1. Introduction
Despite therapeutic advances, colorectal cancer (CRC) remains a major cause of mortality worldwide [
1]. Early diagnosis plays a crucial role in the survival of CRC patients. At diagnosis, patients with localized CRC account for 37% of cases, while those with metastatic stage account for 22%, according to the Surveillance, Epidemiology, and End Results (SEER) program [
1]. Importantly, 5-year survival is strongly related to CRC stage. Indeed, the 5-year survival is around 90% for localized CRC, while it decreases sharply for metastatic CRC with only 14% survival. Thus, a better understanding of the cells responsible for CRC progression, metastasis, and treatment resistance is required. Although new treatment options are available, chemotherapy remains a standard therapy for CRC after surgery [
2]. Treatment protocols for patients with localized or advanced CRC include 5-fluorouracil(5-FU)-based chemotherapies—such as FOLFOX (5-FU, leucovorin, and oxaliplatin), FOLFIRI (5-FU, leucovorin, and irinotecan), or FOLFIRINOX (5-FU, leucovorin, oxaliplatin and irinotecan) [
3,
4]. Therapeutic resistance and relapse are responsible for decreased 5-year survival in advanced CRC. One explanation for therapy failure is the presence of a minor cell subpopulation, cancer stem cells (CSCs), which are resistant to conventional therapies and therefore likely to cause tumor relapse [
5]. CSCs are a small population of cancer cells endowed with self-renewal, multi-lineage differentiation, and tumor-initiating capacity in immunocompromised mice [
6,
7]. The main challenge in studying CSC is their identification and isolation. Surface markers were originally and for a long time used to identify CSCs. However, due to CSC plasticity and the shared expression of some markers by intestinal stem cells or cancer cells, the use of markers is no longer sufficient to define a CSC [
8,
9]. Currently, functional characteristics such as tumorigenic potential, relative quiescence, and chemoresistance are more often used to identify CSCs. The CSC state is subject to cellular plasticity and is inherent to intratumoral cellular heterogeneity. A few years ago, our laboratory adapted the sedimentation field–flow fractionation (SdFFF) as a cell sorting technique [
10]. SdFFF is a gentle, non-invasive method that does not require cell labeling or fixation [
10]. Cell separation by SdFFF depends on the differential elution of cells subjected to both the action of a parabolic profile generated by the mobile phase in the separation channel and a multi-gravitational external field generated by rotation of this channel [
9]. The separation of cells is based on their biophysical properties, such as size and density, using the hyperlayer elution mode of SdFFF [
11].
In previous work, our laboratory used SdFFF as a tool to isolate cell subpopulations independently of surface marker expression from CRC cell lines [
10]. Results showed that one of the cell subpopulations sorted by SdFFF exhibited CSC-like features in two early-stage CRC cell lines [
10,
12]. However, the ability of this cell subpopulation to initiate tumors in mice, which is the gold standard for defining a CSC, remains to be determined. In our study, we therefore further characterized the phenotypic and functional characterization of the sorted cell subpopulations and assessed their ability to generate tumors in vivo. Furthermore, because of the involvement of CSCs in tumor progression and metastasis, we performed these experiments using late-stage CRC cell lines, in addition to early-stage ones, as well as primary cultures. Finally, as CSCs play a key role in resistance to conventional therapies, we explored the response of SdFFF-sorted cell subpopulations to chemotherapies. Thus, our study aimed to highlight the impact of intratumoral cellular heterogeneity on treatment resistance in order to better understand the mechanisms involved and to provide leads for more personalized therapies. In agreement with previous results, we reported here that SdFFF-sorted cell subpopulations have distinct phenotypic and functional characteristics [
10]. Nevertheless, in our study, the expression of CSC markers did not necessarily correlate with their functional characteristics. The characterization results showed that a cell subpopulation exhibited the functional features of CSCs, including in vivo tumorigenicity and resistance to conventional therapies. This observation prevailed for both early- and late-stage cell lines as well as for primary cultures. Therefore, we highlighted a label-free cell sorting approach to study the individualized response of each tumor cell subpopulation in order to understand the complexity of intra-tumor cell heterogeneity and to identify chemoresistance. Ultimately, this approach could provide valuable information from CRC patient samples, offering new perspectives for more personalized therapy.
