
Piezo1 Channel Activation Reverses Pulmonary Artery Vasoconstriction in an Early Rat Model of Pulmonary Hypertension: The Role of Ca2+ Influx and Akt-eNOS Pathway

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Hemodynamic Measurement and Fulton’s Index
2.3. Vascular Reactivity on IPAs from Rats
2.4. Isolation of PASMCs or PAECs from Rat IPAs
2.5. Measurement of [Ca2+]i and Transmembrane Potential in PAECs and PASMCs
2.6. Electrophysiological Recording
2.7. Western Blot Analysis
2.8. Immunostaining
2.9. Drugs
2.10. Data Analysis
3. Results
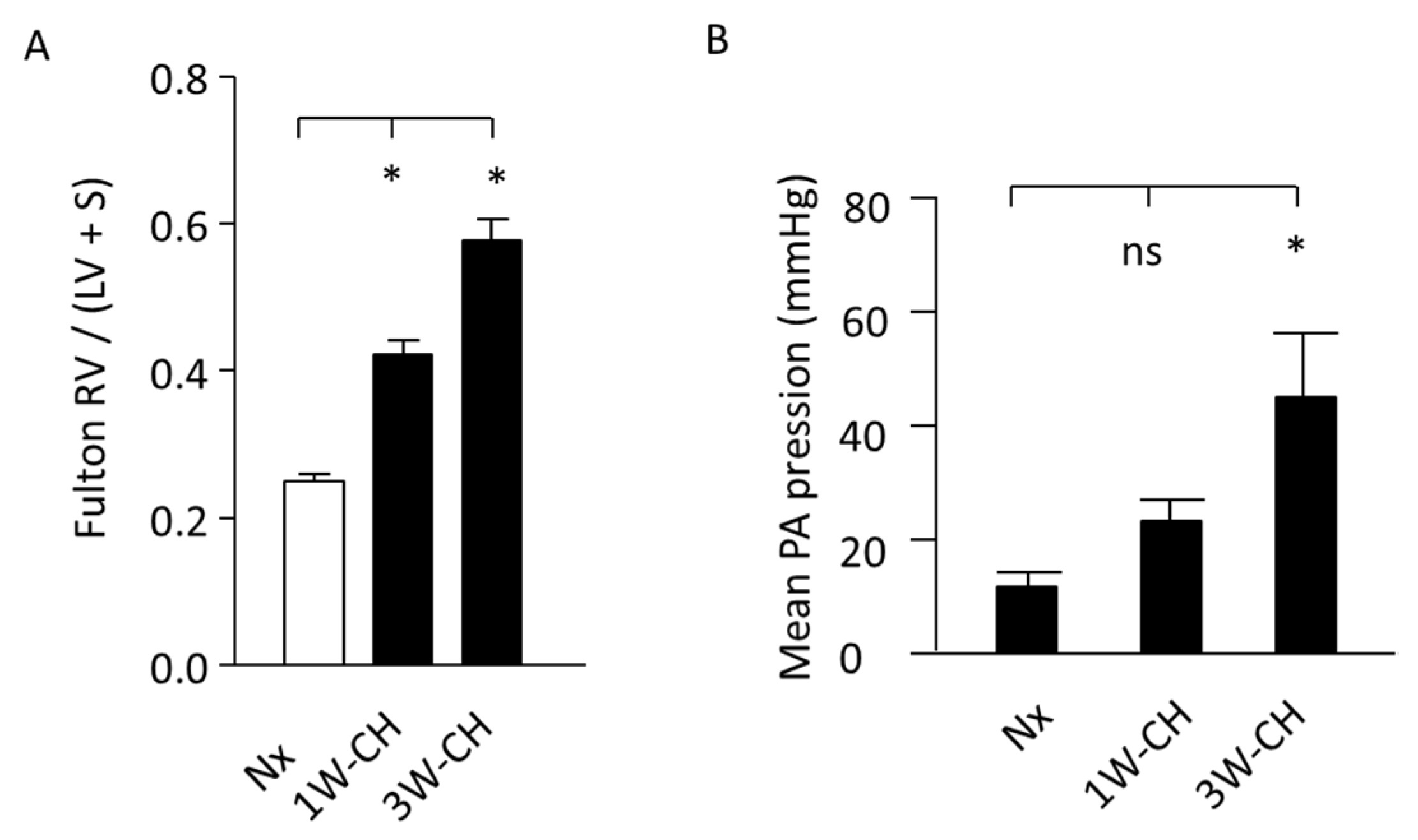
3.1. Increase in Mean Pulmonary Artery Pressure (mPAP) and Right Ventricular Hypertrophy Induced by CH
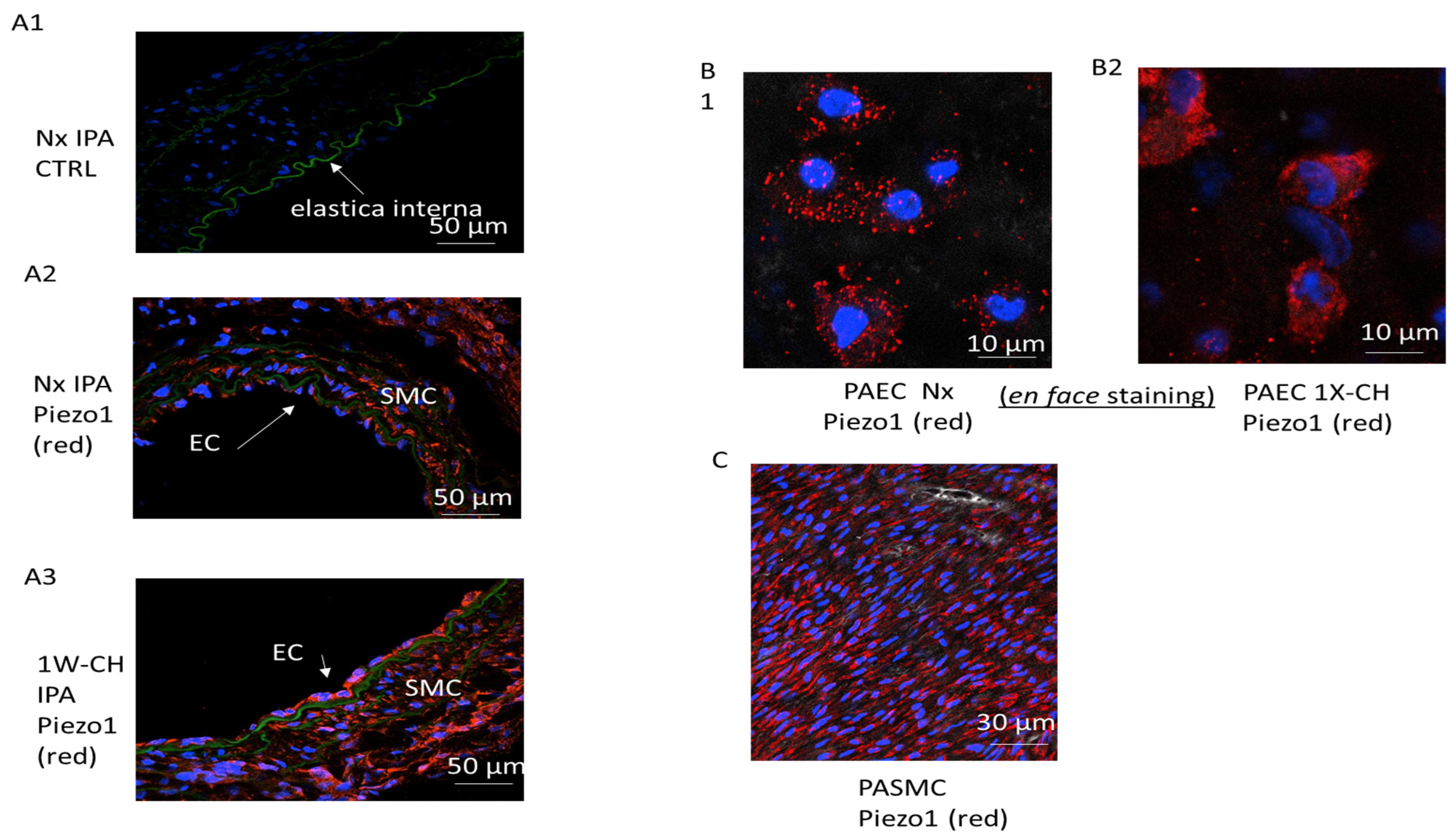
3.2. Piezo1 Channels Are Expressed in PAECs and PASMCs in an Early Stage of CH-PH
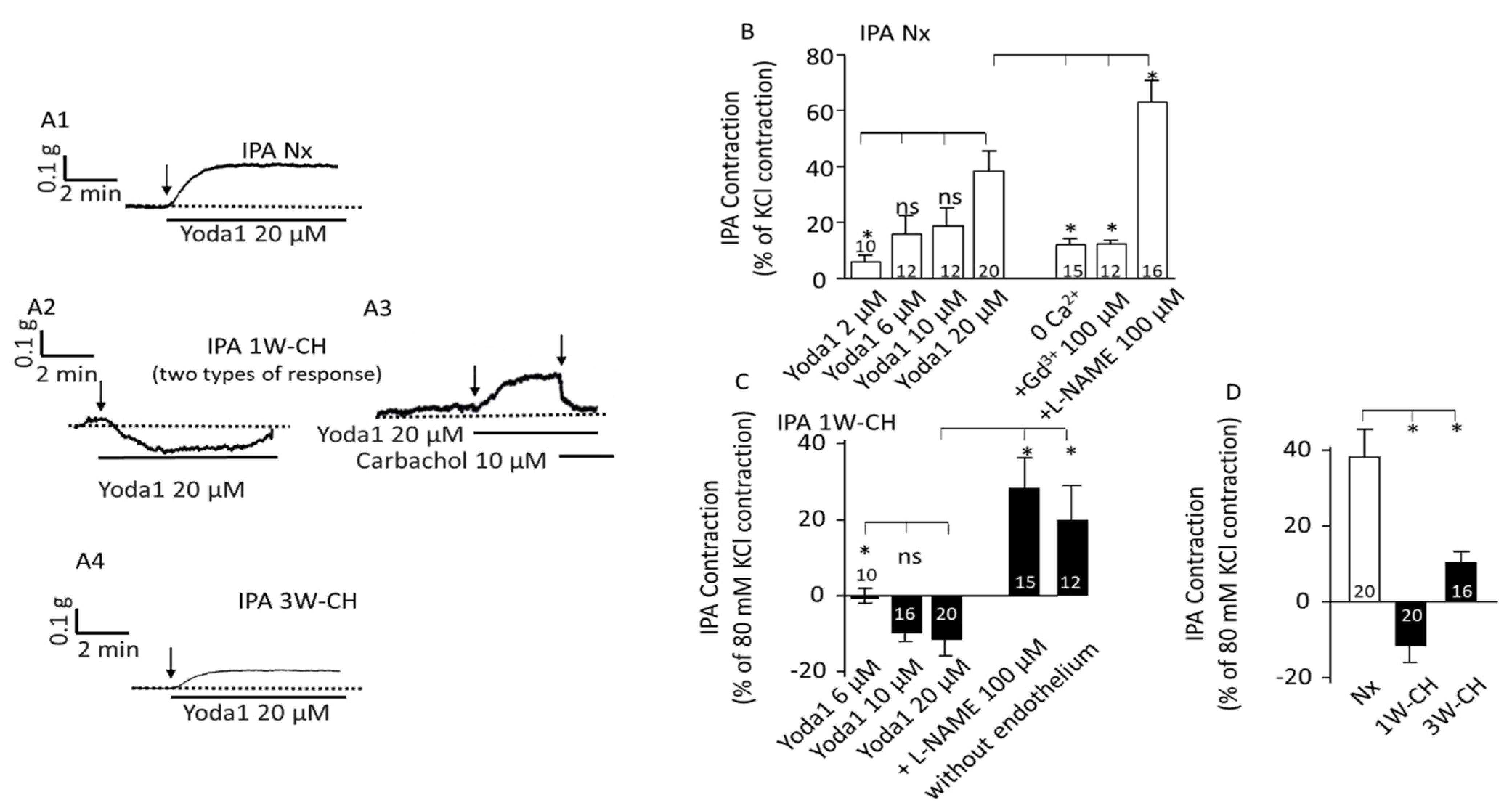
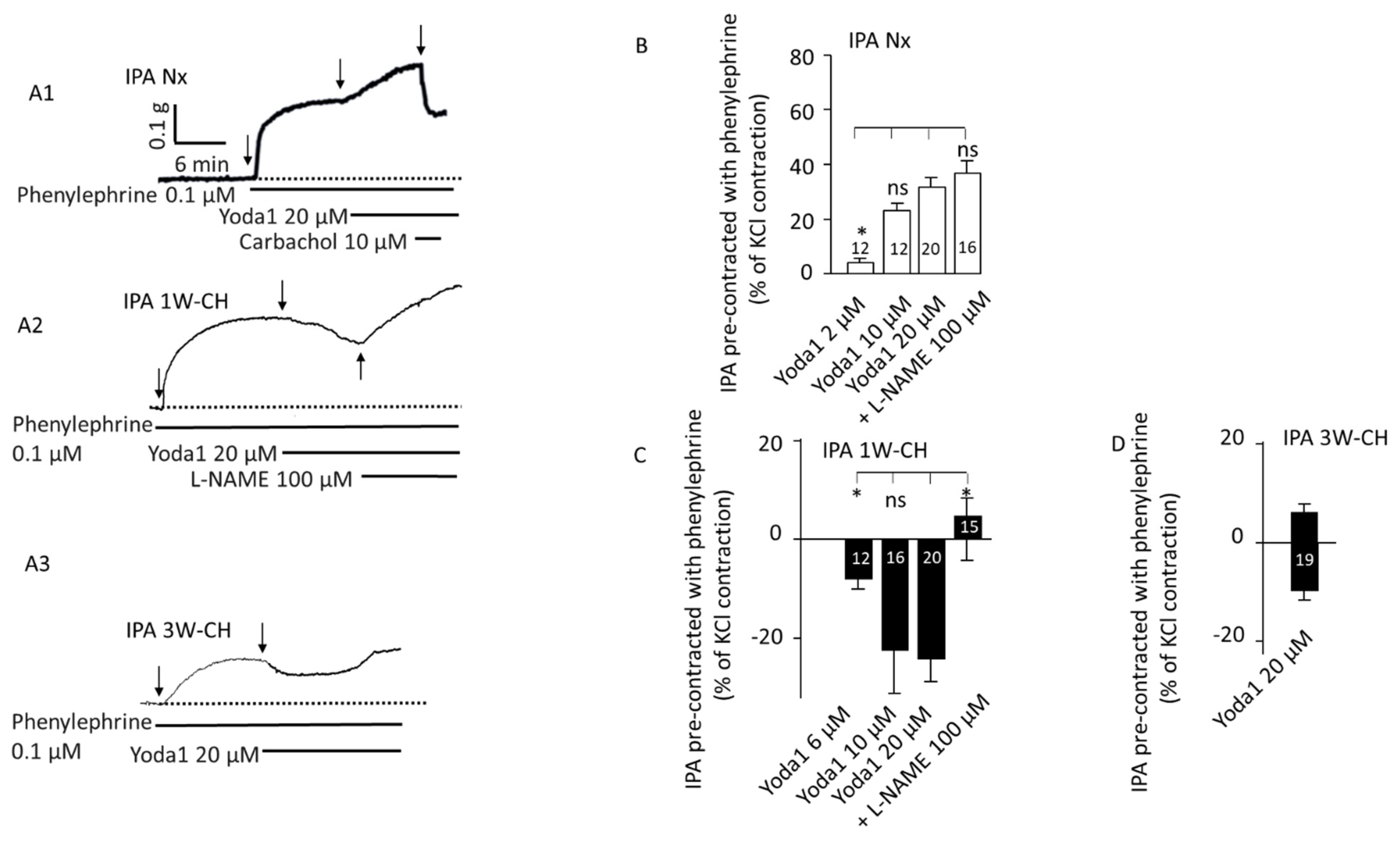
3.3. Piezo1 Activation Changes Vascular Tone Response in IPA
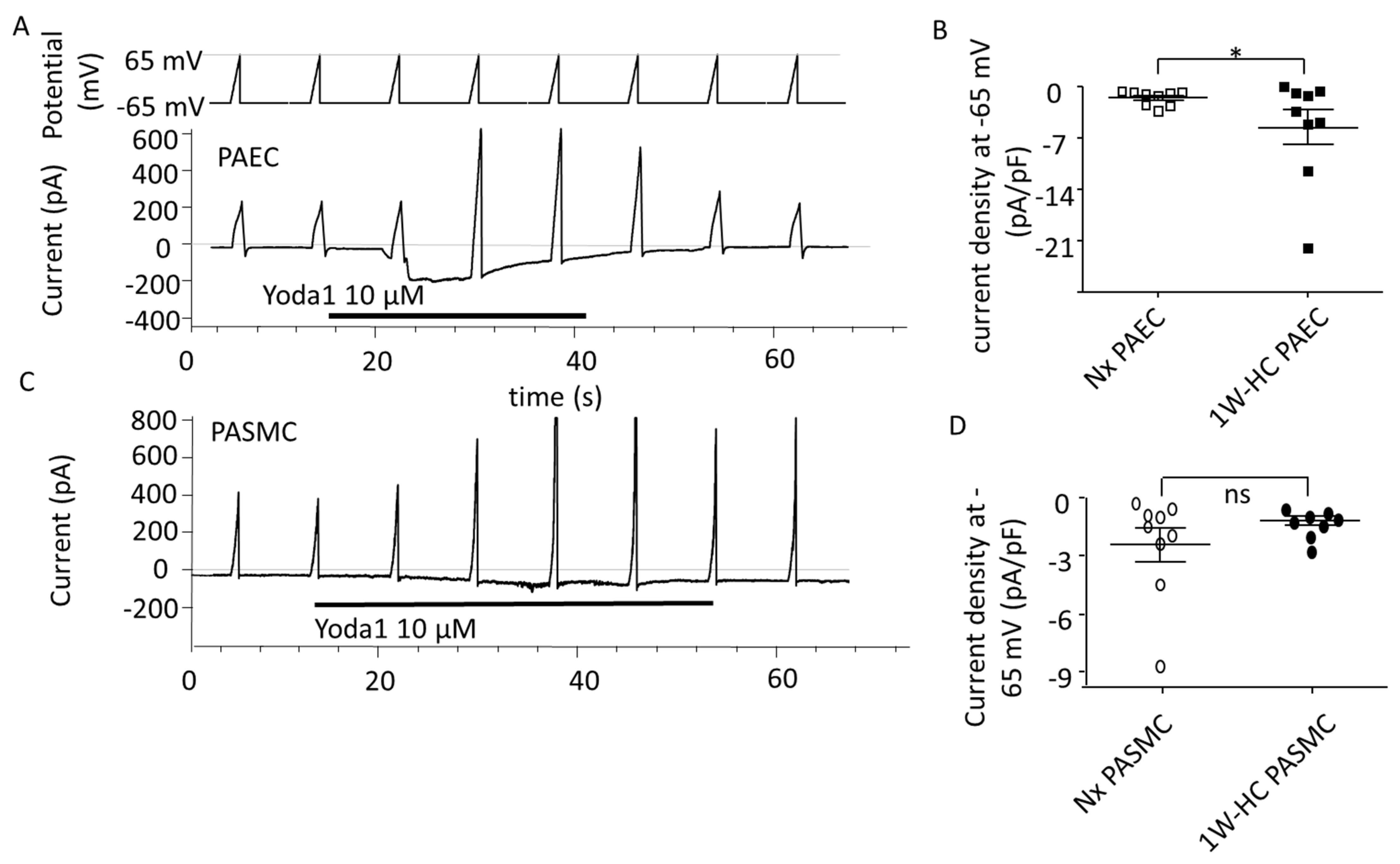
3.4. PAEC and PASMC Piezo1 Current in an Early Stage of CH-PH
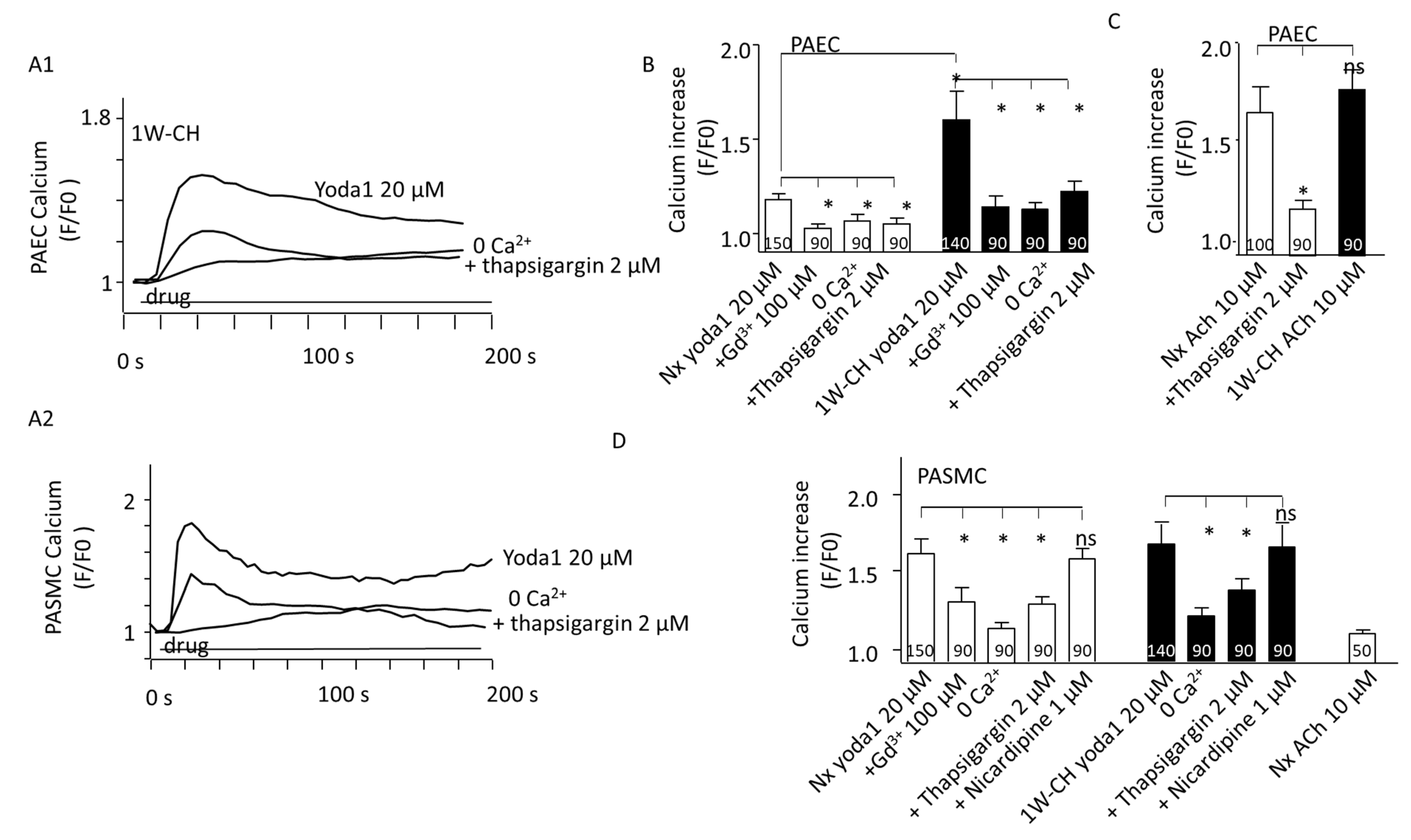
3.5. PAEC and PASMC Intracellular Calcium Concentration
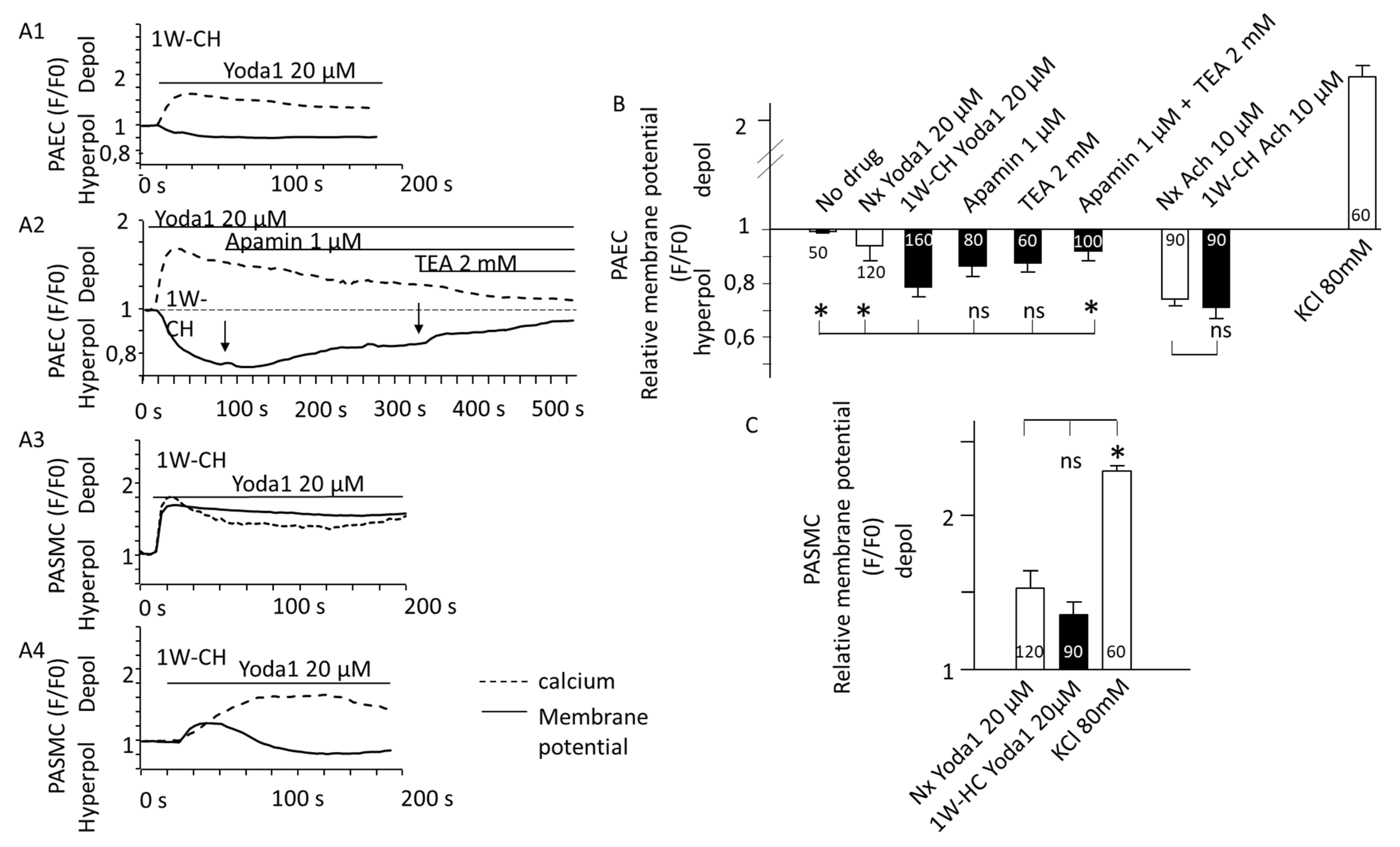
3.6. Transmembrane Potential and Piezo1 in PAECs and PASMCs
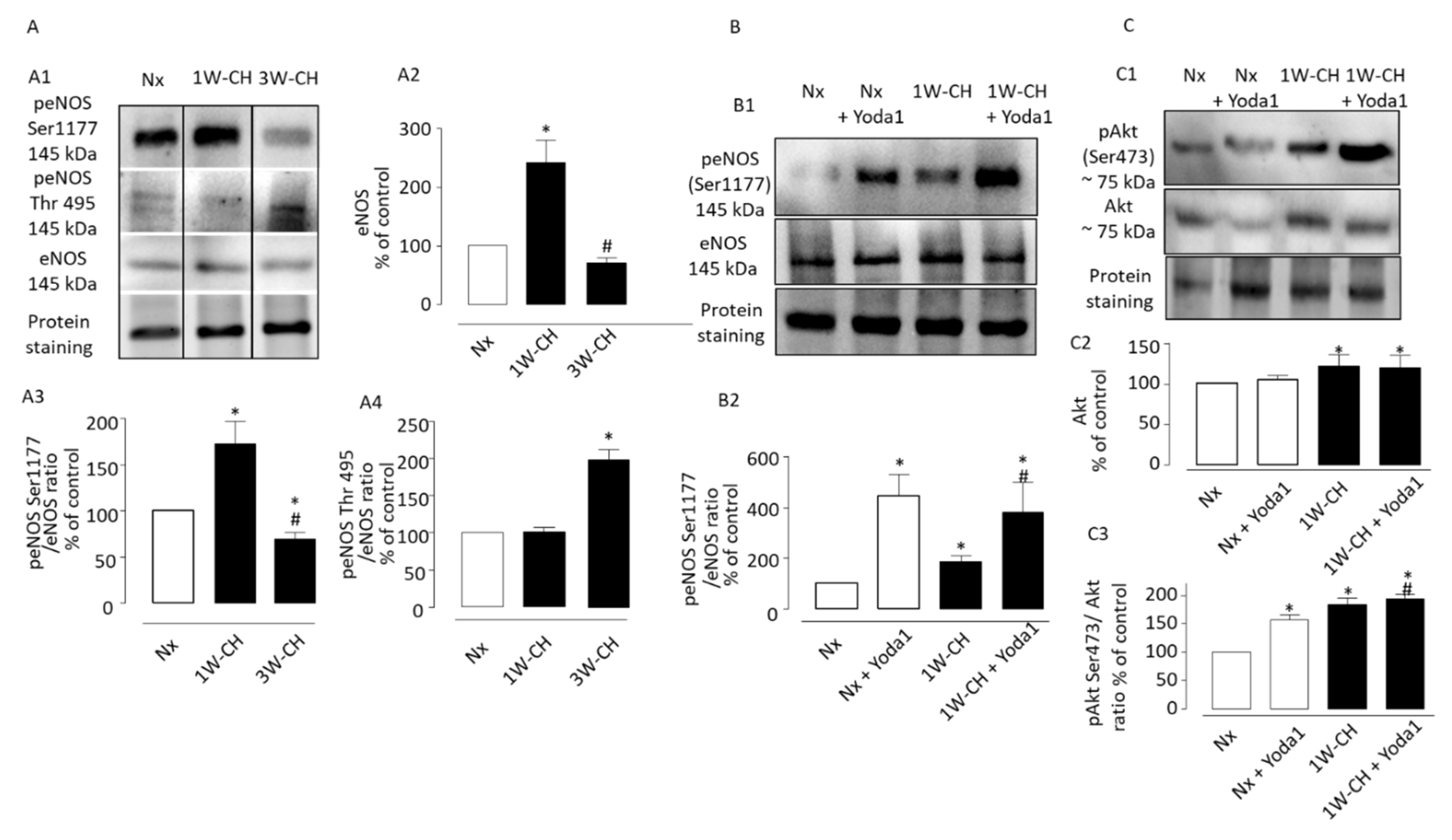
3.7. Yoda1 Induces Signaling Pathways Leading to eNOS Activation via Akt Signal Pathways in IPA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simonneau, G.; Montani, D.; Celermajer, D.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Freund-Michel, V.; Guibert, C.; Dubois, M.; Courtois, A.; Marthan, R.; Savineau, J.-P.; Muller, B. Reactive oxygen species as therapeutic targets in pulmonary hypertension. Ther. Adv. Respir. Dis. 2013, 7, 175–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condon, D.; Nickel, N.P.; Anderson, R.; Mirza, S.; Perez, V.A.D.J. The 6th World Symposium on Pulmonary Hypertension: What’s old is new. F1000Research 2019, 8, 888. [Google Scholar] [CrossRef] [PubMed]
