
RNA-Binding Proteins: Emerging Therapeutics for Vascular Dysfunction


Abstract
:1. Introduction
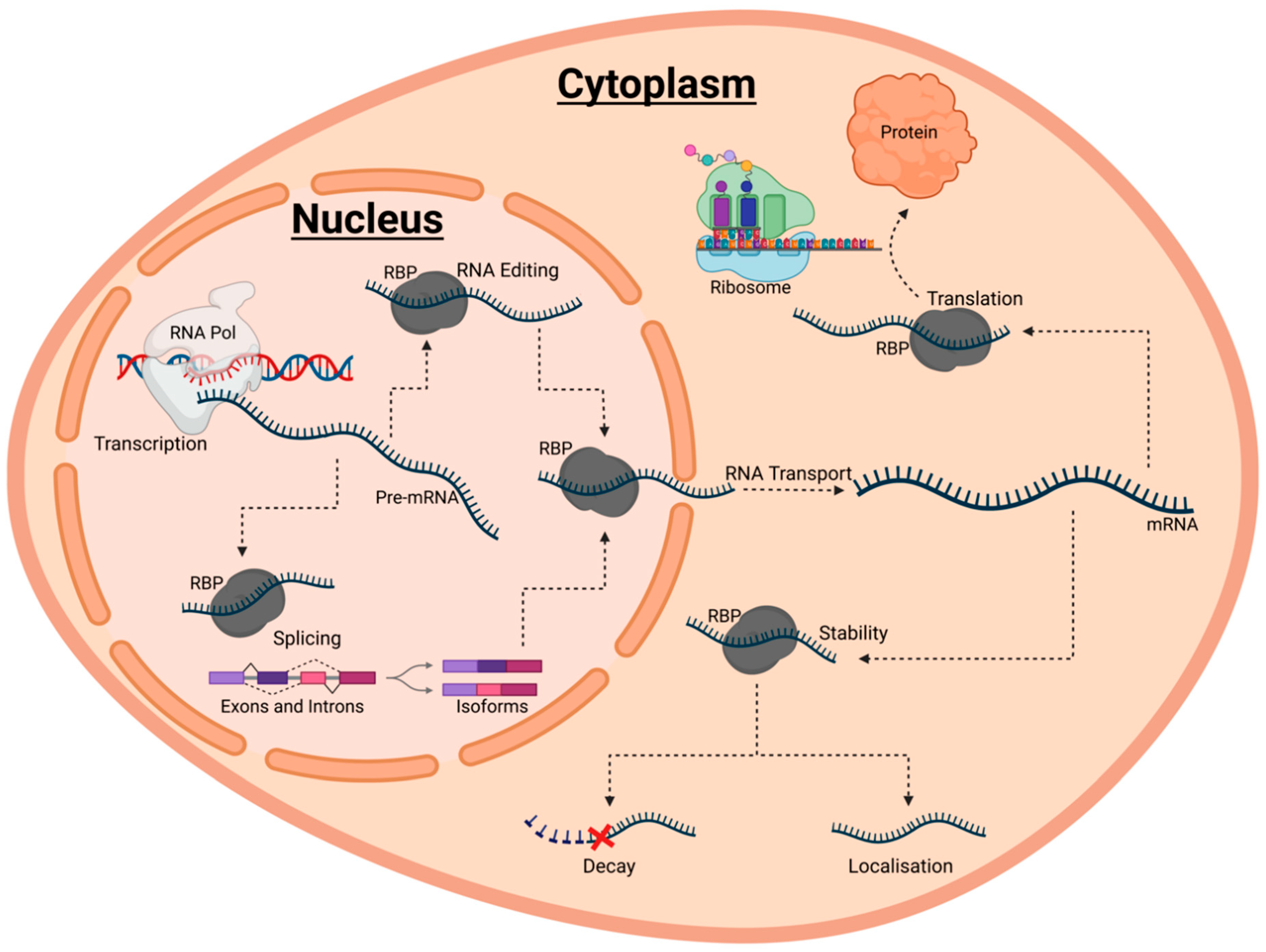
2. RBPs Are Fundamental Regulators of RNA Fate
Uncovering the Roles of RBPs