2. Materials and Methods
2.1. Cell Cultures
Four colorectal cancer cell lines were used. The two early-stage CRC cell lines, labeled “primary tumors” in the graphs, WiDr (CCL-218 ™) and SW480 (CCL-228 ™); as well as the two metastatic cell lines, labeled “metastasis” in the graphs, SW620 (CCL-227 ™) and T84 (CCL-248 ™), were obtained from the American Type Culture Collection (ATCC/LGC Promochem, Molsheim, France). SW480 and SW620 were established from the same patient, SW480 was from the patient’s primary tumor while SW620 was from lymph node metastases. The T84 cell line was derived from lung metastasis of a CRC patient. Cells were maintained at 37 °C under humidified 5% CO
2 in MEM medium (no. 31095-029, Gibco ™, Thermo Fisher Scientific Inc., Waltham, MA, USA) for WiDr and RPMI GlutaMAX medium (no. 61870-010, Thermo Fisher) for SW480, SW620, and T84 cell lines, both supplemented with 10% fetal bovine serum (FBS), 1 mM sodium pyruvate (no. 11360-039, Thermo Fisher), 100 IU/mL penicillin and 100 mg/mL streptomycin (no. 15140-122, Thermo Fisher), and only for WiDr 1% non-essential amino acids (no. 11140-035, Thermo Fisher) [
13].
Two primary cultures from CRC patients (CPP14 and CPP35) were obtained from the Institute of Functional Genomics (Univ. Montpellier, CNRS, INSERM, Montpellier, France), after informed consent of patients (Material Transfer Agreement CNRS 190287). The primary culture CPP14, labeled “early-stage tumor” in the graphs, was derived from a patient with T2N0M0 CRC—i.e., early stage (stage I)—whereas CPP35, labeled “tumor-invaded peritoneum” in the graphs, was derived from a patient with T4aN0M0 CRC—i.e., a stage at which the tumor had invaded the peritoneum (stage IIB)—as indicated in
Table 1 [
14]. Both primary cultures are treatment-naive. The culture conditions for these primary cultures were DMEM GlutaMAX medium (no. 61965059, Thermo Fisher) supplemented with 10% FBS, 100 IU/mL penicillin and 100 mg/mL streptomycin, at 37 °C under a humidified atmosphere of 5% CO
2.
2.2. SdFFF Cell Sorting
The SdFFF technique used enables sorting of cell subpopulations as described and schematized previously [
9,
10]. Cell-solid phase interactions in SdFFF are limited due to the use of a ribbon-like empty channel with no stationary phase and a size/density-based separation mechanism through the hyperlayer elution mode. SdFFF parameters used during cell sorting are field (units of gravity, g), flow rate of the mobile phase which is sterile phosphate-buffered saline (PBS, pH 7.4, no. 14190-094, Thermo Fisher) and the rotation speed of the channel (revolutions per minute, rpm) which is related to the field. Adjustment steps were performed in order to choose the SdFFF parameters that allow an optimal separation between the void volume peak and the peak containing the cells, and to define the best elution conditions for all cell lines and primary cultures used. Elution conditions are summarized in
Table 2, with a check of these parameters before and during each cell sorting.
The injected volume of the cell suspension, 100 µL, as well as the detection wavelength, 254 nm, are common for each cell line and primary culture. A cleaning and decontamination procedure was performed at the end of each cell sorting [
15]. Once sorted, cells can be recultured in vitro and characterized, as no cell fixation or labeling is required for SdFFF. In order to perform experiments with the sorted cells, successive injections and collections of the same cell suspension (>10) are required to obtain a sufficient quantity of cells.