- Fessart, D.; Martin-Negrier, M.-L.; Claverol, S.; Thiolat, M.-L.; Crevel, H.; Toussaint, C.; Bonneu, M.; Muller, B.; Savineau, J.-P.; Delom, F. Proteomic remodeling of proteasome in right heart failure. J. Mol. Cell. Cardiol. 2014, 66, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.; Dumas-De-La-Roque, E.; Bégueret, H.; Marthan, R.; Fayon, M.; Dos Santos, P.; Savineau, J.-P.; Baulieu, E.-E. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2003, 100, 9488–9493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, S.; Hyvelin, J.-M.; Bonnet, P.; Marthan, R.; Savineau, J.-P. Chronic hypoxia-induced spontaneous and rhythmic contractions in the rat main pulmonary artery. Am. J. Physiol. Cell. Mol. Physiol. 2001, 281, L183–L192. [Google Scholar] [CrossRef] [PubMed]
- Barbeau, S.; Gilbert, G.; Cardouat, G.; Baudrimont, I.; Freund-Michel, V.; Guibert, C.; Marthan, R.; Vacher, P.; Quignard, J.-F.; Ducret, T. Mechanosensitivity in Pulmonary Circulation: Pathophysiological Relevance of Stretch-Activated Channels in Pulmonary Hypertension. Biomolecules 2021, 11, 1389. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, M.; Gilbert, G.; Roux, E.; Lory, P.; Marthan, R.; Savineau, J.-P.; Quignard, J.-F. T-type calcium channels are involved in hypoxic pulmonary hypertension. Cardiovasc. Res. 2014, 103, 597–606. [Google Scholar] [CrossRef] [Green Version]
- Lhomme, A.; Gilbert, G.; Pele, T.; Deweirdt, J.; Henrion, D.; Baudrimont, I.; Campagnac, M.; Marthan, R.; Guibert, C.; Ducret, T.; et al. Stretch-activated Piezo1 Channel in Endothelial Cells Relaxes Mouse Intrapulmonary Arteries. Am. J. Respir. Cell Mol. Biol. 2019, 60, 650–658. [Google Scholar] [CrossRef]
- Yamashiro, Y.; Yanagisawa, H. The molecular mechanism of mechanotransduction in vascular homeostasis and disease. Clin. Sci. 2020, 134, 2399–2418. [Google Scholar] [CrossRef]
