Therapeutics for SBMA/Kennedy’s Disease. Neurotherapeutics 2019, 16, 928–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monks, D.A.; Holmes, M.M. Androgen receptors and muscle: A key mechanism underlying life history trade-offs. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 2018, 204, 51–60. [Google Scholar] [CrossRef]
- Pronsato, L.; Boland, R.; Milanesi, L. Non-classical localization of androgen receptor in the C2C12 skeletal muscle cell line. Arch. Biochem. Biophys. 2013, 530, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Borgia, D.; Malena, A.; Spinazzi, M.; Desbats, M.A.; Salviati, L.; Russell, A.P.; Miotto, G.; Tosatto, L.; Pegoraro, E.; Soraru, G.; et al. Increased mitophagy in the skeletal muscle of spinal and bulbar muscular atrophy patients. Hum. Mol. Genet. 2017, 26, 1087–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntosh, C.S.; Li, D.; Wilton, S.D.; Aung-Htut, M.T. Polyglutamine Ataxias: Our Current Molecular Understanding and What the Future Holds for Antisense Therapies. Biomedicines 2021, 9, 1499. [Google Scholar] [CrossRef]
- Sambataro, F.; Pennuto, M. Cell-autonomous and non-cell-autonomous toxicity in polyglutamine diseases. Prog. Neurobiol. 2012, 97, 152–172. [Google Scholar] [CrossRef]
- Huang, S.; Zhu, S.; Li, X.J.; Li, S. The Expanding Clinical Universe of Polyglutamine Disease. Neuroscientist 2019, 25, 512–520. [Google Scholar] [CrossRef]
- Carroll, J.B.; Bates, G.P.; Steffan, J.; Saft, C.; Tabrizi, S.J. Treating the whole body in Huntington’s disease. Lancet Neurol. 2015, 14, 1135–1142. [Google Scholar] [CrossRef]
- Ansved, T.; Lundin, A.; Anvret, M. Larger CAG expansions in skeletal muscle compared with lymphocytes in Kennedy disease but not in Huntington disease. Neurology 1998, 51, 1442–1444. [Google Scholar] [CrossRef]
- Costa de Miranda, R.; Di Lorenzo, N.; Andreoli, A.; Romano, L.; De Santis, G.L.; Gualtieri, P.; De Lorenzo, A. Body composition and bone mineral density in Huntington’s disease. Nutrition 2019, 59, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Djousse, L.; Knowlton, B.; Cupples, L.A.; Marder, K.; Shoulson, I.; Myers, R.H. Weight loss in early stage of Huntington’s disease. Neurology 2002, 59, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.M.; Wolfson, T.; Peavy, G.M.; Jacobson, M.W.; Corey-Bloom, J.; Huntington Study, G. Rate and correlates of weight change in Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2004, 75, 209–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busse, M.E.; Hughes, G.; Wiles, C.M.; Rosser, A.E. Use of hand-held dynamometry in the evaluation of lower limb muscle strength in people with Huntington’s disease. J. Neurol. 2008, 255, 1534–1540. [Google Scholar] [CrossRef] [Green Version]
- Cubo, E.; Rivadeneyra, J.; Gil-Polo, C.; Armesto, D.; Mateos, A.; Mariscal-Perez, N. Body composition analysis as an indirect marker of skeletal muscle mass in Huntington’s disease. J. Neurol. Sci. 2015, 358, 335–338. [Google Scholar] [CrossRef]
- Mielcarek, M. Huntington’s disease is a multi-system disorder. Rare Dis. 2015, 3, e1058464. [Google Scholar] [CrossRef]
- Kosinski, C.M.; Schlangen, C.; Gellerich, F.N.; Gizatullina, Z.; Deschauer, M.; Schiefer, J.; Young, A.B.; Landwehrmeyer, G.B.; Toyka, K.V.; Sellhaus, B.; et al. Myopathy as a first symptom of Huntington’s disease in a Marathon runner. Mov. Disord. 2007, 22, 1637–1640. [Google Scholar] [CrossRef]
- Cox, H.; Costin-Kelly, N.M.; Ramani, P.; Whitehouse, W.P. An established case of dentatorubral pallidoluysian atrophy (DRPLA) with unusual features on muscle biopsy. Eur. J. Paediatr. Neurol. 2000, 4, 119–123. [Google Scholar] [CrossRef]
- Rocchi, A.; Milioto, C.; Parodi, S.; Armirotti, A.; Borgia, D.; Pellegrini, M.; Urciuolo, A.; Molon, S.; Morbidoni, V.; Marabita, M.; et al. Glycolytic-to-oxidative fiber-type switch and mTOR signaling activation are early-onset features of SBMA muscle modified by high-fat diet. Acta Neuropathol. 2016, 132, 127–144. [Google Scholar] [CrossRef]
- Manzano, R.; Soraru, G.; Grunseich, C.; Fratta, P.; Zuccaro, E.; Pennuto, M.; Rinaldi, C. Beyond motor neurons: Expanding the clinical spectrum in Kennedy’s disease. J. Neurol. Neurosurg. Psychiatry 2018, 89, 808–812. [Google Scholar] [CrossRef]