2.3. CSC Marker Expression
After cell sorting by SdFFF, cell subpopulation concentrations were standardized to the same amount of cells in each condition. Anti-CD44, anti-LGR5, anti-CD133/1, and viability marker antibodies were added to the cells and incubated for 30 min at 4 °C and in the dark, antibody references are summarized in
Table S1. The viability marker was used to exclude nonviable cells. Cells were then fixed in 4% paraformaldehyde (PFA, no. 10231622, Thermo Fisher) for 10 min at room temperature and permeabilized with Perm Buffer III (no. 558050, BD Phosflow ™, BD Biosciences, Franklin Lakes, NJ, USA) for 30 min at 4 °C. Next, antibodies recognizing the intracellular marker BMI-1 were added and incubated for 30 min at 4 °C in the dark (reference in
Table S1). As reference controls, anti-IgG2bκ FITC, anti-IgG2bκ PE-Vio 770, anti-IgG1 PE-Vio 615, and mouse anti-IgG1κ PE isotype controls were used under the same conditions and concentrations to ensure specific recognition of our antibodies of interest and set gates (references in
Table S1). Samples were analyzed by the CytoFlex LX and data analysis using Kaluza software v2.1 (Beckman Coulter, Indianapolis, IN, USA)
2.4. Cell Cycle Analysis
Cell concentrations of SdFFF-sorted subpopulations were standardized in each condition. After a centrifugation step, the cells were resuspended with cold PBS, fixed with cold 96% ethanol added slowly and under shaking, and then placed at −20 °C. After a few minutes at room temperature, the cells were washed and then resuspended with PBS and RNAse A (no. R6148, Sigma-Aldrich, Saint-Louis, MO, USA) fo) for 20 min at room temperature. Next, propidium iodide was added 15 min before acquisition on the FACS Calibur. Data analysis was performed with ModFit LTTM software v5.0 (Verity Software House, Topsham, ME, USA)
2.5. Clonogenic Assay: Soft Agar Assay