- Morley, L.C.; Shi, J.; Gaunt, H.J.; Hyman, A.; Webster, P.J.; Williams, C.; Forbes, K.; Walker, J.J.; Simpson, N.A.B.; Beech, D. Piezo1 channels are mechanosensors in human fetoplacental endothelial cells. Mol. Hum. Reprod. 2018, 24, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 Are Essential Components of Distinct Mechanically Activated Cation Channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Syeda, R.; Xu, J.; E Dubin, A.; Coste, B.; Mathur, J.; Huynh, T.; Matzen, J.T.; Lao, J.; Tully, D.C.; Engels, I.H.; et al. Chemical activation of the mechanotransduction channel Piezo1. eLife 2015, 4, e07369. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Lu, W.; Chen, Y.; Duan, X.; Zhang, C.; Luo, X.; Lin, Z.; Chen, J.; Liu, S.; Yan, H.; et al. Upregulation of Piezo1 (Piezo Type Mechanosensitive Ion Channel Component 1) Enhances the Intracellular Free Calcium in Pulmonary Arterial Smooth Muscle Cells From Idiopathic Pulmonary Arterial Hypertension Patients. Hypertension 2021, 77, 1974–1989. [Google Scholar] [CrossRef] [PubMed]
- Barer, G.; Emery, C.; Stewart, A.; Bee, D.; Howard, P. Endothelial control of the pulmonary circulation in normal and chronically hypoxic rats. J. Physiol. 1993, 463, 1–16. [Google Scholar] [CrossRef]
- Cahill, E.; Rowan, S.C.; Sands, M.; Banahan, M.; Ryan, D.; Howell, K.; McLoughlin, P. The pathophysiological basis of chronic hypoxic pulmonary hypertension in the mouse: Vasoconstrictor and structural mechanisms contribute equally. Exp. Physiol. 2012, 97, 796–806. [Google Scholar] [CrossRef]
- A Shimoda, L.; Sham, J.; Sylvester, J.T. Altered pulmonary vasoreactivity in the chronically hypoxic lung. Physiol. Res. 2000, 49. [Google Scholar]