- Dahlqvist, J.R.; Oestergaard, S.T.; Poulsen, N.S.; Knak, K.L.; Thomsen, C.; Vissing, J. Muscle contractility in spinobulbar muscular atrophy. Sci. Rep. 2019, 9, 4680. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.E.; Thomas, P.K.; Baraitser, M.; Bradbury, P.G.; Morgan-Hughes, J.A.; Ponsford, J.R. X-linked recessive bulbospinal neuronopathy: A report of ten cases. J. Neurol. Neurosurg. Psychiatry 1982, 45, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chahin, N.; Sorenson, E.J. Serum creatine kinase levels in spinobulbar muscular atrophy and amyotrophic lateral sclerosis. Muscle Nerve 2009, 40, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, E.J.; Klein, C.J. Elevated creatine kinase and transaminases in asymptomatic SBMA. Amyotroph. Lateral Scler. 2007, 8, 62–64. [Google Scholar] [CrossRef]
- Suzuki, K.; Katsuno, M.; Banno, H.; Takeuchi, Y.; Atsuta, N.; Ito, M.; Watanabe, H.; Yamashita, F.; Hori, N.; Nakamura, T.; et al. CAG repeat size correlates to electrophysiological motor and sensory phenotypes in SBMA. Brain 2008, 131, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lim, Y.M.; Lee, E.J.; Oh, Y.J.; Kim, K.K. Correlation between the CAG repeat size and electrophysiological findings in patients with spinal and bulbar muscular atrophy. Muscle Nerve 2018, 57, 683–686. [Google Scholar] [CrossRef]
- Rhodes, L.E.; Freeman, B.K.; Auh, S.; Kokkinis, A.D.; La Pean, A.; Chen, C.; Lehky, T.J.; Shrader, J.A.; Levy, E.W.; Harris-Love, M.; et al. Clinical features of spinal and bulbar muscular atrophy. Brain 2009, 132, 3242–3251. [Google Scholar] [CrossRef] [Green Version]
- Forouhan, M.; Lim, W.F.; Zanetti-Domingues, L.C.; Tynan, C.J.; Roberts, T.C.; Malik, B.; Manzano, R.; Speciale, A.A.; Ellerington, R.; Garcia-Guerra, A.; et al. AR cooperates with SMAD4 to maintain skeletal muscle homeostasis. Acta Neuropathol. 2022, 143, 713–731. [Google Scholar] [CrossRef]
- Maltecca, F.; Filla, A.; Castaldo, I.; Coppola, G.; Fragassi, N.A.; Carella, M.; Bruni, A.; Cocozza, S.; Casari, G.; Servadio, A.; et al. Intergenerational instability and marked anticipation in SCA-17. Neurology 2003, 61, 1441–1443. [Google Scholar] [CrossRef]
- Koide, R.; Kobayashi, S.; Shimohata, T.; Ikeuchi, T.; Maruyama, M.; Saito, M.; Yamada, M.; Takahashi, H.; Tsuji, S. A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: A new polyglutamine disease? Hum. Mol. Genet. 1999, 8, 2047–2053. [Google Scholar] [CrossRef] [Green Version]
- Valadao, P.A.C.; de Aragao, B.C.; Andrade, J.N.; Magalhaes-Gomes, M.P.S.; Foureaux, G.; Joviano-Santos, J.V.; Nogueira, J.C.; Machado, T.C.G.; de Jesus, I.C.G.; Nogueira, J.M.; et al. Abnormalities in the Motor Unit of a Fast-Twitch Lower Limb Skeletal Muscle in Huntington’s Disease. ASN Neuro. 2019, 11, 1759091419886212. [Google Scholar] [CrossRef] [PubMed]
- Valadao, P.A.C.; Gomes, M.; Aragao, B.C.; Rodrigues, H.A.; Andrade, J.N.; Garcias, R.; Joviano-Santos, J.V.; Luiz, M.A.; Camargo, W.L.; Naves, L.A.; et al. Corrigendum to “Neuromuscular synapse degeneration without muscle function loss in the diaphragm of a murine model for Huntington’s Disease”. Neurochem. Int. 2018, 118, 264. [Google Scholar] [CrossRef] [PubMed]
- de Aragao, B.C.; Rodrigues, H.A.; Valadao, P.A.; Camargo, W.; Naves, L.A.; Ribeiro, F.M.; Guatimosim, C. Changes in structure and function of diaphragm neuromuscular junctions from BACHD mouse model for Huntington’s disease. Neurochem. Int. 2016, 93, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Ciammola, A.; Sassone, J.; Alberti, L.; Meola, G.; Mancinelli, E.; Russo, M.A.; Squitieri, F.; Silani, V. Increased apoptosis, Huntingtin inclusions and altered differentiation in muscle cell cultures from Huntington’s disease subjects. Cell Death Differ. 2006, 13, 2068–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malena, A.; Pennuto, M.; Tezze, C.; Querin, G.; D’Ascenzo, C.; Silani, V.; Cenacchi, G.; Scaramozza, A.; Romito, S.; Morandi, L.; et al. Androgen-dependent impairment of myogenesis in spinal and bulbar muscular atrophy. Acta Neuropathol. 2013, 126, 109–121. [Google Scholar] [CrossRef]