This assay was based on the use of two gels, the first is a 0.5% agar gel that prevents the cells from adhering to the culture plate and the second was a 0.7% agarose gel containing the cells. The agar (no. A7002, Sigma-Aldrich) and agarose (no. A9539, Sigma-Aldrich) solutions were prepared upstream with sterile PBS and autoclaved to limit potential contamination. Agar gel was prepared in advance and plated into wells of 24-well plates at room temperature and under the culture hood. Once the agar gel solidified, the agarose gel was heated and then gently mixed with cell subpopulations sorted by SdFFF to have a cell concentration of 1 × 103 cells per well (12 wells/condition). As soon as the second gel solidified, culture medium was added on top to prevent evaporation and 24-well culture plates were incubated at 37 °C under a humidified atmosphere of 5% CO2 for 30 days. Four weeks later, the formed colonies were fixed in 4% PFA (no. 10231622, Thermo Fischer) for 15 min and then stained with 0.1% crystal violet. Wells of the 24-well plates used were captured with the Leica DFC300 FX Digital Color Camera to allow colony quantification and analyzed by ImageJ software v1.53e (National Institutes of Health, Bethesda, MD, USA)
2.6. In Vivo Tumor Initiation Assay
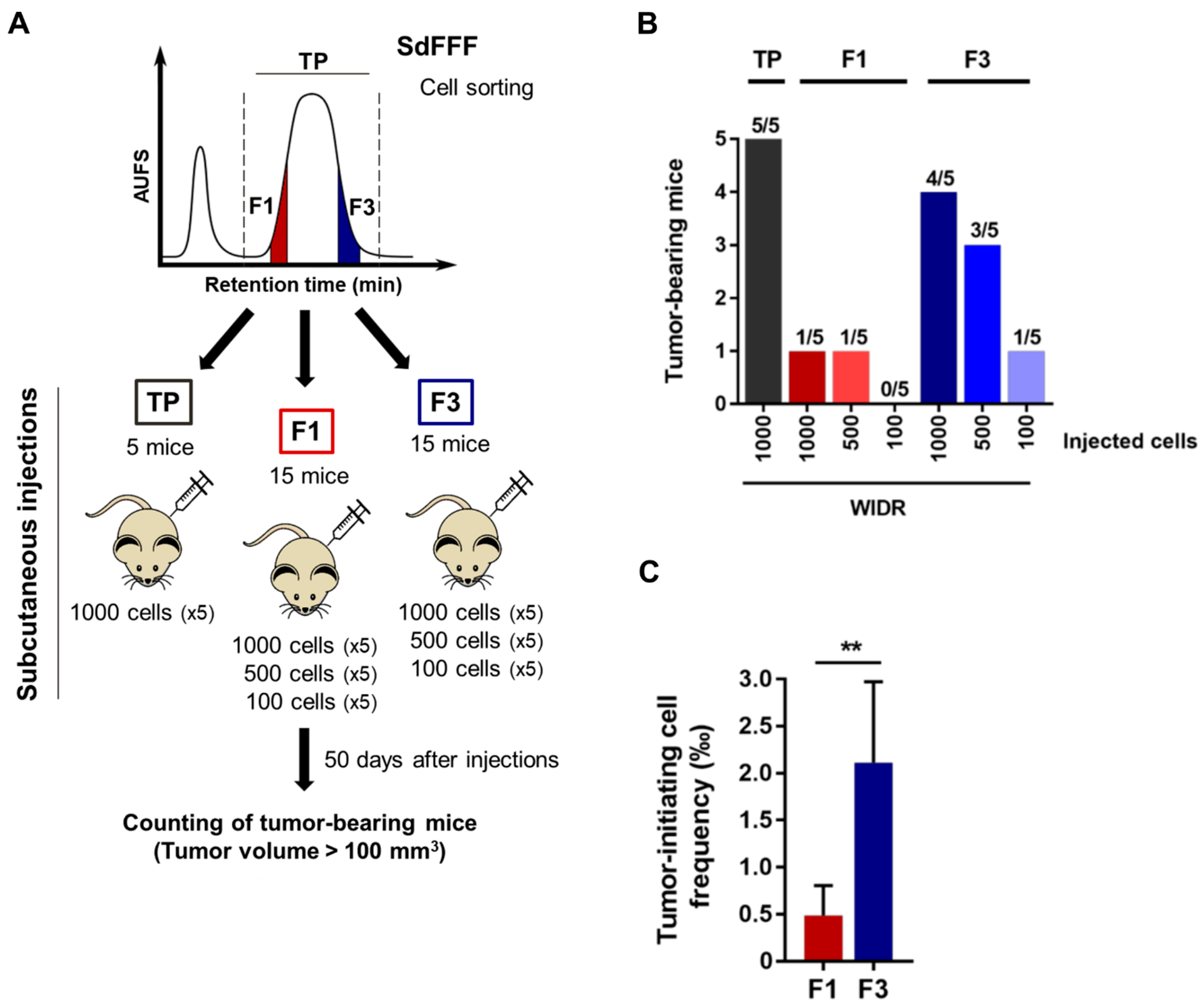
SdFFF-sorted cell subpopulations of the WiDr cell line were injected subcutaneously into nude mice (Hsd:Athymic Nude-Foxn1nu nu/nu, six weeks old, female, five mice per group) in decreasing amounts of cells (1000, 500, and 100) in Matrigel (no. 356237, Corning, New York, NY, USA)–MEM medium. Mice weight and tumor size were measured three times a week for 7 weeks. After 50 days, mice were sacrificed and tumors were collected. The number of tumor-bearing mice with a tumor volume greater than 100 mm
3 was counted. The online software Extreme limiting dilution analysis (ELDA) was used from the in vivo results to determine the frequency of tumor occurrence and thus tumor initiating cells frequency (
https://bioinf.wehi.edu.au/software/elda/, accessed on 28 January 2022) [
16]. This animal experimentation protocol has been approved by the Ethics Committee for Animal Experimentation no. 33 and by the French Ministry of Higher Education, Research and Innovation (protocol code APAFIS no. 3 1963-2021061014298122 v2, approved 21 July 2021).