3. RBPs: Therapeutic Targets for Vascular Disease
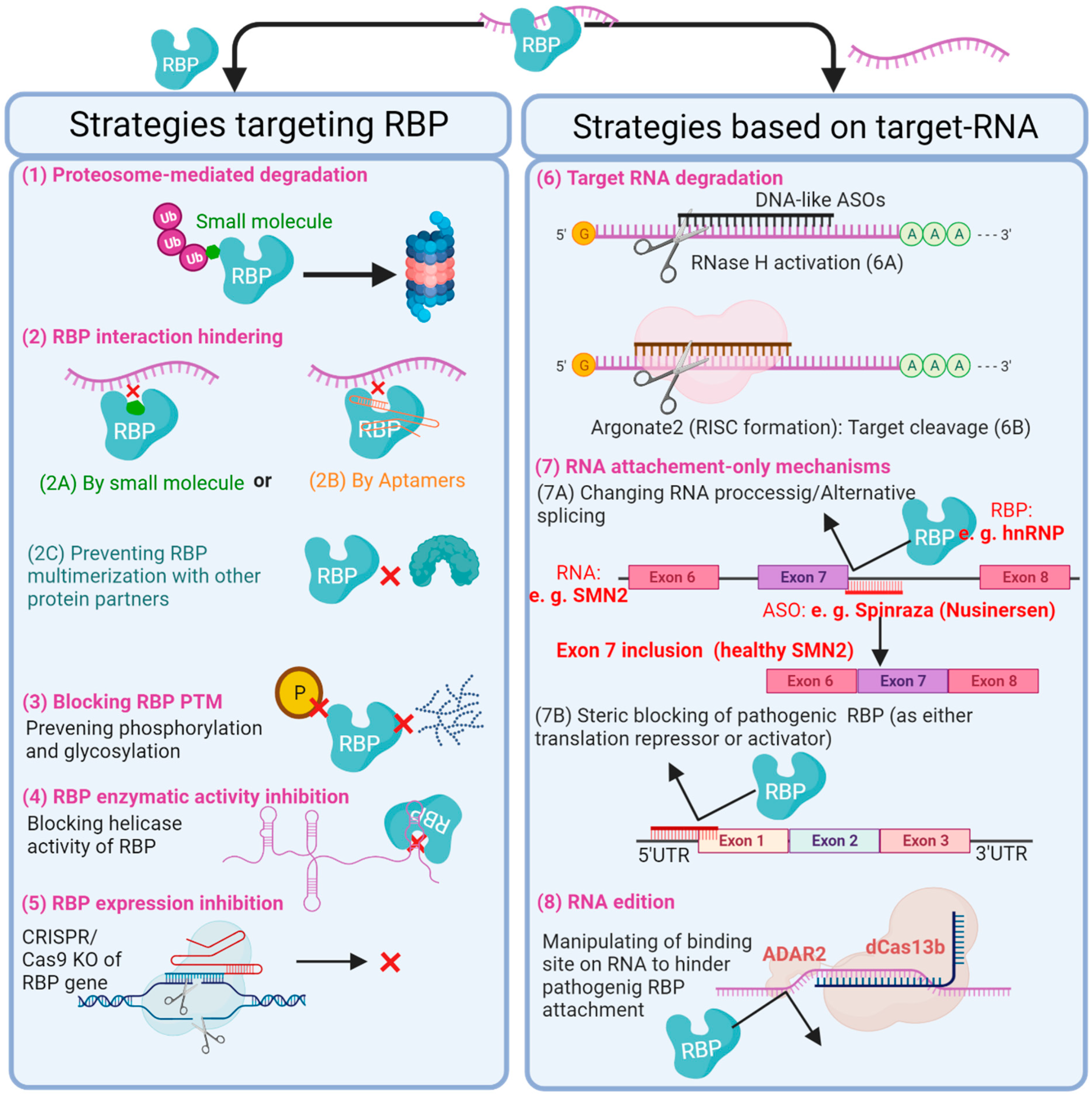
4. RBP-Based Therapeutic Strategies
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 30 June 2022).
