
The Role of Inflammatory Mediators in Colorectal Cancer Hepatic Metastasis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cytokines
3. TNF-α
3.1. TNF-α Discovery
3.2. TNF-α Expression and Regulation in Primary Colon Tumor and Hepatic Metastasis
3.3. TNF-α Function and Therapeutic Targeting Potential
4. Interleukin-6 (IL-6)
4.1. IL-6 Discovery
4.2. IL-6 Expression and Regulation in Primary Colon Tumor and Hepatic Metastasis Tissue
4.3. IL-6 Function and Therapeutically Targeting Potential
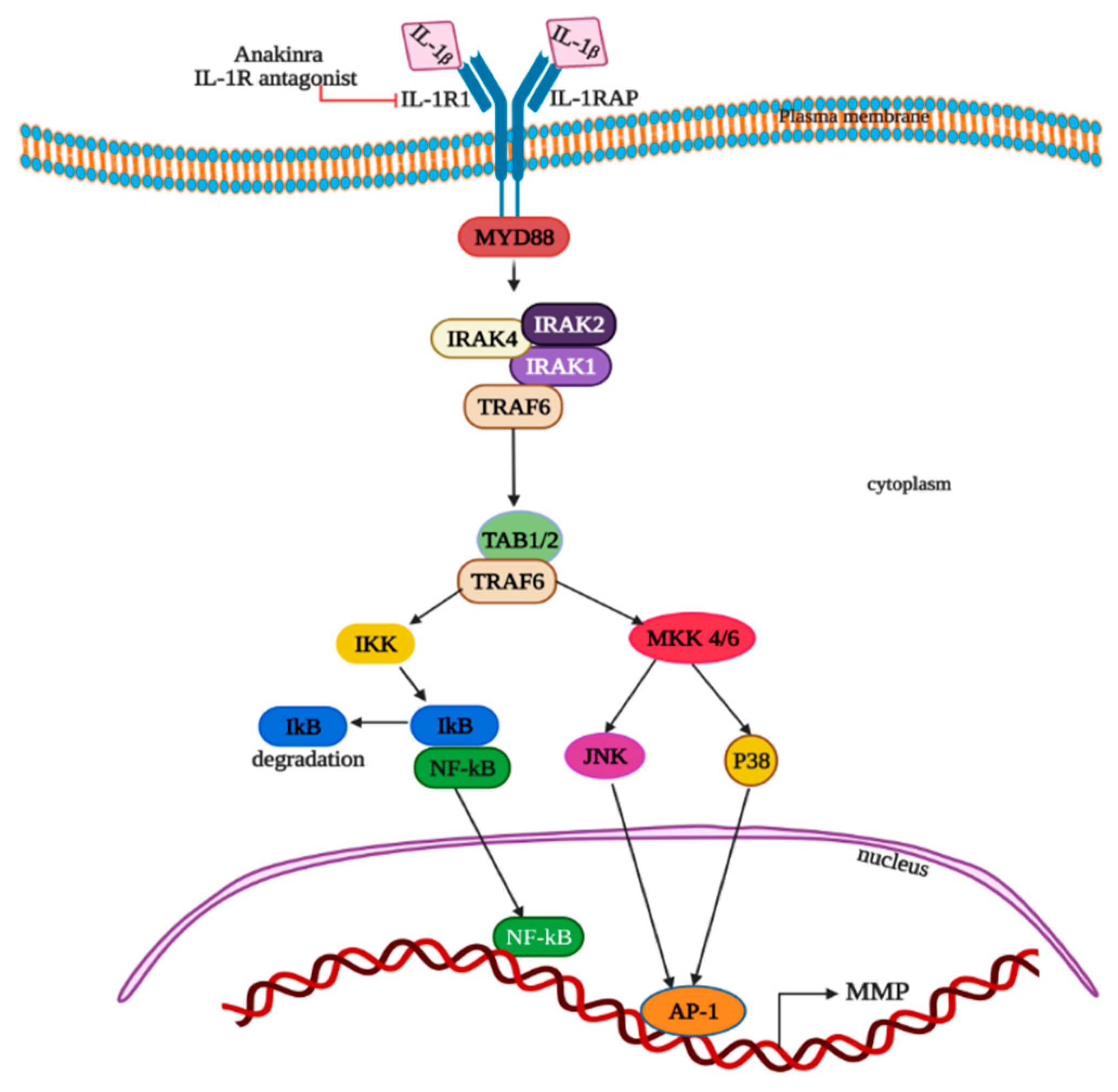
5. Interleukin (IL-1β)
5.1. IL-1β Discovery
5.2. IL-1β Expression and Regulation in Primary Colon Tumor and Hepatic Metastasis Tissue
5.3. IL-1β Function and Therapeutically Targeting Potential
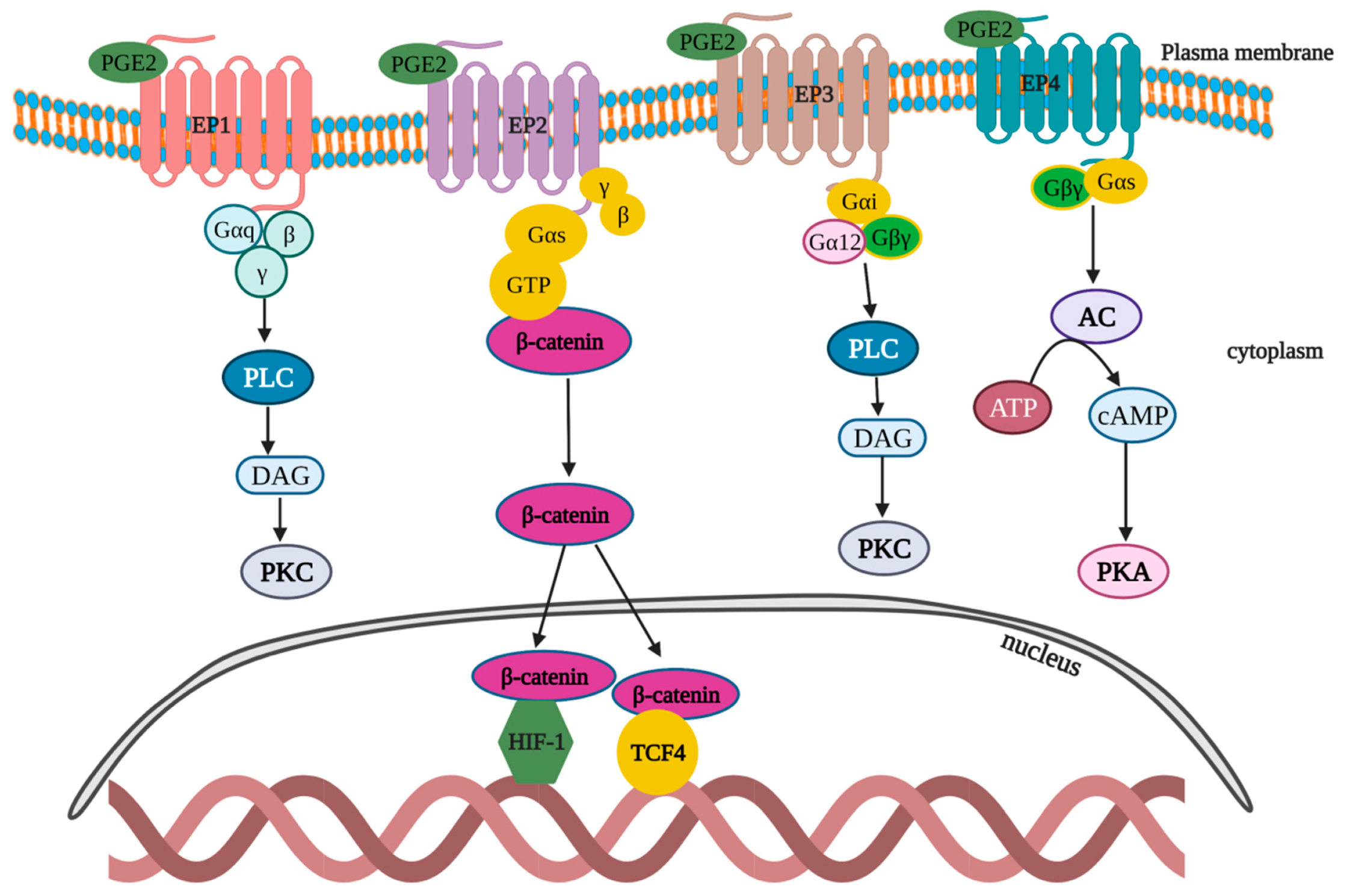
6. Lipid Molecule PGE2
6.1. PGE2 Discovery
6.2. PGE2 Expression and Regulation in Primary Colon Tumor and Hepatic Metastasis
6.3. PGE2 Function and Therapeutic Targeting Potential
7. Chemokines
8. CXCL1
8.1. CXCL1 Discovery
8.2. CXCL1 Expression and Regulation in Primary Colon Tumor and Hepatic Metastasis
8.3. CXCL1 Function and Therapeutic Targeting Potential
9. CXCL2
9.1. CXCL2 Discovery
9.2. CXCL2 Expression and Regulation in Primary Colon Tumor and Hepatic Metastasis
9.3. CXCL2 Function and Therapeutic Targeting Potential
10. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Gastroenterol. Rev./Przegląd Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Vatandoust, S.; Price, T.J.; Karapetis, C.S. Colorectal cancer: Metastases to a single organ. World J. Gastroenterol. 2015, 21, 11767–11776. [Google Scholar] [CrossRef] [PubMed]
- Zarour, L.R.; Anand, S.; Billingsley, K.G.; Bisson, W.H.; Cercek, A.; Clarke, M.F.; Coussens, L.M.; Gast, C.E.; Geltzeiler, C.B.; Hansen, L.; et al. Colorectal Cancer Liver Metastasis: Evolving Paradigms and Future Directions. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Bazzichetto, C.; Conciatori, F.; Falcone, I.; Cognetti, F.; Milella, M.; Ciuffreda, L. Advances in Tumor-Stroma Interactions: Emerging Role of Cytokine Network in Colorectal and Pancreatic Cancer. J. Oncol. 2019, 2019, 5373580. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Inamoto, S.; Yamamoto, T.; Ogawa, R.; Taketo, M.M.; Sakai, Y. The Role of Chemokines in Promoting Colorectal Cancer Invasion/Metastasis. Int. J. Mol. Sci. 2016, 17, 643. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Deng, C.-X. Effect of Stromal Cells in Tumor Microenvironment on Metastasis Initiation. Int. J. Biol. Sci. 2018, 14, 2083–2093. [Google Scholar] [CrossRef]
- Klampfer, L. Cytokines, inflammation and colon cancer. Curr. Cancer Drug Targets 2011, 11, 451–464. [Google Scholar] [CrossRef]
- le Rolle, A.-F.; Chiu, T.K.; Fara, M.; Shia, J.; Zeng, Z.; Weiser, M.R.; Paty, P.B.; Chiu, V.K. The prognostic significance of CXCL1 hypersecretion by human colorectal cancer epithelia and myofibroblasts. J. Transl. Med. 2015, 13, 199. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, H.; Brown, J.; Daikoku, T.; Ning, W.; Shi, Q.; Richmond, A.; Strieter, R.; Dey, S.K.; DuBois, R.N. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J. Exp. Med. 2006, 203, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Moulton, V.R. Chapter 17—Cytokines. In Systemic Lupus Erythematosus; Tsokos, G.C., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 137–141. [Google Scholar]
- Donnelly, R.P.; Young, H.A.; Rosenberg, A.S. An overview of cytokines and cytokine antagonists as therapeutic agents. Ann. N. Y. Acad. Sci. 2009, 1182, 1–13. [Google Scholar] [CrossRef]
- Lee, S.; Margolin, K. Cytokines in cancer immunotherapy. Cancers 2011, 3, 3856–3893. [Google Scholar] [CrossRef]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [Green Version]
- Pennica, D.; Nedwin, G.E.; Hayflick, J.S.; Seeburg, P.H.; Derynck, R.; Palladino, M.A.; Kohr, W.J.; Aggarwal, B.B.; Goeddel, D.V. Human tumour necrosis factor: Precursor structure, expression and homology to lymphotoxin. Nature 1984, 312, 724–729. [Google Scholar] [CrossRef]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Idriss, H.T.; Naismith, J.H. TNFα and the TNF receptor superfamily: Structure-function relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Zins, K.; Abraham, D.; Sioud, M.; Aharinejad, S. Colon Cancer Cell–Derived Tumor Necrosis Factor-α Mediates the Tumor Growth–Promoting Response in Macrophages by Up-regulating the Colony-Stimulating Factor-1 Pathway. Cancer Res. 2007, 67, 1038–1045. [Google Scholar] [CrossRef] [Green Version]
- Cox, G.W.; Melillo, G.; Chattopadhyay, U.; Mullet, D.; Fertel, R.H.; Varesio, L. Tumor necrosis factor-alpha-dependent production of reactive nitrogen intermediates mediates IFN-gamma plus IL-2-induced murine macrophage tumoricidal activity. J. Immunol. 1992, 149, 3290–3296. [Google Scholar]
- Xue, X.; Ramakrishnan, S.; Anderson, E.; Taylor, M.; Zimmermann, E.M.; Spence, J.R.; Huang, S.; Greenson, J.K.; Shah, Y.M. Endothelial PAS Domain Protein 1 Activates the Inflammatory Response in the Intestinal Epithelium to Promote Colitis in Mice. Gastroenterology 2013, 145, 831–841. [Google Scholar] [CrossRef] [Green Version]