- Sathasivam, K.; Hobbs, C.; Turmaine, M.; Mangiarini, L.; Mahal, A.; Bertaux, F.; Wanker, E.E.; Doherty, P.; Davies, S.W.; Bates, G.P. Formation of polyglutamine inclusions in non-CNS tissue. Hum. Mol. Genet. 1999, 8, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Menalled, L.B.; Kudwa, A.E.; Miller, S.; Fitzpatrick, J.; Watson-Johnson, J.; Keating, N.; Ruiz, M.; Mushlin, R.; Alosio, W.; McConnell, K.; et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PLoS ONE 2012, 7, e49838. [Google Scholar] [CrossRef]
- Huang, S.; Yang, S.; Guo, J.; Yan, S.; Gaertig, M.A.; Li, S.; Li, X.J. Large Polyglutamine Repeats Cause Muscle Degeneration in SCA17 Mice. Cell Rep. 2015, 13, 196–208. [Google Scholar] [CrossRef] [Green Version]
- Chivet, M.; Marchioretti, C.; Pirazzini, M.; Piol, D.; Scaramuzzino, C.; Polanco, M.J.; Romanello, V.; Zuccaro, E.; Parodi, S.; D’Antonio, M.; et al. Polyglutamine-Expanded Androgen Receptor Alteration of Skeletal Muscle Homeostasis and Myonuclear Aggregation Are Affected by Sex, Age and Muscle Metabolism. Cells 2020, 9, 325. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Dadgar, N.; Albertelli, M.; Gruis, K.; Jordan, C.; Robins, D.M.; Lieberman, A.P. Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J. Clin. Investig. 2006, 116, 2663–2672. [Google Scholar] [CrossRef]
- Katsuno, M.; Adachi, H.; Kume, A.; Li, M.; Nakagomi, Y.; Niwa, H.; Sang, C.; Kobayashi, Y.; Doyu, M.; Sobue, G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 2002, 35, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Ribchester, R.R.; Thomson, D.; Wood, N.I.; Hinks, T.; Gillingwater, T.H.; Wishart, T.M.; Court, F.A.; Morton, A.J. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington’s disease mutation. Eur. J. Neurosci. 2004, 20, 3092–3114. [Google Scholar] [CrossRef] [PubMed]
- Lodi, R.; Schapira, A.H.; Manners, D.; Styles, P.; Wood, N.W.; Taylor, D.J.; Warner, T.T. Abnormal in vivo skeletal muscle energy metabolism in Huntington’s disease and dentatorubropallidoluysian atrophy. Ann. Neurol. 2000, 48, 72–76. [Google Scholar] [CrossRef]
- Strand, A.D.; Aragaki, A.K.; Shaw, D.; Bird, T.; Holton, J.; Turner, C.; Tapscott, S.J.; Tabrizi, S.J.; Schapira, A.H.; Kooperberg, C.; et al. Gene expression in Huntington’s disease skeletal muscle: A potential biomarker. Hum. Mol. Genet. 2005, 14, 1863–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgetti, E.; Yu, Z.; Chua, J.P.; Shimamura, R.; Zhao, L.; Zhu, F.; Venneti, S.; Pennuto, M.; Guan, Y.; Hung, G.; et al. Rescue of Metabolic Alterations in AR113Q Skeletal Muscle by Peripheral Androgen Receptor Gene Silencing. Cell Rep. 2016, 17, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Braubach, P.; Orynbayev, M.; Andronache, Z.; Hering, T.; Landwehrmeyer, G.B.; Lindenberg, K.S.; Melzer, W. Altered Ca(2+) signaling in skeletal muscle fibers of the R6/2 mouse, a model of Huntington’s disease. J. Gen. Physiol. 2014, 144, 393–413. [Google Scholar] [CrossRef]
- Xu, Y.; Halievski, K.; Henley, C.; Atchison, W.D.; Katsuno, M.; Adachi, H.; Sobue, G.; Breedlove, S.M.; Jordan, C.L. Defects in Neuromuscular Transmission May Underlie Motor Dysfunction in Spinal and Bulbar Muscular Atrophy. J. Neurosci. 2016, 36, 5094–5106. [Google Scholar] [CrossRef] [Green Version]
- Poort, J.E.; Rheuben, M.B.; Breedlove, S.M.; Jordan, C.L. Neuromuscular junctions are pathological but not denervated in two mouse models of spinal bulbar muscular atrophy. Hum. Mol. Genet. 2016, 25, 3768–3783. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.J.; Ling, S.C.; Guo, L.T.; Hung, G.; Tsunemi, T.; Ly, L.; Tokunaga, S.; Lopez, E.; Sopher, B.L.; Bennett, C.F.; et al. Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron 2014, 82, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Monks, D.A.; Johansen, J.A.; Mo, K.; Rao, P.; Eagleson, B.; Yu, Z.; Lieberman, A.P.; Breedlove, S.M.; Jordan, C.L. Overexpression of wild-type androgen receptor in muscle recapitulates polyglutamine disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18259–18264. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, V.; Querin, G.; Ziff, O.J.; Zampedri, L.; Martinelli, I.; Heller, C.; Foiani, M.; Bertolin, C.; Lu, C.H.; Malik, B.; et al. Muscle and not neuronal biomarkers correlate with severity in spinal and bulbar muscular atrophy. Neurology 2019, 92, e1205–e1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman, A.P.; Yu, Z.; Murray, S.; Peralta, R.; Low, A.; Guo, S.; Yu, X.X.; Cortes, C.J.; Bennett, C.F.; Monia, B.P.; et al. Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep. 2014, 7, 774–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milioto, C.; Malena, A.; Maino, E.; Polanco, M.J.; Marchioretti, C.; Borgia, D.; Pereira, M.G.; Blaauw, B.; Lieberman, A.P.; Venturini, R.; et al. Beta-agonist stimulation ameliorates the phenotype of spinal and bulbar muscular atrophy mice and patient-derived myotubes. Sci. Rep. 2017, 7, 41046. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, H.; McPhail, G.D.; Woodman, B.; Hobbs, C.; Bates, G.P. Formation of polyglutamine inclusions in a wide range of non-CNS tissues in the HdhQ150 knock-in mouse model of Huntington’s disease. PLoS ONE 2009, 4, e8025. [Google Scholar] [CrossRef] [PubMed]
- Kojer, K.; Hering, T.; Bazenet, C.; Weiss, A.; Herrmann, F.; Taanman, J.W.; Orth, M. Huntingtin Aggregates and Mitochondrial Pathology in Skeletal Muscle but not Heart of Late-Stage R6/2 Mice. J. Huntingtons Dis. 2019, 8, 145–159. [Google Scholar] [CrossRef]
- Nath, S.R.; Lieberman, M.L.; Yu, Z.; Marchioretti, C.; Jones, S.T.; Danby, E.C.E.; Van Pelt, K.M.; Soraru, G.; Robins, D.M.; Bates, G.P.; et al. MEF2 impairment underlies skeletal muscle atrophy in polyglutamine disease. Acta Neuropathol. 2020, 140, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Badders, N.M.; Korff, A.; Miranda, H.C.; Vuppala, P.K.; Smith, R.B.; Winborn, B.J.; Quemin, E.R.; Sopher, B.L.; Dearman, J.; Messing, J.; et al. Selective modulation of the androgen receptor AF2 domain rescues degeneration in spinal bulbar muscular atrophy. Nat. Med. 2018, 24, 427–437. [Google Scholar] [CrossRef]
- Adachi, H.; Katsuno, M.; Minamiyama, M.; Waza, M.; Sang, C.; Nakagomi, Y.; Kobayashi, Y.; Tanaka, F.; Doyu, M.; Inukai, A.; et al. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain 2005, 128, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Olivé, M.; Winter, L.; Fürst, D.O.; Schröder, R.; Behin, A.; Breukel, A.; Brumhard, M.; Bryson-Richardson, R.; Claeys, K.; Ferreiro, A.; et al. 246th ENMC International Workshop: Protein aggregate myopathies 24–26 May 2019, Hoofddorp, The Netherlands. Neuromuscul. Disord. 2021, 31, 158–166. [Google Scholar] [CrossRef]
- van der Sluijs, B.M.; Raz, V.; Lammens, M.; van den Heuvel, L.P.; Voermans, N.C.; van Engelen, B.G. Intranuclear Aggregates Precede Clinical Onset in Oculopharyngeal Muscular Dystrophy. J. Neuromuscul. Dis. 2016, 3, 101–109. [Google Scholar] [CrossRef]
- Vogler, T.O.; Wheeler, J.R.; Nguyen, E.D.; Hughes, M.P.; Britson, K.A.; Lester, E.; Rao, B.; Betta, N.D.; Whitney, O.N.; Ewachiw, T.E.; et al. TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature 2018, 563, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Becanovic, K.; Asghar, M.; Gadawska, I.; Sachdeva, S.; Walker, D.; Lazarowski, E.R.; Franciosi, S.; Park, K.H.J.; Cote, H.C.F.; Leavitt, B.R. Age-related mitochondrial alterations in brain and skeletal muscle of the YAC128 model of Huntington disease. NPJ Aging Mech. Dis. 2021, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Squitieri, F.; Cannella, M.; Sgarbi, G.; Maglione, V.; Falleni, A.; Lenzi, P.; Baracca, A.; Cislaghi, G.; Saft, C.; Ragona, G.; et al. Severe ultrastructural mitochondrial changes in lymphoblasts homozygous for Huntington disease mutation. Mech. Ageing Dev. 2006, 127, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Saft, C.; Zange, J.; Andrich, J.; Muller, K.; Lindenberg, K.; Landwehrmeyer, B.; Vorgerd, M.; Kraus, P.H.; Przuntek, H.; Schols, L. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington’s disease. Mov. Disord. 2005, 20, 674–679. [Google Scholar] [CrossRef]
- Ranganathan, S.; Harmison, G.G.; Meyertholen, K.; Pennuto, M.; Burnett, B.G.; Fischbeck, K.H. Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2009, 18, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Pourshafie, N.; Masati, E.; Bunker, E.; Nickolls, A.R.; Thepmankorn, P.; Johnson, K.; Feng, X.; Ekins, T.; Grunseich, C.; Fischbeck, K.H. Linking epigenetic dysregulation, mitochondrial impairment, and metabolic dysfunction in SBMA motor neurons. JCI Insight 2020, 5, e136539. [Google Scholar] [CrossRef]