- Bhatnagar, P.; Wickramasinghe, K.; Williams, J.; Rayner, M.; Townsend, N. The epidemiology of cardiovascular disease in the UK 2014. Heart 2015, 101, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Endothelial Ca2+ Signaling, Angiogenesis and Vasculogenesis: Just What It Takes to Make a Blood Vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Belair, D.; Whisler, J.A.; Valdez, J.; Velazquez, J.J.; Molenda, J.A.; Vickerman, V.; Lewis, R.; Daigh, C.; Hansen, T.D.; Mann, D.A.; et al. Human Vascular Tissue Models Formed from Human Induced Pluripotent Stem Cell Derived Endothelial Cells. Stem Cell Rev. Rep. 2015, 11, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Vilà-González, M.; Kelaini, S.; Magee, C.; Caines, R.; Campbell, D.; Eleftheriadou, M.; Cochrane, A.; Drehmer, D.; Tsifaki, M.; O’Neill, K.; et al. Enhanced Function of Induced Pluripotent Stem Cell-Derived Endothelial Cells Through ESM1 Signaling. Stem Cells 2019, 37, 226–239. [Google Scholar] [CrossRef]
- Ishigami, M.; Masumoto, H.; Ikuno, T.; Aoki, T.; Kawatou, M.; Minakata, K.; Ikeda, T.; Sakata, R.; Yamashita, J.K.; Minatoya, K. Human iPS cell-derived cardiac tissue sheets for functional restoration of infarcted porcine hearts. PLoS ONE 2018, 13, e0201650. [Google Scholar] [CrossRef] [PubMed]
- Orlova, V.V.; Drabsch, Y.; Freund, C.; Petrus-Reurer, S.; Hil, F.E.V.D.; Muenthaisong, S.; Dijke, P.T.; Mummery, C.L. Functionality of Endothelial Cells and Pericytes From Human Pluripotent Stem Cells Demonstrated in Cultured Vascular Plexus and Zebrafish Xenografts. Arter. Thromb. Vasc. Biol. 2014, 34, 177–186. [Google Scholar] [CrossRef]
- Orlova, V.V.; Hil, F.E.V.D.; Petrus-Reurer, S.; Drabsch, Y.; Dijke, P.T.; Mummery, C.L. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat. Protoc. 2014, 9, 1514–1531. [Google Scholar] [CrossRef]
- Kusuma, S.; Shen, Y.-I.; Hanjaya-Putra, D.; Mali, P.; Cheng, L.; Gerecht, S. Self-organized vascular networks from human pluripotent stem cells in a synthetic matrix. Proc. Natl. Acad. Sci. USA 2013, 110, 12601–12606. [Google Scholar] [CrossRef]
- Chan, X.Y.; Black, R.; Dickerman, K.; Federico, J.; Lévesque, M.; Mumm, J.; Gerecht, S. Three-Dimensional Vascular Network Assembly From Diabetic Patient-Derived Induced Pluripotent Stem Cells. Arter. Thromb. Vasc. Biol. 2015, 35, 2677–2685. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Weiße, J.; Rosemann, J.; Krauspe, V.; Kappler, M.; Eckert, A.W.; Haemmerle, M.; Gutschner, T. RNA-Binding Proteins as Regulators of Migration, Invasion and Metastasis in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2020, 21, 6835. [Google Scholar] [CrossRef]
- Wang, J.; Wang, B.; Bi, J.; Zhang, C. Cytoplasmic HuR expression correlates with angiogenesis, lymphangiogenesis, and poor outcome in lung cancer. Med. Oncol. 2011, 28, 577–585. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, R.G.; Rabelink, T.J.; van Zonneveld, A.J.; van der Veer, E.P. Emerging roles for RNA-binding proteins as effectors and regulators of cardiovascular disease. Eur. Heart J. 2017, 38, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Sachse, M.; Mareti, A.; Georgiopoulos, G.; Sopova, K.; Vlachogiannis, N.; Tual-Chalot, S.; Kritsioti, C.; Laina, A.; Kontogiannis, C.; Zaman, A.; et al. P4492Peripheral blood mononuclear cell expression of the stabilizing RNA-binding protein HuR is associated with incidence and extent of human atherosclerotic cardiovascular disease. Eur. Heart J. 2019, 40, 2713. [Google Scholar] [CrossRef]
- Dong, R.; Yang, G.-D.; Luo, N.-A.; Qu, Y.-Q. HuR: A promising therapeutic target for angiogenesis. Gland Surg. 2014, 3, 203–206. [Google Scholar] [PubMed]
- Liu, S.; Jiang, X.; Lu, H.; Xing, M.; Qiao, Y.; Zhang, C.; Zhang, W. HuR (Human Antigen R) Regulates the Contraction of Vascular Smooth Muscle and Maintains Blood Pressure. Arter. Thromb. Vasc. Biol. 2020, 40, 943–957. [Google Scholar] [CrossRef]
- Amadio, M.; Bucolo, C.; Leggio, G.M.; Drago, F.; Govoni, S.; Pascale, A. The PKCbeta/HuR/VEGF pathway in diabetic retinopathy. Biochem. Pharm. 2010, 80, 1230–1237. [Google Scholar] [CrossRef]