- Al Obeed, O.A.; Alkhayal, K.A.; Al Sheikh, A.; Zubaidi, A.M.; Vaali-Mohammed, M.-A.; Boushey, R.; McKerrow, J.H.; Abdulla, M.-H. Increased expression of tumor necrosis factor-α is associated with advanced colorectal cancer stages. World J. Gastroenterol. 2014, 20, 18390–18396. [Google Scholar] [CrossRef]
- Stanilov, N.; Miteva, L.; Dobreva, Z.; Stanilova, S. Colorectal cancer severity and survival in correlation with tumour necrosis factor-alpha. Biotechnol. Biotechnol. Equip. 2014, 28, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.E.; Reddy, A.B.M.; Dietzmann, K.; Suriano, A.R.; Kocieda, V.P.; Stewart, M.; Bhatia, M. Epigenetic regulation of tumor necrosis factor alpha. Mol. Cell. Biol. 2007, 27, 5147–5160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, W.; Gencheng, H.; Yu, C.; Ke, W.; Guijun, L.; Renxi, W.; He, X.; Xinying, L.; Chunmei, H.; Beifen, S.; et al. Protective role of tumor necrosis factor (TNF) receptors in chronic intestinal inflammation: TNFR1 ablation boosts systemic inflammatory response. Lab. Investig. 2013, 93, 1024–1035. [Google Scholar]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef]
- Mager, L.F.; Wasmer, M.-H.; Rau, T.T.; Krebs, P. Cytokine-Induced Modulation of Colorectal Cancer. Front. Oncol. 2016, 6, 96. [Google Scholar] [CrossRef]
- Tam, S.Y.; Law, H.K.-W. JNK in Tumor Microenvironment: Present Findings and Challenges in Clinical Translation. Cancers 2021, 13, 2196. [Google Scholar] [CrossRef]
- Mohebali, N.; Pandurangan, A.K.; Mustafa, M.R.; Anandasadagopan, S.K.; Alagumuthu, T. Vernodalin induces apoptosis through the activation of ROS/JNK pathway in human colon cancer cells. J. Biochem. Mol. Toxicol. 2020, 34, e22587. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, Z. TNF-α promotes colon cancer cell migration and invasion by upregulating TROP-2. Oncol. Lett. 2018, 15, 3820–3827. [Google Scholar] [CrossRef]
- Hamilton, K.E.; Simmons, J.G.; Ding, S.; Van Landeghem, L.; Lund, P.K. Cytokine induction of tumor necrosis factor receptor 2 is mediated by STAT3 in colon cancer cells. Mol. Cancer Res. MCR 2011, 9, 1718–1731. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Rong, L.; Li, X.; Liu, X.; Deng, J.; Wu, H.; Xu, X.; Erben, U.; Wu, P.; Syrbe, U.; et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J. Clin. Investig. 2012, 122, 4094–4104. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Bi, J.; Liang, Q.; Wang, S.; Zhang, L.; Han, F.; Li, S.; Qiu, B.; Fan, X.; Chen, W.; et al. VCAM1 Promotes Tumor Cell Invasion and Metastasis by Inducing EMT and Transendothelial Migration in Colorectal Cancer. Front. Oncol. 2020, 10, 1066. [Google Scholar] [CrossRef]
- Hale, L.P.; Greer, P.K. A novel murine model of inflammatory bowel disease and inflammation-associated colon cancer with ulcerative colitis-like features. PLoS ONE 2012, 7, e41797. [Google Scholar] [CrossRef]
- Reissfelder, C.; Stamova, S.; Gossmann, C.; Braun, M.; Bonertz, A.; Walliczek, U.; Grimm, M.; Rahbari, N.N.; Koch, M.; Saadati, M.; et al. Tumor-specific cytotoxic T lymphocyte activity determines colorectal cancer patient prognosis. J. Clin. Investig. 2015, 125, 739–751. [Google Scholar] [CrossRef]
- Zhao, X.; Mohaupt, M.; Jiang, J.; Liu, S.; Li, B.; Qin, Z. Tumor Necrosis Factor Receptor 2–Mediated Tumor Suppression Is Nitric Oxide Dependent and Involves Angiostasis. Cancer Res. 2007, 67, 4443–4450. [Google Scholar] [CrossRef] [Green Version]
- Blatner, N.R.; Mulcahy, M.F.; Dennis, K.L.; Scholtens, D.; Bentrem, D.J.; Phillips, J.D.; Ham, S.; Sandall, B.P.; Khan, M.W.; Mahvi, D.M.; et al. Expression of RORgammat marks a pathogenic regulatory T cell subset in human colon cancer. Sci. Transl. Med. 2012, 4, 164ra59. [Google Scholar] [CrossRef] [Green Version]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-α in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar] [CrossRef]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek Mark, A.; Seki, E.; Hidalgo, J.; et al. ER Stress Cooperates with Hypernutrition to Trigger TNF-Dependent Spontaneous HCC Development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Ham, B.; Fernandez, M.C.; D’Costa, Z.; Brodt, P. The diverse roles of the TNF axis in cancer progression and metastasis. Trends Cancer Res. 2016, 11, 1–27. [Google Scholar]
- Nyboe Andersen, N.; Pasternak, B.; Basit, S.; Andersson, M.; Svanström, H.; Caspersen, S.; Munkholm, P.; Hviid, A.; Jess, T. Association between tumor necrosis factor-α antagonists and risk of cancer in patients with inflammatory bowel disease. JAMA 2014, 311, 2406–2413. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.-F.; Sun, K.; Chen, X.-J.; Zhao, X.; Cai, N.; Liu, Y.-J.; Xu, L.-M.; Kong, X.-M.; Wei, L.-X. Inhibition of tumor necrosis factor alpha reduces the outgrowth of hepatic micrometastasis of colorectal tumors in a mouse model of liver ischemia-reperfusion injury. J. Biomed. Sci. 2014, 21, 1. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.R.; Charles, K.A.; Hoare, S.A.; Rye, R.L.; Jodrell, D.I.; Aird, R.E.; Vora, R.; Prabhakar, U.; Nakada, M.; Corringham, R.E.; et al. A clinical study assessing the tolerability and biological effects of infliximab, a TNF-alpha inhibitor, in patients with advanced cancer. Ann. Oncol. 