2.7. Cytotoxicity Assay
To compare results between cell lines, we defined the same cell concentration for all cell lines: 1.5 × 10
3 cells per well of 96-well plate, after optimization. Once sorted and seeded, cells were then treated for 72 h with 5-FU using a range from 0.16 to 250 µM. Three days later, the MTS reagent was added according to the manufacturer’s instructions (no. G3580, CellTiter96 AQueous One solution Cell Poliferation assay, Promega, Madison, WI, USA) and incubated for 3 h at 37 °C under a humidified atmosphere of 5% CO
2. Absorbance was then measured at 490 nm with the Multiskan ™ FC 96-well plate reader (Thermo Fisher) and the results were expressed as a percentage comparing the treated condition to the untreated condition defined as 100%. Generation of drug response curves and determination of IC50 values were performed using GraphPad Prism software (San Diego, CA, USA). We compared the obtained IC50s with those reported at
https://www.cancerrxgene.org/. These experiments were also performed with oxaliplatin and irinotecan. The concentration range used for oxaliplatin was 0.13 to 200 µM and for irinotecan it was 0.4 to 650 µM. The three chemotherapies were provided by the anticancer preparation unit of the University Hospital of Limoges.
2.8. Proliferation Assay
Once sorted by SdFFF, cells were seeded at the same concentration as for the MTS assay, 1.5 × 103 cells per well in 96-well plates. Cells were then treated for 72 h with the average IC50 values obtained for 5-FU from the cell lines and primary cultures used. Cell proliferation was assessed using the BrdU cell proliferation assay kit (no. 6813, Cell signaling technology, Danvers, MA, USA) ac) according to the manufacturer’s instructions. Absorbance was then measured at 450 nm with the Multiskan ™ FC 96-well plate reader (Thermo Fisher) and the results were expressed as a ratio comparing the treated condition to the untreated condition defined as 1.
2.9. Cell Death Assay
From the same cell sorting by SdFFF as for the proliferation analysis, an apoptosis analysis was performed. Cell concentration and 5-FU dose and incubation time were the same as for the cell proliferation assay. Apoptosis rate was measured using the Cell Death Detection ELISAPLUS kit (no. 11774425001, Roche, Bâle, Suisse), according to the manufacturer’s instructions. Absorbance was then measured at 405nm with the Multiskan ™ FC 96-well plate reader (Thermo Fisher) and the results were expressed as a ratio comparing the treated condition to the untreated condition defined as 1.
2.10. Tumorsphere Assay
Five hundred cells were seeded in nonadherent 96-well culture plates previously coated with a 10% solution of poly-2-hydroxyethylmethacrylate (no. P3932, Sigma Aldrich) in 95% ethanol and dried overnight at 56 °C [
17]. These cells were cultured in defined medium: with serum-free GlutaMAX-DMEM/F12 (no. 10565018, Thermo Fisher) medium supplemented with 20 ng/mL epidermal growth factor (no. PHG0314, Thermo Fisher), 10 ng/mL basic fibroblast growth factor (no. PHG0264, Thermo Fisher), 0.3% glucose (no. 49163, Sigma-Aldrich), 20 μg/mL insulin (no. 12585-014, Thermo Fisher), 1:100 N2 supplement (no. 17502-001, Thermo Fisher) [
18] and incubated at 37 °C in a humidified atmosphere of 5% CO
2 for one week. Seven days later, spheres larger than 50 μm in diameter were counted using the Leica DMi8 microscope. Then, to study chemotherapy response in a 3D culture model, chemotherapies were added after the seven days of incubation that allow colonosphere formation, and incubated for three days. Chemotherapy doses used in the 3D chemosensitivity assays were defined from the results of the 2D cytotoxicity assays that allowed calculation of the average IC50 for all cell matrices, including cell lines and primary cultures. Colonospheres were treated using the average IC50 obtained (0.7 µM for 5-FU, 1.9 µM for oxaliplatin and 22.8 µM for irinotecan) for both single and combination therapy. Colonospheres were counted three days after treatment, imaged using the Leica DMi8 microscope, and their size was measured using ImageJ software v1.53e (National Institutes of Health).