- Tanaka, S.; Shiroto, T.; Godo, S.; Saito, H.; Ikumi, Y.; Ito, A.; Kajitani, S.; Sato, S.; Shimokawa, H. Important role of endothelium-dependent hyperpolarization in the pulmonary microcirculation in male mice: Implications for hypoxia-induced pulmonary hypertension. Am. J. Physiol. Circ. Physiol. 2018, 314, H940–H953. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial dysfunction and vascular disease - a 30th anniversary update. Acta Physiol. 2015, 219, 22–96. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Babicheva, A.; Jain, P.P.; Rodriguez, M.; Ayon, R.J.; Ravellette, K.S.; Wu, L.; Balistrieri, F.; Tang, H.; et al. Endothelial upregulation of mechanosensitive channel Piezo1 in pulmonary hypertension. Am. J. Physiol. Physiol. 2021, 321, C1010–C1027. [Google Scholar] [CrossRef]
- Dahan, D.; Ducret, T.; Quignard, J.-F.; Marthan, R.; Savineau, J.-P.; Estève, E. Implication of the ryanodine receptor in TRPV4-induced calcium response in pulmonary arterial smooth muscle cells from normoxic and chronically hypoxic rats. Am. J. Physiol. Cell. Mol. Physiol. 2012, 303, L824–L833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Liu, Z.H.; Chen, J.W.; Shu, X.Y.; Shen, Y.; Ding, F.H.; Zhang, R.Y.; Shen, W.F.; Lu, L.; Wang, X.Q. Using En Face Immunofluorescence Staining to Observe Vascular Endothelial Cells Directly. J. Vis. Exp. 2019, e59325. [Google Scholar] [CrossRef] [PubMed]
- Kohler, R.; Distler, A.; Hoyer, J. Increased mechanosensitive currents in aortic endothelial cells from genetically hypertensive rats. J. Hypertens. 1999, 17, 365–371. [Google Scholar] [CrossRef]
- Wan, J.; Yamamura, A.; Zimnicka, A.M.; Voiriot, G.; Smith, K.A.; Tang, H.; Ayon, R.J.; Choudhury, M.S.R.; Ko, E.A.; Wang, C.; et al. Chronic hypoxia selectively enhances L- and T-type voltage-dependent Ca2+ channel activity in pulmonary artery by upregulating Cav1.2 and Cav3.2. Am. J. Physiol. Cell. Mol. Physiol. 2013, 305, L154–L164. [Google Scholar] [CrossRef] [Green Version]