2008, 19, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T. The biology of interleukin-6. Blood 1989, 74, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauldie, J.; Richards, C.; Harnish, D.; Lansdorp, P.; Baumann, H. Interferon beta 2/B-cell stimulatory factor type 2 shares identity with monocyte-derived hepatocyte-stimulating factor and regulates the major acute phase protein response in liver cells. Proc. Natl. Acad. Sci. USA 1987, 84, 7251–7255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andus, T.; Geiger, T.; Hirano, T.; Northoff, H.; Ganter, U.; Bauer, J.; Kishimoto, T.; Heinrich, P.C. Recombinant human B cell stimulatory factor 2 (BSF-2/IFN-beta 2) regulates beta-fibrinogen and albumin mRNA levels in Fao-9 cells. FEBS Lett. 1987, 221, 18–22. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Jawa, R.S.; Anillo, S.; Huntoon, K.; Baumann, H.; Kulaylat, M. Analytic review: Interleukin-6 in surgery, trauma, and critical care: Part I: Basic science. J. Intensive Care Med. 2011, 26, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Denson, J.L.; Brüning, J.C. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 2015, 36, 92–101. [Google Scholar] [CrossRef]
- Xu, H.; Lai, W.; Zhang, Y.; Liu, L.; Luo, X.; Zeng, Y.; Wu, H.; Lan, Q.; Chu, Z. Tumor-associated macrophage-derived IL-6 and IL-8 enhance invasive activity of LoVo cells induced by PRL-3 in a KCNN4 channel-dependent manner. BMC Cancer 2014, 14, 330. [Google Scholar] [CrossRef] [Green Version]
- Belluco, C.; Nitti, D.; Frantz, M.; Toppan, P.; Basso, D.; Plebani, M.; Lise, M.; Jessup, J.M. Interleukin-6 Blood Level Is Associated with Circulating Carcinoembryonic Antigen and Prognosis in Patients with Colorectal Cancer. Ann. Surg. Oncol. 2000, 7, 133–138. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Kitamura, H.; Xiang, H.; Ohno, Y.; Homma, S.; Kawamura, H.; Takahashi, N.; Kamiyama, T.; Tanino, M.; Taketomi, A. IL6 Modulates the Immune Status of the Tumor Microenvironment to Facilitate Metastatic Colonization of Colorectal Cancer Cells. Cancer Immunol. Res. 2019, 7, 1944–1957. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.; Kirkeby, L.T.; Olsen, J.; Eiholm, S.; Jess, P.; Gögenur, I.; Troelsen, J.T. High interleukin-6 mRNA expression is a predictor of relapse in colon cancer. Anticancer Res. 2015, 35, 2235–2240. [Google Scholar]
- Saji, H.; Koike, M.; Yamori, T.; Saji, S.; Seiki, M.; Matsushima, K.; Toi, M. Significant correlation of monocyte chemoattractant protein-1 expression with neovascularization and progression of breast carcinoma. Cancer 2001, 92, 1085–1091. [Google Scholar] [CrossRef]
- Yoshidome, H.; Kohno, H.; Shida, T.; Kimura, F.; Shimizu, H.; Ohtsuka, M.; Nakatani, Y.; Miyazaki, M. Significance of monocyte chemoattractant protein-1 in angiogenesis and survival in colorectal liver metastases. Int. J. Oncol. 2009, 34, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Knupfer, H.; Preiss, R. Serum interleukin-6 levels in colorectal cancer patients—A summary of published results. Int. J. Colorectal Dis. Clin. Mol. Gastroenterol. 2010, 25, 135–140. [Google Scholar] [CrossRef]
- Tommelein, J.; Verset, L.; Boterberg, T.; Demetter, P.; Bracke, M.; De Wever, O. Cancer-associated fibroblasts connect metastasis-promoting communication in colorectal cancer. Front. Oncol. 2015, 5, 63. [Google Scholar] [CrossRef] [Green Version]
- Mueller, L.; von Seggern, L.; Schumacher, J.; Goumas, F.; Wilms, C.; Braun, F.; Broering, D.C. TNF-α similarly induces IL-6 and MCP-1 in fibroblasts from colorectal liver metastases and normal liver fibroblasts. Biochem. Biophys. Res. Commun. 2010, 397, 586–591. [Google Scholar] [CrossRef]
- Xu, K.; Zhan, Y.; Yuan, Z.; Qiu, Y.; Wang, H.; Fan, G.; Wang, J.; Li, W.; Cao, Y.; Shen, X.; et al. Hypoxia Induces Drug Resistance in Colorectal Cancer through the HIF-1α/miR-338-5p/IL-6 Feedback Loop. Mol. Ther. 2019, 27, 1810–1824. [Google Scholar] [CrossRef]
- De Simone, V.; Franzè, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-α synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef]
- Multhoff, G.; Molls, M.; Radons, J. Chronic Inflammation in Cancer Development. Front. Immunol. 2012, 2, 98. [Google Scholar] [CrossRef] [Green Version]
- Holmer, R.; Wätzig, G.H.; Tiwari, S.; Rose-John, S.; Kalthoff, H. Interleukin-6 trans-signaling increases the expression of carcinoembryonic antigen-related cell adhesion molecules 5 and 6 in colorectal cancer cells. BMC Cancer 2015, 15, 975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Song, P.; Zhong, T.; Wang, X.; Xiang, X.; Liu, Q.; Chen, H.; Xia, T.; Liu, H.; Niu, Y.; et al. The inflammatory cytokine IL-6 induces FRA1 deacetylation promoting colorectal cancer stem-like properties. Oncogene 2019, 38, 4932–4947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.-C.; Chang, Y.-F. Serum interleukin-6 levels reflect the disease status of colorectal cancer. J. Surg. Oncol. 2003, 83, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Huot, J.R.; Novinger, L.J.; Pin, F.; Bonetto, A. HCT116 colorectal liver metastases exacerbate muscle wasting in a mouse model for the study of colorectal cancer cachexia. Dis. Models Mech. 2020, 13, dmm043166. [Google Scholar] [CrossRef] [Green Version]