2.11. Statistical Analysis
All bar plots are represented by the mean ± S.E.M. of results obtained from at least three independent experiments. Flow cytometry results were represented as histograms from one representative biological replicate among the three independent experiments performed (n = 3). Significance of results was specified by stars: * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001. The presence of a star without a bar below indicates that the result is significantly different from the control condition: TP. Significance between the three sorted cell subpopulations was represented using the star and a bar below to allow identification of the subpopulations involved in these significantly different results. Non-significance of results is indicated by ‘ns’ for not significant, underlining that no significant differences is observed either between the sorted subpopulations or between F1/F2/F3 and the TP control. Results were analyzed by the Kruskal–Wallis test for clonogenicity assay because the results did not follow a normal distribution, verified using the Shapiro–Wilk test. Chi-squared test was used to identify the frequency of tumor-initiating cells from in vivo experiment results. The one-way ANOVA test was used for all other experiments because the results followed a normal distribution (Shapiro–Wilk test) and the comparison was between the three sorted cell subpopulations and the control. Statistical tests were performed using PAST software, version 2.17c.
4. Discussion
Our study addressed the impact of intratumoral cellular heterogeneity in the context of treatment resistance, without relying on surface marker expression to sort cell subpopulations. Regardless of cell line and stage, the SdFFF technique allowed isolation of cell subpopulations with distinct phenotypical and functional characteristics. Compared to previous results [
10], we showed here the presence of two therapeutically relevant cell subpopulations based on their CSCs characteristics: F1 and F3. This was achieved by in vivo tumor initiation assay and in vitro chemosensitivity assays. The F1 subpopulation was proliferative and chemosensitive, while the F3 subpopulation exhibited CSC functional hallmarks, including the ability to initiate tumors in mice and chemoresistance. These two cell subpopulations of interest were both identified from early- and late-stage cell lines. From the primary cultures, we also identified the F3 subpopulation as a CSC-enriched fraction due to its clonogenicity and quiescence. Thus, the results of the functional characterizations obtained with the primary cultures mirrored those obtained for the cell lines. Furthermore, we demonstrated the chemoresistance of the F3 CSC-like subpopulation in the two primary cultures, as observed in the cell lines. However, the mechanisms developed by F3 cells to resist chemotherapies are different between cell lines and primary cultures. Indeed, the F3 subpopulation of cell lines induce increased proliferation after 5-FU treatment without modifying apoptosis, while F3 from primary cultures does not significantly change its proliferation rate but decreases its apoptosis rate compared to F1. Therefore, thanks to our label-free approach, we highlighted a cell subpopulation with a CSC-like phenotype that may play a crucial role in tumor progression and metastasis. Furthermore, we were able to identify different mechanisms used by these cells to resist chemotherapies.