- Dubois, M.; Delannoy, E.; Duluc, L.; Closs, E.; Li, H.; Toussaint, C.; Gadeau, A.-P.; Gödecke, A.; Freund-Michel, V.; Courtois, A.; et al. Biopterin Metabolism and eNOS Expression during Hypoxic Pulmonary Hypertension in Mice. PLoS ONE 2013, 8, e82594. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, G.; Ducret, T.; Marthan, R.; Savineau, J.-P.; Quignard, J.-F. Stretch-induced Ca2+ signalling in vascular smooth muscle cells depends on Ca2+ store segregation. Cardiovasc. Res. 2014, 103, 313–323. [Google Scholar] [CrossRef] [Green Version]
- McHugh, B.J.; Buttery, R.; Lad, Y.; Banks, S.; Haslett, C.; Sethi, T. Integrin activation by Fam38A uses a novel mechanism of R-Ras targeting to the endoplasmic reticulum. J. Cell Sci. 2010, 123, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Rode, B.; Shi, J.; Endesh, N.; Drinkhill, M.J.; Webster, P.J.; Lotteau, S.J.; Bailey, M.A.; Yuldasheva, N.Y.; Ludlow, M.J.; Cubbon, R.M.; et al. Piezo1 channels sense whole body physical activity to reset cardiovascular homeostasis and enhance performance. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Barbeau, S.; Joushomme, A.; Chappe, Y.; Cardouat, G.; Baudrimont, I.; Freund-Michel, V.; Guibert, C.; Marthan, R.; Berger, P.; Vacher, P. Cell Confluence Modulates TRPV4 Channel Activity in Response to Hypoxia. Biomolecules 2022, 12, 954. [Google Scholar] [CrossRef]
- Sanderson, M.J.; Bai, Y.; Perez-Zoghbi, J.F. Ca2+ Oscillations Regulate Contraction Of Intrapulmonary Smooth Muscle Cells. In Membrane Receptors, Channels and Transporters in Pulmonary Circulation. Advances in Experimental Medicine and Biology; Humana Press: Totowa, NJ, USA, 2009; Volume 661, pp. 77–96. [Google Scholar] [CrossRef]
- Wang, H.; Yuan, Z.; Wang, B.; Li, B.; Lv, H.; He, J.; Huang, Y.; Cui, Z.; Ma, Q.; Li, T.; et al. COMP (Cartilage Oligomeric Matrix Protein), a Novel PIEZO1 Regulator That Controls Blood Pressure. Hypertension 2022, 79, 549–561. [Google Scholar] [CrossRef]