- Heichler, C.; Scheibe, K.; Schmied, A.; Geppert, C.I.; Schmid, B.; Wirtz, S.; Thoma, O.-M.; Kramer, V.; Waldner, M.J.; Büttner, C.; et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut 2020, 69, 1269–1282. [Google Scholar] [CrossRef]
- Han, J.; Xi, Q.; Meng, Q.; Liu, J.; Zhang, Y.; Han, Y.; Zhuang, Q.; Jiang, Y.; Ding, Q.; Wu, G. Interleukin-6 promotes tumor progression in colitis-associated colorectal cancer through HIF-1α regulation. Oncol. Lett. 2016, 12, 4665–4670. [Google Scholar] [CrossRef] [Green Version]
- Angevin, E.; Tabernero, J.; Elez, E.; Cohen, S.J.; Bahleda, R.; Van Laethem, J.-L.; Ottensmeier, C.; Lopez-Martin, J.A.; Clive, S.; Joly, F. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 2192–2204. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.A.; Scheller, J.; Rose-John, S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Investig. 2011, 121, 3375–3383. [Google Scholar] [CrossRef] [Green Version]
- Seavey, M.M.; Lu, L.D.; Stump, K.L.; Wallace, N.H.; Hockeimer, W.; O’Kane, T.M.; Ruggeri, B.A.; Dobrzanski, P. Therapeutic efficacy of CEP-33779, a novel selective JAK2 inhibitor, in a mouse model of colitis-induced colorectal cancer. Mol. Cancer Ther. 2012, 11, 984–993. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. The history of fever, leukocytic pyrogen and interleukin-1. Temperature 2015, 2, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. IL-1: Discoveries, controversies and future directions. Eur. J. Immunol. 2010, 40, 599–606. [Google Scholar] [CrossRef]
- Libby, P. Interleukin-1 beta as a target for atherosclerosis therapy: Biological basis of CANTOS and beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef]
- Ozato, K.; Tsujimura, H.; Tamura, T. Toll-like receptor signaling and regulation of cytokine gene expression in the immune system. Biotechniques 2002, 33, S66–S75. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, K.; Han, G.C.; Wang, R.X.; Xiao, H.; Hou, C.M.; Guo, R.F.; Dou, Y.; Shen, B.F.; Li, Y.; et al. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunol. 2014, 7, 1106–1115. [Google Scholar] [CrossRef]
- Kim, J.; Bae, J.-S. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediat. Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Irizarry-Caro, R.A.; McDaniel, M.M.; Chawla, A.S.; Carroll, K.R.; Overcast, G.R.; Philip, N.H.; Oberst, A.; Chervonsky, A.V.; Katz, J.D.; et al. T cells instruct myeloid cells to produce inflammasome-independent IL-1β and cause autoimmunity. Nat. Immunol. 2020, 21, 65–74. [Google Scholar] [CrossRef]
- Lorenz, J.J.; Furdon, P.J.; Taylor, J.D.; Verghese, M.W.; Chandra, G.; Kost, T.A.; Haneline, S.A.; Roner, L.A.; Gray, J.G. A cyclic adenosine 3′,5′-monophosphate signal is required for the induction of IL-1 beta by TNF-alpha in human monocytes. J. Immunol. 1995, 155, 836–844. [Google Scholar]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.L.; Berggren, K.L.; Restrepo Cruz, S.; Gan, G.N.; Beswick, E.J. Inhibition of MK2 suppresses IL-1β, IL-6, and TNF-α-dependent colorectal cancer growth. Int. J. Cancer 2018, 142, 1702–1711. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.; Zhang, D.; Bao, C. Two variants of Interleukin-1B gene are associated with the decreased risk, clinical features, and better overall survival of colorectal cancer: A two-center case-control study. Aging 2018, 10, 4084–4092. [Google Scholar] [CrossRef]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, M.; Bennett, N.; Standifer, C.; Smith, A.; Han, A.; Bettaieb, A.; Whelan, J.; Donohoe, D.R. Characterization of the Pro-Inflammatory Cytokine IL-1β on Butyrate Oxidation in Colorectal Cancer Cells. J. Cell. Biochem. 2017, 118, 1614–1621. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Wang, F.; Chen, H.M.; Yan, X.J. κ-carrageenan induces the disruption of intestinal epithelial Caco-2 monolayers by promoting the interaction between intestinal epithelial cells and immune cells. Mol. Med. Rep. 2013, 8, 1635–1642. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 36107. [Google Scholar] [CrossRef]
- Kaler, P.; Galea, V.; Augenlicht, L.; Klampfer, L. Tumor associated macrophages protect colon cancer cells from TRAIL-induced apoptosis through IL-1beta-dependent stabilization of Snail in tumor cells. PLoS ONE 2010, 5, e11700. [Google Scholar] [CrossRef]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Macrophage-derived IL-1β stimulates Wnt signaling and growth of colon cancer cells: A crosstalk interrupted by vitamin D 3. Oncogene 2009, 28, 3892–3902. [Google Scholar] [CrossRef] [Green Version]
- Kapral, M.; Wawszczyk, J.; Jurzak, M.; Hollek, A.; Węglarz, L. The effect of inositol hexaphosphate on the expression of selected metalloproteinases and their tissue inhibitors in IL-1β-stimulated colon cancer cells. Int. J. Colorectal Dis. 2012, 27, 1419–1428. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, L.; Pappan, L.; Galliher-Beckley, A.; Shi, J. IL-1β promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer 2012, 11, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakae, S.; Asano, M.; Horai, R.; Iwakura, Y. Interleukin-1 beta, but not interleukin-1 alpha, is required for T-cell-dependent antibody production. Immunology 2001, 104, 402–409. [Google Scholar] [CrossRef]