- Beauchemin, A.M.; Gottlieb, B.; Beitel, L.K.; Elhaji, Y.A.; Pinsky, L.; Trifiro, M.A. Cytochrome c oxidase subunit Vb interacts with human androgen receptor: A potential mechanism for neurotoxicity in spinobulbar muscular atrophy. Brain Res. Bull. 2001, 56, 285–297. [Google Scholar] [CrossRef]
- Orsucci, D.; Rocchi, A.; Caldarazzo Ienco, E.; Ali, G.; LoGerfo, A.; Petrozzi, L.; Scarpelli, M.; Filosto, M.; Carlesi, C.; Siciliano, G.; et al. Myopathic involvement and mitochondrial pathology in Kennedy disease and in other motor neuron diseases. Curr. Mol. Med. 2014, 14, 598–602. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Adhihetty, P.; Shukla, S.; Hennessy, T.; Calingasan, N.; Yang, L.; Starkov, A.; Kiaei, M.; Cannella, M.; Sassone, J.; et al. Impaired PGC-1alpha function in muscle in Huntington’s disease. Hum. Mol. Genet. 2009, 18, 3048–3065. [Google Scholar] [CrossRef] [Green Version]
- Johri, A.; Calingasan, N.Y.; Hennessey, T.M.; Sharma, A.; Yang, L.; Wille, E.; Chandra, A.; Beal, M.F. Pharmacologic activation of mitochondrial biogenesis exerts widespread beneficial effects in a transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 2012, 21, 1124–1137. [Google Scholar] [CrossRef] [Green Version]
- Bajpai, P.; Koc, E.; Sonpavde, G.; Singh, R.; Singh, K.K. Mitochondrial localization, import, and mitochondrial function of the androgen receptor. J. Biol. Chem. 2019, 294, 6621–6634. [Google Scholar] [CrossRef] [PubMed]
- Luthi-Carter, R.; Hanson, S.A.; Strand, A.D.; Bergstrom, D.A.; Chun, W.; Peters, N.L.; Woods, A.M.; Chan, E.Y.; Kooperberg, C.; Krainc, D.; et al. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: Parallel changes in muscle and brain. Hum. Mol. Genet. 2002, 11, 1911–1926. [Google Scholar] [CrossRef] [PubMed]
- Hering, T.; Braubach, P.; Landwehrmeyer, G.B.; Lindenberg, K.S.; Melzer, W. Fast-to-Slow Transition of Skeletal Muscle Contractile Function and Corresponding Changes in Myosin Heavy and Light Chain Formation in the R6/2 Mouse Model of Huntington’s Disease. PLoS ONE 2016, 11, e0166106. [Google Scholar] [CrossRef] [PubMed]
- Palamiuc, L.; Schlagowski, A.; Ngo, S.T.; Vernay, A.; Dirrig-Grosch, S.; Henriques, A.; Boutillier, A.L.; Zoll, J.; Echaniz-Laguna, A.; Loeffler, J.P.; et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 2015, 7, 526–546. [Google Scholar] [CrossRef]
- Hardee, J.P.; Martins, K.J.B.; Miotto, P.M.; Ryall, J.G.; Gehrig, S.M.; Reljic, B.; Naim, T.; Chung, J.D.; Trieu, J.; Swiderski, K.; et al. Metabolic remodeling of dystrophic skeletal muscle reveals biological roles for dystrophin and utrophin in adaptation and plasticity. Mol. Metab. 2021, 45, 101157. [Google Scholar] [CrossRef]
- She, P.; Zhang, Z.; Marchionini, D.; Diaz, W.C.; Jetton, T.J.; Kimball, S.R.; Vary, T.C.; Lang, C.H.; Lynch, C.J. Molecular characterization of skeletal muscle atrophy in the R6/2 mouse model of Huntington’s disease. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E49–E61. [Google Scholar] [CrossRef] [Green Version]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330. [Google Scholar] [CrossRef]
- Cortes, C.J.; Miranda, H.C.; Frankowski, H.; Batlevi, Y.; Young, J.E.; Le, A.; Ivanov, N.; Sopher, B.L.; Carromeu, C.; Muotri, A.R.; et al. Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat. Neurosci. 2014, 17, 1180–1189. [Google Scholar] [CrossRef] [Green Version]
- Magnusson-Lind, A.; Davidsson, M.; Silajdzic, E.; Hansen, C.; McCourt, A.C.; Tabrizi, S.J.; Bjorkqvist, M. Skeletal muscle atrophy in R6/2 mice—altered circulating skeletal muscle markers and gene expression profile changes. J. Huntingt. Dis. 2014, 3, 13–24. [Google Scholar] [CrossRef]
- Mielcarek, M.; Toczek, M.; Smeets, C.J.; Franklin, S.A.; Bondulich, M.K.; Jolinon, N.; Muller, T.; Ahmed, M.; Dick, J.R.; Piotrowska, I.; et al. HDAC4-myogenin axis as an important marker of HD-related skeletal muscle atrophy. PLoS Genet. 2015, 11, e1005021. [Google Scholar] [CrossRef]