- Wang, S.; Chennupati, R.; Kaur, H.; Iring, A.; Wettschureck, N.; Offermanns, S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Investig. 2016, 126, 4527–4536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Fagan, K.; Morrissey, B.; Fouty, B.W.; Sato, K.; Harral, J.W.; Morris, K.G.; Hoedt-Miller, M.; Vidmar, S.; McMurtry, I.F.; Rodman, D.M. Upregulation of nitric oxide synthase in mice with severe hypoxia-induced pulmonary hypertension. Respir. Res. 2001, 2, 306–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, C.; Johns, R.A. Upregulation of Nitric Oxide Synthase Correlates Temporally With Onset of Pulmonary Vascular Remodeling in the Hypoxic Rat. Hypertension 1996, 28, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Blum-Johnston, C.; Thorpe, R.B.; Wee, C.; Opsahl, R.; Romero, M.; Murray, S.; Brunelle, A.; Blood, Q.; Wilson, R.; Blood, A.B.; et al. Long-term hypoxia uncouples Ca2+ and eNOS in bradykinin-mediated pulmonary arterial relaxation. Am. J. Physiol. Integr. Comp. Physiol. 2018, 314, R870–R882. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.-J.; Chennupati, R.; Li, R.; Liang, G.; Wang, S.; Iring, A.; Graumann, J.; Wettschureck, N.; Offermanns, S. Protein kinase N2 mediates flow-induced endothelial NOS activation and vascular tone regulation. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, X.; Wang, Y.; King, J.A.C.; Xie, P.; Wu, S. CaMK4 is a downstream effector of the α1G T-type calcium channel to determine the angiogenic potential of pulmonary microvascular endothelial cells. Am. J. Physiol. Physiol. 2021, 321, C964–C977. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P.B.L.; De Mey, J.G.R.; Vanhoutte, P.M. Endothelium-dependent hyperpolarizations in health and disease. Acta Physiol. 2016, 219, 97–99. [Google Scholar] [CrossRef] [Green Version]