- Postlethwaite, A.E.; Raghow, R.; Stricklin, G.P.; Poppleton, H.; Seyer, J.M.; Kang, A.H. Modulation of fibroblast functions by interleukin 1: Increased steady-state accumulation of type I procollagen messenger RNAs and stimulation of other functions but not chemotaxis by human recombinant interleukin 1 alpha and beta. J. Cell. Biol. 1988, 106, 311–318. [Google Scholar] [CrossRef]
- Duhen, T.; Campbell, D.J. IL-1β promotes the differentiation of polyfunctional human CCR6+CXCR3+ Th1/17 cells that are specific for pathogenic and commensal microbes. J. Immunol. 2014, 193, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, K.; Yoshimoto, T.; Torigoe, K.; Kurimoto, M.; Matsui, K.; Hada, T.; Okamura, H.; Nakanishi, K. IL-12 synergizes with IL-18 or IL-1beta for IFN-gamma production from human T cells. Int. Immunol. 2000, 12, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.J.; Houston, A.; Brint, E. IL-1 Family Members in Cancer; Two Sides to Every Story. Front. Immunol. 2019, 10, 1197. [Google Scholar] [CrossRef] [Green Version]
- Dmitrieva-Posocco, O.; Dzutsev, A.; Posocco, D.F.; Hou, V.; Yuan, W.; Thovarai, V.; Mufazalov, I.A.; Gunzer, M.; Shilovskiy, I.P.; Khaitov, M.R.; et al. Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal Cancer. Immunity 2019, 50, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Litmanovich, A.; Khazim, K.; Cohen, I. The Role of Interleukin-1 in the Pathogenesis of Cancer and its Potential as a Therapeutic Target in Clinical Practice. Oncol. Ther. 2018, 6, 109–127. [Google Scholar] [CrossRef] [Green Version]
- Isambert, N.; Hervieu, A.; Rébé, C.; Hennequin, A.; Borg, C.; Zanetta, S.; Chevriaux, A.; Richard, C.; Derangère, V.; Limagne, E.; et al. Fluorouracil and bevacizumab plus anakinra for patients with metastatic colorectal cancer refractory to standard therapies (IRAFU): A single-arm phase 2 study. Oncoimmunology 2018, 7, e1474319. [Google Scholar] [CrossRef]
- Saha, S. mPGES-1: A key Regulator of Fever and Neonatal Respiratory Depression; Karolinska Institutet: Solna City, Sweden, 2006. [Google Scholar]
- Marks, F.; Fürstenberger, G. Prostaglandins, Leukotrienes and Other Eicosanoids: From Biogenesis to Clinical Application; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Yoo, Y.S.; Lim, S.C.; Kim, K.J. Prognostic significance of cytosolic phospholipase A2 expression in patients with colorectal cancer. J. Korean Surg. Soc. 2011, 80, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; He, X.; Xie, C.; Hua, S.; Li, J.; Wang, T.; Yao, M.; Vignarajan, S.; Teng, Y.; Hejazi, L.; et al. Targeting cytosolic phospholipase A2 α in colorectal cancer cells inhibits constitutively activated protein kinase B (AKT) and cell proliferation. Oncotarget 2014, 5, 12304–12316. [Google Scholar] [CrossRef]
- Buhmeida, A.; Bendardaf, R.; Hilska, M.; Laine, J.; Collan, Y.; Laato, M.; Syrjänen, K.; Pyrhönen, S. PLA2 (group IIA phospholipase A2) as a prognostic determinant in stage II colorectal carcinoma. Ann. Oncol. 2009, 20, 1230–1235. [Google Scholar] [CrossRef]
- Murase, R.; Taketomi, Y.; Miki, Y.; Nishito, Y.; Saito, M.; Fukami, K.; Yamamoto, K.; Murakami, M. Group III phospholipase A2 promotes colitis and colorectal cancer. Sci. Rep. 2017, 7, 12261. [Google Scholar] [CrossRef] [Green Version]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Park, J.Y.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clin. Immunol. 2006, 119, 229–240. [Google Scholar] [CrossRef]
- Kaidi, A.; Qualtrough, D.; Williams, A.C.; Paraskeva, C. Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res. 2006, 66, 6683–6691. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Shah, Y.M. Hypoxia-inducible factor-2α is essential in activating the COX2/mPGES-1/PGE 2 signaling axis in colon cancer. Carcinogenesis 2012, 34, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, K.; Hamaguchi, A.; Fukushima, K.; Nakano, Y.; Regan, J.W.; Mashimo, M.; Fujino, H. Down-regulation of the expression of cyclooxygenase-2 and prostaglandin E(2) by interleukin-4 is mediated via a reduction in the expression of prostanoid EP4 receptors in HCA-7 human colon cancer cells. Eur. J. Pharmacol. 2022, 920, 174863. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawada, K.; Sakai, Y. Prostaglandin E2/EP Signaling in the Tumor Microenvironment of Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 6254. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.; Kim, M.; Hwang, S.W. Molecular mechanisms underlying the actions of arachidonic acid-derived prostaglandins on peripheral nociception. J. Neuroinflamm. 2020, 17, 30. [Google Scholar] [CrossRef]
- Greenhough, A.; Smartt, H.J.M.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE 2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivia, M. COX-2/PGE2 Signaling: A Target for Colorectal Cancer Prevention; Cayman Chemicls: Ann Arbor, MI, USA, 2009. [Google Scholar]