- Miranda, D.R.; Wong, M.; Romer, S.H.; McKee, C.; Garza-Vasquez, G.; Medina, A.C.; Bahn, V.; Steele, A.D.; Talmadge, R.J.; Voss, A.A. Progressive Cl- channel defects reveal disrupted skeletal muscle maturation in R6/2 Huntington’s mice. J. Gen. Physiol. 2017, 149, 55–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, D.R.; Reed, E.; Jama, A.; Bottomley, M.; Ren, H.; Rich, M.M.; Voss, A.A. Mechanisms of altered skeletal muscle action potentials in the R6/2 mouse model of Huntington’s disease. Am. J. Physiol. Cell Physiol. 2020, 319, C218–C232. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.R.; Yu, Z.; Gipson, T.A.; Marsh, G.B.; Yoshidome, E.; Robins, D.M.; Todi, S.V.; Housman, D.E.; Lieberman, A.P. Androgen receptor polyglutamine expansion drives age-dependent quality control defects and muscle dysfunction. J. Clin. Investig. 2018, 128, 3630–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.M.; Mihaylova, V.; Frese, S.; Petersen, J.A.; Ligon-Auer, M.; Aguayo, D.; Fluck, M.; Jung, H.H.; Toigo, M. Satellite cell content in Huntington’s disease patients in response to endurance training. Orphanet. J. Rare Dis. 2019, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Al Tanoury, Z.; Rao, J.; Tassy, O.; Gobert, B.; Gapon, S.; Garnier, J.M.; Wagner, E.; Hick, A.; Hall, A.; Gussoni, E.; et al. Differentiation of the human PAX7-positive myogenic precursors/satellite cell lineage in vitro. Development 2020, 147, dev187344. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.B.; Vankara, A.; Ettyreddy, A.R.; Bohning, J.D.; Gersbach, C.A. Myogenic Progenitor Cell Lineage Specification by CRISPR/Cas9-Based Transcriptional Activators. Stem Cell Rep. 2020, 14, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Ito, A.; Kawabe, Y.; Kamihira, M. Novel neuromuscular junction model in 2D and 3D myotubes co-cultured with induced pluripotent stem cell-derived motor neurons. J. Biosci. Bioeng. 2020, 129, 486–493. [Google Scholar] [CrossRef]
- Rao, L.; Qian, Y.; Khodabukus, A.; Ribar, T.; Bursac, N. Engineering human pluripotent stem cells into a functional skeletal muscle tissue. Nat. Commun. 2018, 9, 126. [Google Scholar] [CrossRef]
- Maffioletti, S.M.; Sarcar, S.; Henderson, A.B.H.; Mannhardt, I.; Pinton, L.; Moyle, L.A.; Steele-Stallard, H.; Cappellari, O.; Wells, K.E.; Ferrari, G.; et al. Three-Dimensional Human iPSC-Derived Artificial Skeletal Muscles Model Muscular Dystrophies and Enable Multilineage Tissue Engineering. Cell Rep. 2018, 23, 899–908. [Google Scholar] [CrossRef] [Green Version]
- Jiwlawat, S.; Lynch, E.; Glaser, J.; Smit-Oistad, I.; Jeffrey, J.; Van Dyke, J.M.; Suzuki, M. Differentiation and sarcomere formation in skeletal myocytes directly prepared from human induced pluripotent stem cells using a sphere-based culture. Differentiation 2017, 96, 70–81. [Google Scholar] [CrossRef]
- O’Regan, G.C.; Farag, S.H.; Casey, C.S.; Wood-Kaczmar, A.; Pocock, J.M.; Tabrizi, S.J.; Andre, R. Human Huntington’s disease pluripotent stem cell-derived microglia develop normally but are abnormally hyper-reactive and release elevated levels of reactive oxygen species. J. Neuroinflamm. 2021, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Sheila, M.; Narayanan, G.; Ma, S.; Tam, W.L.; Chai, J.; Stanton, L.W. Phenotypic and molecular features underlying neurodegeneration of motor neurons derived from spinal and bulbar muscular atrophy patients. Neurobiol. Dis. 2019, 124, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Burman, R.J.; Watson, L.M.; Smith, D.C.; Raimondo, J.V.; Ballo, R.; Scholefield, J.; Cowley, S.A.; Wood, M.J.A.; Kidson, S.H.; Greenberg, L.J. Molecular and electrophysiological features of spinocerebellar ataxia type seven in induced pluripotent stem cells. PLoS ONE 2021, 16, e0247434. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.G.; Quan, C.; Reyes-Ortiz, A.M.; Lutz, S.E.; Kedaigle, A.J.; Gipson, T.A.; Wu, J.; Vatine, G.D.; Stocksdale, J.; Casale, M.S.; et al. Huntington’s Disease iPSC-Derived Brain Microvascular Endothelial Cells Reveal WNT-Mediated Angiogenic and Blood-Brain Barrier Deficits. Cell Rep. 2017, 19, 1365–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattis, V.B.; Tom, C.; Akimov, S.; Saeedian, J.; Ostergaard, M.E.; Southwell, A.L.; Doty, C.N.; Ornelas, L.; Sahabian, A.; Lenaeus, L.; et al. HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum. Mol. Genet. 