- Ottolini, M.; Daneva, Z.; Chen, Y.; Cope, E.L.; Kasetti, R.B.; Zode, G.S.; Sonkusare, S.K. Mechanisms underlying selective coupling of endothelial Ca 2+ signals with eNOS vs. IK/SK channels in systemic and pulmonary arteries. J. Physiol. 2020, 598, 3577–3596. [Google Scholar] [CrossRef]
- Kroigaard, C.; Dalsgaard, T.; Nielsen, G.; E Laursen, B.; Pilegaard, H.; Köhler, R.; Simonsen, U. Activation of endothelial and epithelial KCa2.3 calcium-activated potassium channels by NS309 relaxes human small pulmonary arteries and bronchioles. J. Cereb. Blood Flow Metab. 2012, 167, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Beech, D.J. Endothelial Piezo1 channels as sensors of exercise. J. Physiol. 2018, 596, 979–984. [Google Scholar] [CrossRef] [Green Version]
- Marthan, R.; Savineau, J.-P. Modulation of Ion Channels in Pulmonary Arterial Hypertension. Curr. Pharm. Des. 2007, 13, 2443–2455. [Google Scholar] [CrossRef]
- Klinger, J.R.; Abman, S.H.; Gladwin, M.T. Nitric Oxide Deficiency and Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2013, 188, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.P.; Seccia, T.M.; Nussdorfer, G.G. Reciprocal regulation of endothelin-1 and nitric oxide: Relevance in the physiology and pathology of the cardiovascular system. Int. Rev. Cytol. 2001, 209, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, D.; Zhang, C.; Yang, W.; Li, C.; Gao, Z.; Pei, K.; Li, Y. Piezo1 mediates endothelial atherogenic inflammatory responses via regulation of YAP/TAZ activation. Hum. Cell 2021, 35, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.Y.; Juang, W.C.; Tsai, C.T.; Tseng, C.J.; Lee, W.H.; Chang, S.N.; Cheng, P.W. Mechanical Stretching Simulates Cardiac Physiology and Pathology through Mechanosensor Piezo1. J. Clin. Med. 2018, 7, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resta, T.C.; Chicoine, L.G.; Omdahl, J.L.; Walker, B.R. Maintained upregulation of pulmonary eNOS gene and protein expression during recovery from chronic hypoxia. Am. J. Physiol. 1999, 276, H699–H708. [Google Scholar] [CrossRef]