- Shoji, Y.; Takahashi, M.; Kitamura, T.; Watanabe, K.; Kawamori, T.; Maruyama, T.; Sugimoto, Y.; Negishi, M.; Narumiya, S.; Sugimura, T.; et al. Downregulation of prostaglandin E receptor subtype EP3 during colon cancer development. Gut 2004, 53, 1151–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, U.; Iwatsubo, K.; Umemura, M.; Fujita, T.; Ishikawa, Y. The Prostanoid EP4 Receptor and Its Signaling Pathway. Pharmacol. Rev. 2013, 65, 1010–1052. [Google Scholar] [CrossRef] [Green Version]
- Ganea, D.; Kocieda, V.; Kong, W.; Yen, J.-H. Modulation of dendritic cell function by PGE2 and DHA: A framework for understanding the role of dendritic cells in neuroinflammation. Clin. Lipidol. 2011, 6, 277–291. [Google Scholar] [CrossRef] [Green Version]
- Hooper, K.M.; Yen, J.-H.; Kong, W.; Rahbari, K.M.; Kuo, P.-C.; Gamero, A.M.; Ganea, D. Prostaglandin E2 Inhibition of IL-27 Production in Murine Dendritic Cells: A Novel Mechanism That Involves IRF1. J. Immunol. 2017, 198, 1521–1530. [Google Scholar] [CrossRef] [Green Version]
- Van Elssen, C.H.M.J.; Vanderlocht, J.; Oth, T.; Senden-Gijsbers, B.L.M.G.; Germeraad, W.T.V.; Bos, G.M.J. Inflammation restraining effects of prostaglandin E2 on natural killer–dendritic cell (NK-DC) interaction are imprinted during DC maturation. Blood 2011, 118, 2473–2482. [Google Scholar] [CrossRef] [Green Version]
- Harizi, H.; Juzan, M.; Grosset, C.; Rashedi, M.; Gualde, N. Dendritic cells issued in vitro from bone marrow produce PGE2 that contributes to the immunomodulation induced by antigen-presenting cells. Cell. Immunol. 2001, 209, 19–28. [Google Scholar] [CrossRef]
- Wang, D.; Fu, L.; Sun, H.; Guo, L.; DuBois, R.N. Prostaglandin E2 Promotes Colorectal Cancer Stem Cell Expansion and Metastasis in Mice. Gastroenterology 2015, 149, 1884–1895. [Google Scholar] [CrossRef] [Green Version]
- Obermajer, N.; Muthuswamy, R.; Lesnock, J.; Edwards, R.P.; Kalinski, P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 2011, 118, 5498–5505. [Google Scholar] [CrossRef]
- O’Callaghan, G.; Kelly, J.; Shanahan, F.; Houston, A. Prostaglandin E2 stimulates Fas ligand expression via the EP1 receptor in colon cancer cells. Br. J. Cancer 2008, 99, 502–512. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Löffler, I.; Grün, M.; Böhmer, F.D.; Rubio, I. Role of cAMP in the promotion of colorectal cancer cell growth by Prostaglandin E2. BMC Cancer 2008, 8, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes-Madrid, D.L.; Nagi, S.; Asting Gustafsson, A. FosB transcription factor regulates COX-2 expression in colorectal cancer cells without affecting PGE2 expression. Oncol. Lett. 2017, 13, 1411–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, A.; Yaghoobi, M.M.; Gholamhoseinian Najar, A.; Kalantari-Khandani, B.; Sharifi, H.; Saravani, M. HSP90 Inhibition Suppresses PGE2 Production via Modulating COX-2 and 15-PGDH Expression in HT-29 Colorectal Cancer Cells. Inflammation 2016, 39, 1116–1123. [Google Scholar] [CrossRef]
- Kasai, T.; Nakanishi, T.; Ohno, Y.; Shimada, H.; Nakamura, Y.; Arakawa, H.; Tamai, I. Role of OATP2A1 in PGE2 secretion from human colorectal cancer cells via exocytosis in response to oxidative stress. Exp. Cell Res. 2016, 341, 123–131. [Google Scholar] [CrossRef]
- Walz, D.A.; Wu, V.Y.; de Lamo, R.; Dene, H.; McCoy, L.E. Primary structure of human platelet factor 4. Thromb. Res. 1977, 11, 893–898. [Google Scholar] [CrossRef]
- Richmond, A.; Balentien, E.; Thomas, H.G.; Flaggs, G.; Barton, D.E.; Spiess, J.; Bordoni, R.; Francke, U.; Derynck, R. Molecular characterization and chromosomal mapping of melanoma growth stimulatory activity, a growth factor structurally related to beta-thromboglobulin. EMBO J. 1988, 7, 2025–2033. [Google Scholar] [CrossRef]
- Richmond, A.; Thomas, H.G. Melanoma growth stimulatory activity: Isolation from human melanoma tumors and characterization of tissue distribution. J. Cell. Biochem. 1988, 36, 185–198. [Google Scholar] [CrossRef]
- Martins-Green, M.; Petreaca, M.; Wang, L. Chemokines and Their Receptors Are Key Players in the Orchestra That Regulates Wound Healing. Adv. Wound Care 2013, 2, 327–347. [Google Scholar] [CrossRef] [Green Version]
- Girbl, T.; Lenn, T.; Perez, L.; Rolas, L.; Barkaway, A.; Thiriot, A.; Del Fresno, C.; Lynam, E.; Hub, E.; Thelen, M.; et al. Distinct Compartmentalization of the Chemokines CXCL1 and CXCL2 and the Atypical Receptor ACKR1 Determine Discrete Stages of Neutrophil Diapedesis. Immunity 2018, 49, 1062–1076. [Google Scholar] [CrossRef] [Green Version]
- De Filippo, K.; Dudeck, A.; Hasenberg, M.; Nye, E.; van Rooijen, N.; Hartmann, K.; Gunzer, M.; Roers, A.; Hogg, N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 2013, 121, 4930–4937. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Liu, W.; Zheng, Y.; Wang, S.; Yang, B.; Li, M.; Song, J.; Zhang, F.; Zhang, X.; Wang, Q.; et al. CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-κB/SOX4 signaling. Cell Death Dis. 2018, 9, 880. [Google Scholar] [CrossRef]