2015, 24, 3257–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, Q.; Miao, Y.; Tang, L.; Huang, M.; Yang, Y.; Ba, W.; Liu, Y.; Chi, S.; Li, C. Rab23 promotes squamous cell carcinoma cell migration and invasion via integrin beta1/Rac1 pathway. Oncotarget 2016, 7, 5342–5352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juopperi, T.A.; Kim, W.R.; Chiang, C.H.; Yu, H.; Margolis, R.L.; Ross, C.A.; Ming, G.L.; Song, H. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol. Brain 2012, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Uchimura, T.; Otomo, J.; Sato, M.; Sakurai, H. A human iPS cell myogenic differentiation system permitting high-throughput drug screening. Stem Cell Res. 2017, 25, 98–106. [Google Scholar] [CrossRef]
- Lilja, K.C.; Zhang, N.; Magli, A.; Gunduz, V.; Bowman, C.J.; Arpke, R.W.; Darabi, R.; Kyba, M.; Perlingeiro, R.; Dynlacht, B.D. Pax7 remodels the chromatin landscape in skeletal muscle stem cells. PLoS ONE 2017, 12, e0176190. [Google Scholar] [CrossRef]
- Maffioletti, S.M.; Gerli, M.F.; Ragazzi, M.; Dastidar, S.; Benedetti, S.; Loperfido, M.; VandenDriessche, T.; Chuah, M.K.; Tedesco, F.S. Efficient derivation and inducible differentiation of expandable skeletal myogenic cells from human ES and patient-specific iPS cells. Nat. Protoc. 2015, 10, 941–958. [Google Scholar] [CrossRef]
- Abujarour, R.; Bennett, M.; Valamehr, B.; Lee, T.T.; Robinson, M.; Robbins, D.; Le, T.; Lai, K.; Flynn, P. Myogenic differentiation of muscular dystrophy-specific induced pluripotent stem cells for use in drug discovery. Stem Cells Transl. Med. 2014, 3, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Borchin, B.; Chen, J.; Barberi, T. Derivation and FACS-mediated purification of PAX3+/PAX7+ skeletal muscle precursors from human pluripotent stem cells. Stem Cell Rep. 2013, 1, 620–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoyama, T.; McGivern, J.V.; Van Dyke, J.M.; Ebert, A.D.; Suzuki, M. Derivation of myogenic progenitors directly from human pluripotent stem cells using a sphere-based culture. Stem Cells Transl. Med. 2014, 3, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Al Tanoury, Z.; Hestin, M.; Gobert, B.; Aivio, S.; Hick, A.; Cherrier, T.; Nesmith, A.P.; Parker, K.K.; Pourquie, O. Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat. Protoc. 2016, 11, 1833–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Jang, J. Recent Trends in Biofabrication Technologies for Studying Skeletal Muscle Tissue-Related Diseases. Front. Bioeng. Biotechnol. 2021, 9, 782333. [Google Scholar] [CrossRef]
- Lu, K.; Seidel, T.; Cao-Ehlker, X.; Dorn, T.; Batcha, A.M.N.; Schneider, C.M.; Semmler, M.; Volk, T.; Moretti, A.; Dendorfer, A.; et al. Progressive stretch enhances growth and maturation of 3D stem-cell-derived myocardium. Theranostics 2021, 11, 6138–6153. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, I.; Seol, Y.J.; Ko, I.K.; Yoo, J.J.; Atala, A.; Lee, S.J. Neural cell integration into 3D bioprinted skeletal muscle constructs accelerates restoration of muscle function. Nat. Commun. 2020, 11, 1025. [Google Scholar] [CrossRef]
- Gasparotto, M.; Bellet, P.; Scapin, G.; Busetto, R.; Rampazzo, C.; Vitiello, L.; Shah, D.I.; Filippini, F. 3D Printed Graphene-PLA Scaffolds Promote Cell Alignment and Differentiation. Int. J. Mol. Sci. 2022, 23, 1736. [Google Scholar] [CrossRef]
- Ong, C.S.; Yesantharao, P.; Huang, C.Y.; Mattson, G.; Boktor, J.; Fukunishi, T.; Zhang, H.; Hibino, N. 3D bioprinting using stem cells. Pediatr. Res. 2018, 83, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Bondulich, M.K.; Jolinon, N.; Osborne, G.F.; Smith, E.J.; Rattray, I.; Neueder, A.; Sathasivam, K.; Ahmed, M.; Ali, N.; Benjamin, A.C.; et al. Myostatin inhibition prevents skeletal muscle pathophysiology in Huntington’s disease mice. Sci. Rep. 2017, 7, 14275. [Google Scholar] [CrossRef] [Green Version]