Initially identified in acute myeloid leukemia, CSCs were later discovered in many solid cancers such as CRC based on the expression of surface markers [
22]. In CRC, early publications on CSCs used surface markers as a prerequisite to identify these cells [
23,
24,
25,
26,
27,
28,
29]. Many CSC markers have been identified in CRC and are reviewed in Hervieu et al. [
9]. Our phenotypic characterization results were initially disappointing due to the lack of significant differences between the cell subpopulations sorted for three of the four cell lines studied, although a trend did emerge. However, several publications have questioned the use of these markers since they are also expressed by intestinal stem cells and cancer cells [
20,
30,
31]. Furthermore, Prasetyanti and Medema point out that CSC markers can be considered as a highly context-dependent property of cells [
32]. Furthermore, the use of surface markers is hampered by the plasticity of CSCs, which is another obstacle to their identification and isolation. Shimokawa et al. and de Sousa e Melo et al. showed that ablation of LGR5
+ CSCs limits tumor growth but does not prevent tumor recurrence due to re-emergence of LGR5
+ CSCs from proliferating LGR5
- cells [
33,
34]. Intriguingly, another study demonstrated that dissemination and metastatic colonization were carried out by LGR5
- cells in CRC, with subsequent re-emergence of LGR5
+ CSCs at the metastatic site [
7]. Thus, the results of these publications suggest that LGR5
- non-CSCs are able to transform into LGR5-expressing CSCs, highlighting the phenotypic plasticity of these cells that appears to be crucial for primary tumor growth and metastasis. These results highlight the complexity of identifying as well as isolating these CSCs due to their shared expression with non-CSCs and their cellular plasticity. All of these data converge on evidence that stemness is not as hierarchical and fixed as originally thought, but rather dynamic and endowed with considerable plasticity. It is this polydispersity of CSCs that justified the SdFFF label-free cell sorting approach, offering new perspectives to isolate CSC-enriched subpopulations.
A consensus has emerged suggesting that their functional capabilities, particularly their tumorigenic potential and chemoresistance, are more reliable for identifying CSCs than surface markers [
35]. Our results showed that cells with the ability to initiate tumors in mice and chemoresistance were not necessarily correlated with cells expressing CSC markers. The publication of Lenos et al. is in agreement with our observations, demonstrating that there is a divergence between cells positive for CSC markers and cells with CSC functionality [
36]. In our study, the results of the in vivo tumor initiation assay provide evidence that F3 is a subpopulation of CSCs with high tumorigenicity (8 out of 15 tumor-bearing mice) as well as a high frequency of tumor-initiating cells compared to F1. Nevertheless, F1 cells also possess tumor-initiating capacity, but much less efficiently than F3 cells, with only 2 out of 15 mice developing a tumor. These results as well as the results of the characterization of the sorted cell subpopulations suggest that F3 cells are the CSCs at the top of the cell hierarchy, whereas F1 cells have already started to differentiate and lose their CSC characteristics. F1 cells can be considered as transit-amplifying cells that are known to be highly proliferative, which is consistent with our cell cycle results, and engaged in a differentiation process that explains the maintenance of some CSC characteristics but much less efficient than F3. Accumulating evidence suggests that cell–cell interactions and crosstalk within the tumor microenvironment (TME) can modulate cellular state, stemness, and many fundamental characteristics of CSC [
32,
37,
38]. Our in vivo results suggested cooperation of non-CSC cancer cells with CSCs. Indeed, we noticed that for the TP control, five mice carried tumors while the CSC-enriched subpopulation had only four, which may indicate that the presence of both F3 cells (CSCs) and F1 cells (non-CSCs) in TP promoted tumor development. As F3 cells are CSCs, when injected alone, these cells are able to initiate a tumor and promote its growth through their ability to differentiate multi-lineage in order to recreate tumor heterogeneity. For F1 cells already engaged in a differentiation process, tumor formation is significantly less efficient and takes longer to reach the threshold tumor volume of 100 mm
3, approximately 46 days for F1 versus 37 days for F3. Interestingly, for TP, the threshold volume is reached very slightly before the F3 subpopulation, which demonstrates the importance of cellular heterogeneity in tumor development with the requirement of CSCs for tumor initiation, as well as the presence of non-CSCs to enhance tumor growth.