- Ma, K.; Yang, L.; Shen, R.; Kong, B.; Chen, W.; Liang, J.; Tang, G.; Zhang, B. Th17 cells regulate the production of CXCL1 in breast cancer. Int. Immunopharmacol. 2018, 56, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Bandapalli, O.R.; Ehrmann, F.; Ehemann, V.; Gaida, M.; Macher-Goeppinger, S.; Wente, M.; Schirmacher, P.; Brand, K. Down-regulation of CXCL1 inhibits tumor growth in colorectal liver metastasis. Cytokine 2012, 57, 46–53. [Google Scholar] [CrossRef]
- Katoh, H.; Wang, D.; Daikoku, T.; Sun, H.; Dey, S.K.; Dubois, R.N. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013, 24, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Divella, R.; Daniele, A.; de Luca, R.; Simone, M.; Naglieri, E.; Savino, E.; Abbate, I.; Gadaleta, C.D.; Ranieri, G. Circulating Levels of VEGF and CXCL1 Are Predictive of Metastatic Organotropismin in Patients with Colorectal Cancer. Anticancer Res. 2017, 37, 4867–4871. [Google Scholar]
- Zhuo, C.; Wu, X.; Li, J.; Hu, D.; Jian, J.; Chen, C.; Zheng, X.; Yang, C. Chemokine (C-X-C motif) ligand 1 is associated with tumor progression and poor prognosis in patients with colorectal cancer. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.-L.; Chen, Y.-J.; Chang, W.-A.; Jian, S.-F.; Fan, H.-L.; Wang, J.-Y.; Kuo, P.-L. Interaction between Tumor-Associated Dendritic Cells and Colon Cancer Cells Contributes to Tumor Progression via CXCL1. Int. J. Mol. Sci. 2018, 19, 2427. [Google Scholar] [CrossRef] [Green Version]
- Lukaszewicz-Zając, M.; Pączek, S.; Mroczko, P.; Kulczyńska-Przybik, A. The Significance of CXCL1 and CXCL8 as Well as Their Specific Receptors in Colorectal Cancer. Cancer Manag. Res. 2020, 12, 8435–8443. [Google Scholar] [CrossRef]
- Lo, H.-M.; Lai, T.-H.; Li, C.-H.; Wu, W.-B. TNF-α induces CXCL1 chemokine expression and release in human vascular endothelial cells in vitro via two distinct signaling pathways. Acta Pharmacol. Sin. 2014, 35, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, R.; Yamamoto, T.; Hirai, H.; Hanada, K.; Kiyasu, Y.; Nishikawa, G.; Mizuno, R.; Inamoto, S.; Itatani, Y.; Sakai, Y.; et al. Loss of SMAD4 Promotes Colorectal Cancer Progression by Recruiting Tumor-Associated Neutrophils via the CXCL1/8–CXCR2 Axis. Clin. Cancer Res. 2019, 25, 2887–2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triner, D.; Xue, X.; Schwartz, A.J.; Jung, I.; Colacino, J.A.; Shah, Y.M. Epithelial Hypoxia-Inducible Factor 2α Facilitates the Progression of Colon Tumors through Recruiting Neutrophils. Mol. Cell. Biol. 2017, 37, e00481-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowser, J.L.; Phan, L.H.; Eltzschig, H.K. The Hypoxia-Adenosine Link during Intestinal Inflammation. J. Immunol. 2018, 200, 897–907. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Xu, X.; Xu, Q.; Ren, J.; Shen, S.; Fan, C.; Hou, Y. miR-19a promotes colitis-associated colorectal cancer by regulating tumor necrosis factor alpha-induced protein 3-NF-κB feedback loops. Oncogene 2017, 36, 3240–3251. [Google Scholar] [CrossRef]
- Zhai, Z.; Liu, W.; Kaur, M.; Luo, Y.; Domenico, J.; Samson, J.M.; Shellman, Y.G.; Norris, D.A.; Dinarello, C.A.; Spritz, R.A.; et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 2017, 36, 3820–3830. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Kikuchi, H.; Ohta, M.; Kawabata, T.; Hiramatsu, Y.; Kondo, K.; Baba, M.; Kamiya, K.; Tanaka, T.; Kitagawa, M.; et al. TSU68 prevents liver metastasis of colon cancer xenografts by modulating the premetastatic niche. Cancer Res. 2008, 68, 9754–9762. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Sun, H.; Wei, J.; Cen, B.; DuBois, R.N. CXCL1 Is Critical for Premetastatic Niche Formation and Metastasis in Colorectal Cancer. Cancer Res. 2017, 77, 3655–3665. [Google Scholar] [CrossRef] [Green Version]
- Rubie, C.; Frick, V.O.; Wagner, M.; Schuld, J.; Gräber, S.; Brittner, B.; Bohle, R.M.; Schilling, M.K. ELR+ CXC chemokine expression in benign and malignant colorectal conditions. BMC Cancer 2008, 8, 178. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Giardina, S.F.; Hamming, D.; Greenman, J.; Zachariah, E.; Bacolod, M.D.; Liu, H.; Shia, J.; Amenta, P.S.; Barany, F.; et al. GROalpha is highly expressed in adenocarcinoma of the colon and down-regulates fibulin-1. Clin. Cancer Res. 2006, 12, 5951–5959. [Google Scholar] [CrossRef] [Green Version]
- Kuo, P.-L.; Shen, K.-H.; Hung, S.-H.; Hsu, Y.-L. CXCL1/GROα increases cell migration and invasion of prostate cancer by decreasing fibulin-1 expression through NF-κB/HDAC1 epigenetic regulation. Carcinogenesis 2012, 33, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.A.; Schwarzbauer, J.E. A shared mechanism of adhesion modulation for tenascin-C and fibulin-1. Mol. Biol. Cell 2009, 20, 1141–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Ma, X.L.; Wei, Y.Q.; Wei, X.W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim. Biophys. Acta (BBA) Rev. Cancer 2019, 1871, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Varney, M.L.; Singh, S.; Li, A.; Mayer-Ezell, R.; Bond, R.; Singh, R.K. Small molecule antagonists for CXCR2 and CXCR1 inhibit human colon cancer liver metastases. Cancer Lett. 2011, 300, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.-C.; Liu, Y.-N.; Hu, Y.; Yang, Y.; Chen, Z. Macrophage inflammatory protein-2 as mediator of inflammation in acute liver injury. World J. Gastroenterol. 2017, 23, 3043–3052. [Google Scholar] [CrossRef]