- Sjogren, M.; Duarte, A.I.; McCourt, A.C.; Shcherbina, L.; Wierup, N.; Bjorkqvist, M. Ghrelin rescues skeletal muscle catabolic profile in the R6/2 mouse model of Huntington’s disease. Sci. Rep. 2017, 7, 13896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, C.; Bott, L.C.; Chen, K.L.; Harmison, G.G.; Katsuno, M.; Sobue, G.; Pennuto, M.; Fischbeck, K.H. Insulinlike growth factor (IGF)-1 administration ameliorates disease manifestations in a mouse model of spinal and bulbar muscular atrophy. Mol. Med. 2012, 18, 1261–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querin, G.; D’Ascenzo, C.; Peterle, E.; Ermani, M.; Bello, L.; Melacini, P.; Morandi, L.; Mazzini, L.; Silani, V.; Raimondi, M.; et al. Pilot trial of clenbuterol in spinal and bulbar muscular atrophy. Neurology 2013, 80, 2095–2098. [Google Scholar] [CrossRef] [PubMed]
- Soraru, G.; Pegoraro, E.; Spinella, P.; Turra, S.; D’Ascenzo, C.; Baggio, L.; Mantovan, M.C.; Vergani, L.; Angelini, C. A pilot trial with clenbuterol in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2006, 7, 246–248. [Google Scholar] [CrossRef]


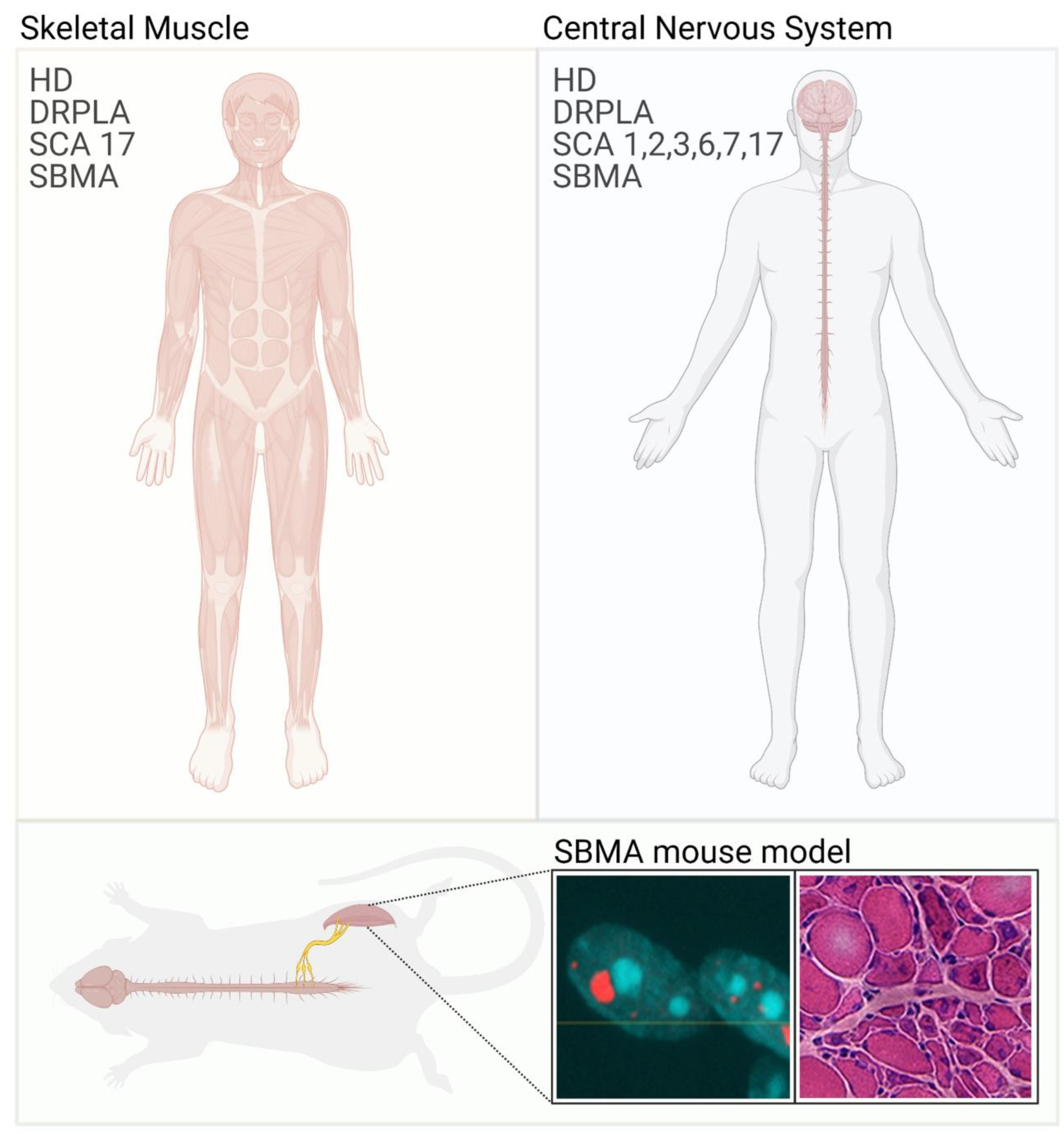{kind=link}
| Disease | Gene | Normal (CAG) n | Pathogenic (CAG) n | Expression | Subcellular Localization | Function | Knock Out Mice | Vulnerable Neurons |
|---|---|---|---|---|---|---|---|---|
| HD [12] | HTT | 6–35 | 36–39 incomplete penetrance 40–250 >75 juvenile forms | Ubiquitous | C > N | Axonal vesicular trafficking, ciliogenesis, regulation of autophagy, regulation of transcription | Embryonic lethal | Medium-sized spiny neurons in the striatum, cortical projection neurons |
| SBMA [13] | AR | 5–35 | 36–37 low penetrance 38–72 | Central nervous system, skeletal muscle, liver, adipose tissue, testis & prostate, and other tissues | Cytosolic Nuclear translocation induced by androgen binding | Steroid hormone receptor: Androgen-activated transcription factor | Viable Feminization | Brainstem and spinal cord motor neurons |
| DRPLA [14] | ATN1 | 7–34 | 49–88 | Ubiquitous | C > N | Involved in protein trafficking and degradation | Viable Normal phenotype | Brainstem, cerebellum, deep midbrain |
| SCA1 [15] | ATX1 | 6–44 | >39 | Central nervous system, skeletal muscle, liver, kidney and other tissues | N in neurons, C in nonneuronal cells | Transcriptional regulation and RNA metabolism | Viable No Purkinjie cell degeneration (altered hippocampal synaptic plasticity) | Purkinje cells in the cerebellum, upper motor neurons |
| SCA2 [16] | ATX2 | 13–33 | 32–77 | Brain, heart, skeletal muscle, liver, pancreas, placenta | C, ER/Golgi | RNA processing and metabolism | Viable Adult-onset obesity | Purkinje cells, brainstem and spinal cord, substantia nigra |
| SCA3 [17] | ATX3 | 12–40 | 54–89 | Ubiquitous | C | Isopeptidase and deubiquitinating activity, proteasomal degradation, regulation of misfolded proteins | Viable Increased protein ubiquitination | Purkinje cells |
| SCA6 [18] | CACNA1 | 4–18 | 21–33 | Neurons | PM | Subunit of voltage-gated P/Q calcium channel | Viable Ataxia, seizures, dystonia | Purkinje cells |
| SCA7 [19] | ATX7 | 7–19 | 20–35 incomplete penetrance 36 to >400 | Brain, retina | N | Member of the transcriptional coactivator STAGA complex | Viable | Purkinje cells, photoreceptor cells of the retina |
| SCA17 [20] | TBP | 25–44 | 47–66 | Ubiquitous | N | Universal basal transcription factor | Embryonic lethal | Purkinje cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchioretti, C.; Zuccaro, E.; Pandey, U.B.; Rosati, J.; Basso, M.; Pennuto, M. Skeletal Muscle Pathogenesis in Polyglutamine Diseases. Cells 2022, 11, 2105. https://doi.org/10.3390/cells11132105
Marchioretti C, Zuccaro E, Pandey UB, Rosati J, Basso M, Pennuto M. Skeletal Muscle Pathogenesis in Polyglutamine Diseases. Cells. 2022; 11(13):2105. https://doi.org/10.3390/cells11132105
Chicago/Turabian StyleMarchioretti, Caterina, Emanuela Zuccaro, Udai Bhan Pandey, Jessica Rosati, Manuela Basso, and Maria Pennuto. 2022. "Skeletal Muscle Pathogenesis in Polyglutamine Diseases" Cells 11, no. 13: 2105. https://doi.org/10.3390/cells11132105
APA StyleMarchioretti, C., Zuccaro, E., Pandey, U. B., Rosati, J., Basso, M., & Pennuto, M. (2022). Skeletal Muscle Pathogenesis in Polyglutamine Diseases. Cells, 11(13), 2105. https://doi.org/10.3390/cells11132105