- Fleming, I. Molecular mechanisms underlying the activation of eNOS. Pflug. Arch.-Eur. J. Physiol. 2010, 459, 793–806. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porto Ribeiro, T.; Barbeau, S.; Baudrimont, I.; Vacher, P.; Freund-Michel, V.; Cardouat, G.; Berger, P.; Guibert, C.; Ducret, T.; Quignard, J.-F. Piezo1 Channel Activation Reverses Pulmonary Artery Vasoconstriction in an Early Rat Model of Pulmonary Hypertension: The Role of Ca2+ Influx and Akt-eNOS Pathway. Cells 2022, 11, 2349. https://doi.org/10.3390/cells11152349
Porto Ribeiro T, Barbeau S, Baudrimont I, Vacher P, Freund-Michel V, Cardouat G, Berger P, Guibert C, Ducret T, Quignard J-F. Piezo1 Channel Activation Reverses Pulmonary Artery Vasoconstriction in an Early Rat Model of Pulmonary Hypertension: The Role of Ca2+ Influx and Akt-eNOS Pathway. Cells. 2022; 11(15):2349. https://doi.org/10.3390/cells11152349
Chicago/Turabian StylePorto Ribeiro, Thais, Solène Barbeau, Isabelle Baudrimont, Pierre Vacher, Véronique Freund-Michel, Guillaume Cardouat, Patrick Berger, Christelle Guibert, Thomas Ducret, and Jean-François Quignard. 2022. "Piezo1 Channel Activation Reverses Pulmonary Artery Vasoconstriction in an Early Rat Model of Pulmonary Hypertension: The Role of Ca2+ Influx and Akt-eNOS Pathway" Cells 11, no. 15: 2349. https://doi.org/10.3390/cells11152349
APA StylePorto Ribeiro, T., Barbeau, S., Baudrimont, I., Vacher, P., Freund-Michel, V., Cardouat, G., Berger, P., Guibert, C., Ducret, T., & Quignard, J. -F. (2022). Piezo1 Channel Activation Reverses Pulmonary Artery Vasoconstriction in an Early Rat Model of Pulmonary Hypertension: The Role of Ca2+ Influx and Akt-eNOS Pathway. Cells, 11(15), 2349. https://doi.org/10.3390/cells11152349