- Kollmar, O.; Scheuer, C.; Menger, M.D.; Schilling, M.K. Macrophage Inflammatory Protein-2 Promotes Angiogenesis, Cell Migration, and Tumor Growth in Hepatic Metastasis. Ann. Surg. Oncol. 2006, 13, 263–275. [Google Scholar] [CrossRef]
- Lou, N.; Lennard Richard, M.L.; Yu, J.; Kindy, M.; Zhang, X.K. The Fli-1 transcription factor is a critical regulator for controlling the expression of chemokine C-X-C motif ligand 2 (CXCL2). Mol. Immunol. 2017, 81, 59–66. [Google Scholar] [CrossRef]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. Chapter 3—CXC Chemokines in Cancer Angiogenesis and Metastases. In Advances in Cancer Research; Vande Woude, G.F., Klein, G., Eds.; Academic Press: Cambridge, MA, USA, 2010; pp. 91–111. [Google Scholar]
- Gulati, K.; Jamsandekar, M.; Poluri, K.M. Mechanistic insights into molecular evolution of species-specific differential glycosaminoglycan binding surfaces in growth-related oncogene chemokines. R. Soc. Open Sci. 2017, 4, 171059. [Google Scholar] [CrossRef] [Green Version]
- Wolpe, S.D.; Sherry, B.; Juers, D.; Davatelis, G.; Yurt, R.W.; Cerami, A. Identification and characterization of macrophage inflammatory protein 2. Proc. Natl. Acad. Sci. USA 1989, 86, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Iida, N.; Grotendorst, G.R. Cloning and sequencing of a new gro transcript from activated human monocytes: Expression in leukocytes and wound tissue. Mol. Cell. Biol. 1990, 10, 5596–5599. [Google Scholar]
- Doll, D.; Keller, L.; Maak, M.; Boulesteix, A.-L.; Siewert, J.R.; Holzmann, B.; Janssen, K.-P. Differential expression of the chemokines GRO-2, GRO-3, and interleukin-8 in colon cancer and their impact on metastatic disease and survival. Int. J. Colorectal Dis. 2010, 25, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Shang, K.; Bai, Y.-P.; Wang, C.; Wang, Z.; Gu, H.-Y.; Du, X.; Zhou, X.-Y.; Zheng, C.-L.; Chi, Y.-Y.; Mukaida, N.; et al. Crucial Involvement of Tumor-Associated Neutrophils in the Regulation of Chronic Colitis-Associated Carcinogenesis in Mice. PLoS ONE 2012, 7, e51848. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Tan, X.; Luo, J.; Cui, B.; Lei, S.; Si, Z.; Shen, L.; Yao, H. GNA13 promotes tumor growth and angiogenesis by upregulating CXC chemokines via the NF-κB signaling pathway in colorectal cancer cells. Cancer Med. 2018, 7, 5611–5620. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.-L.; Lan, H.-Y.; Cheng, W.-C.; Huang, S.-C.; Yang, M.-H. Tumor stem-like cell-derived exosomal RNAs prime neutrophils for facilitating tumorigenesis of colon cancer. J. Hematol. Oncol. 2019, 12, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.C.; Baskaran, R.; Lee, N.H.; Hsu, H.H.; Ho, T.J.; Tu, C.C.; Lin, Y.M.; Viswanadha, V.P.; Kuo, W.W.; Huang, C.Y. CXCL2/CXCR2 axis induces cancer stem cell characteristics in CPT-11-resistant LoVo colon cancer cells via Gαi-2 and Gαq/11. J. Cell. Physiol. 2019, 234, 11822–11834. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ye, Y.L.; Li, M.X.; Ye, S.B.; Huang, W.R.; Cai, T.T.; He, J.; Peng, J.Y.; Duan, T.H.; Cui, J.; et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene 2017, 36, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, Z.; Jin, Y.; Wang, Y.; Duan, W. Loss of miR-532-5p in vitro promotes cell proliferation and metastasis by influencing CXCL2 expression in HCC. Am. J. Transl. Res. 2015, 7, 2254–2261. [Google Scholar]
- Kollmar, O.; Menger, M.D.; Schilling, M.K. Role of CXC Chemokines and Receptors in Liver Metastasis—Impact on Liver Resection-Induced Engraftment and Tumor Growth. In Liver Metastasis: Biology and Clinical Management; Brodt, P., Ed.; Springer: Dordrecht, The Netherlands, 2011; pp. 129–154. [Google Scholar]
- Pagès, F.; Galon, J.; Dieu-Nosjean, M.C.; Tartour, E.; Sautès-Fridman, C.; Fridman, W.H. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene 2010, 29, 1093–1102. [Google Scholar] [CrossRef] [Green Version]
- Kartikasari, A.E.R.; Huertas, C.S.; Mitchell, A.; Plebanski, M. Tumor-Induced Inflammatory Cytokines and the Emerging Diagnostic Devices for Cancer Detection and Prognosis. Front. Oncol. 2021, 11, 692142. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goodla, L.; Xue, X. The Role of Inflammatory Mediators in Colorectal Cancer Hepatic Metastasis. Cells 2022, 11, 2313. https://doi.org/10.3390/cells11152313
Goodla L, Xue X. The Role of Inflammatory Mediators in Colorectal Cancer Hepatic Metastasis. Cells. 2022; 11(15):2313. https://doi.org/10.3390/cells11152313
Chicago/Turabian StyleGoodla, Lavanya, and Xiang Xue. 2022. "The Role of Inflammatory Mediators in Colorectal Cancer Hepatic Metastasis" Cells 11, no. 15: 2313. https://doi.org/10.3390/cells11152313
APA StyleGoodla, L., & Xue, X. (2022). The Role of Inflammatory Mediators in Colorectal Cancer Hepatic Metastasis. Cells, 11(15), 2313. https://doi.org/10.3390/cells11152313