CSCs are of particularly therapeutic interest. Although chemotherapies kill most tumor cells, CSCs are able to escape the lethal effect of these drugs, which can lead to tumor recurrence. One of the main reasons for treatment failure is that anticancer drugs often only target actively cycling tumor cells and therefore do not affect CSCs, which are frequently in a quiescent or minimally proliferative state. The results of our study support this hypothesis: the cell subpopulation enriched in CSC features was quiescent/low proliferative and chemoresistant, while the subpopulation of actively cycling cells was more sensitive. Similar results have been reported in Kreso et al. [
39]. Remarkably, our study showed that only the CSC-enriched cell subpopulation escaped chemotherapy by significantly increasing treatment-induced cell proliferation without any change in cell death, demonstrating cell plasticity. These 2D results show that the chemoresistance of F3-CSCs corresponds to the exit of their quiescent state and the transition to a proliferative state, allowing the reconstitution of cellular heterogeneity. This is also illustrated by our results in the 3D model of the WiDr cell line in response to 5-FU-based chemotherapies. Furthermore, our results highlight that resistance and therapeutic escape are a functional property of CSCs, reinforcing the relevance of our approach based on label-free cell sorting by SdFFF. Collectively, our in vitro results reflect what frequently occurs in CRC patients: chemotherapies kill proliferative cells (i.e., the F1 subpopulation), resulting in tumor regression; but fail to target CSCs (i.e., the F3 subpopulation), which are resistant and evade therapy, resulting in cancer relapse in patients.
Therapies targeting CSCs are a promising therapeutic approach. However, the development of anticancer agents capable of specifically targeting CSCs has proven very difficult or has shown limited efficacy [
40]. These disappointing results may be explained by the fact that the study models used often fail to reproduce the heterogeneous tumor architecture of patients [
32]. One of our original hypotheses was that cell subpopulations isolated from early-stage CRC cell lines might behave differently to chemotherapies than those from metastatic stages. Although one of the two metastatic cell lines had the highest IC50 for each of the chemotherapies tested, the results were not as pronounced as initially expected. Cancer cell lines grown in 2D have traditionally been used to model cancer, but the absence of the microenvironment may impact response to treatment [
32]. Cellular heterogeneity and plasticity may also compromise treatment efficacy, as highlighted in the studies of Shimokawa et al. and de Sousa e Melo et al. [
32,
33,
34,
38]. CRC patient-derived cultures, such as primary cultures, better model the heterogeneity and complexity of patient tumors, which are essential to consider when studying response to therapy. In our study, the IC50 differences are more pronounced between the two different stage primary cultures, especially for oxaliplatin and irinotecan, which may provide insights for more personalized therapy in patients from whom the primary cultures were derived. In addition to optimizing the biological model used, 3D cell culture increases the sensitivity of chemotherapy response studies [
32]. Our 3D chemosensitivity results from microtumor-like spheroids confirmed and refined those obtained in 2D. Importantly, the CSC-enriched subpopulation sorted by SdFFF has the ability to survive in suspension in serum-free media and proliferate even in the presence of chemotherapy. The transition from a quiescent state under physiological conditions to a proliferative state after stress may suggest a dormant CSC state. Indeed, F3 CSCs leave the cell cycle and become quiescent/dormant (G0/G1 arrest), allowing them to escape chemotherapies targeting highly proliferative cells, while retaining the potential to reiterate proliferative expansion [
41]. Thus, cell proliferation plays a crucial role for this subpopulation in response to stress such as chemotherapies. Therefore, our study models based on label-free cell sorting provide new insights to study responses to therapies and resistance mechanisms developed by CSCs. Nevertheless, key components of TEM must be incorporated into our model to mimic patient tumors [
32]. Future cancer therapies will have to take into account both the CSCs and non-CSCs that form the tumor mass, as well as the surrounding TEM.
In summary, we have demonstrated the relevance of using SdFFF to obtain a chemoresistance signature. Thus, the CSC-enriched subpopulation will allow to specifically explore the signaling pathways associated with therapeutic resistance. Since this technique provides a model reflecting intratumoral cellular heterogeneity, it will be valued in the study of the response to therapies of each cell subpopulation of a biological sample. Our study thus opens new perspectives for the understanding of CSC-related resistance and offer solutions specifically adapted to a more personalized and targeted therapy of colorectal cancer.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}