- Yu, C.; Xin, W.; Zhen, J.; Liu, Y.; Javed, A.; Wang, R.; Wan, Q. Human antigen R mediated post-transcriptional regulation of epithelial-mesenchymal transition related genes in diabetic nephropathy. J. Diabetes 2015, 7, 562–572. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Q.; Wang, X.; Duan, Y.; Wang, Z.; Wei, X.; Zhang, Y.; Wang, H.; Wang, R.; Yi, F. Identification of NOD2 as a novel target of RNA-binding protein HuR: Evidence from NADPH oxidase-mediated HuR signaling in diabetic nephropathy. Free Radic. Biol. Med. 2014, 79, 217–227. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Lambers, E.; Verma, S.; Thorne, T.; Qin, G.; Losordo, D.W.; Kishore, R. Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. FASEB J. 2010, 24, 2484–2494. [Google Scholar] [CrossRef] [PubMed]
- Amadio, M.; Pascale, A.; Cupri, S.; Pignatello, R.; Osera, C.; Agata, V.D.; Amico, A.G.D.; Leggio, G.M.; Ruozi, B.; Govoni, S.; et al. Nanosystems based on siRNA silencing HuR expression counteract diabetic retinopathy in rat. Pharmacol. Res. 2016, 111, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Yi, C. Chemical Modifications to RNA: A New Layer of Gene Expression Regulation. ACS Chem. Biol. 2017, 12, 316–325. [Google Scholar] [CrossRef]
- Chothani, S.; Schäfer, S.; Adami, E.; Viswanathan, S.; Widjaja, A.A.; Langley, S.R.; Tan, J.; Wang, M.; Quaife, N.M.; Pua, C.J.; et al. Widespread Translational Control of Fibrosis in the Human Heart by RNA-Binding Proteins. Circulation 2019, 140, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.P.; Nagel, R.J.; Fagg, W.S.; Shiue, L.; Cline, M.S.; Perriman, R.J.; Donohue, J.P.; Ares, M., Jr. Quaking and PTB control overlapping splicing regulatory networks during muscle cell differentiation. RNA 2013, 19, 627–638. [Google Scholar] [CrossRef]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Dominguez, D.; Freese, P.; Alexis, M.S.; Su, A.; Hochman, M.; Palden, T.; Bazile, C.; Lambert, N.J.; Van Nostrand, E.L.; Pratt, G.A.; et al. Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol. Cell 2018, 70, 854–867.e9. [Google Scholar] [CrossRef]
- Hong, S. RNA Binding Protein as an Emerging Therapeutic Target for Cancer Prevention and Treatment. J. Cancer Prev. 2017, 22, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Dorn, L.E.; Tual-Chalot, S.; Stellos, K.; Accornero, F. RNA epigenetics and cardiovascular diseases. J. Mol. Cell. Cardiol. 2019, 129, 272–280. [Google Scholar] [CrossRef]
- Kadumuri, R.V.; Janga, S.C. Epitranscriptomic Code and Its Alterations in Human Disease. Trends Mol. Med. 2018, 24, 886–903. [Google Scholar] [CrossRef]
- Gatsiou, A.; Stellos, K. Dawn of Epitranscriptomic Medicine. Circ. Genom. Precis. Med. 2018, 11, e001927. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, V.A.; Fulton, J.R.; Margariti, A. Alternative Splicing: A Key Mediator of Diabetic Vasculopathy. Genes 2021, 12, 1332. [Google Scholar] [CrossRef] [PubMed]
- Minvielle-Sebastia, L.; Keller, W. mRNA polyadenylation and its coupling to other RNA processing reactions and to transcription. Curr. Opin. Cell Biol. 1999, 11, 352–357. [Google Scholar] [CrossRef]
- Brennicke, A.; Marchfelder, A.; Binder, S. RNA editing. FEMS Microbiol. Rev. 1999, 23, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.M.; Feagin, J.E.; Stuart, K.; Simpson, L. Editing of kinetoplastid mitochondrial mRNAs by uridine addition and deletion generates conserved amino acid sequences and AUG initiation codons. Cell 1988, 53, 401–411. [Google Scholar] [CrossRef]
- Quinones-Valdez, G.; Tran, S.S.; Jun, H.-I.; Bahn, J.H.; Yang, E.-W.; Zhan, L.; Brümmer, A.; Wei, X.; Van Nostrand, E.; Pratt, G.A.; et al. Regulation of RNA editing by RNA-binding proteins in human cells. Commun. Biol. 2019, 2, 19. [Google Scholar] [CrossRef]
- Beckmann, B.M.; Horos, R.; Fischer, B.; Castello, A.; Eichelbaum, K.; Alleaume, A.-M.; Schwarzl, T.; Curk, T.; Foehr, S.; Huber, W.; et al. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat. Commun. 2015, 6, 10127. [Google Scholar] [CrossRef]
- Butter, F.; Scheibe, M.; Mörl, M.; Mann, M. Unbiased RNA–protein interaction screen by quantitative proteomics. Proc. Natl. Acad. Sci. USA 2009, 106, 10626–10631. [Google Scholar] [CrossRef] [PubMed]
- Treiber, T.; Treiber, N.; Plessmann, U.; Harlander, S.; Daiß, J.-L.; Eichner, N.; Lehmann, G.; Schall, K.; Urlaub, H.; Meister, G. A Compendium of RNA-Binding Proteins that Regulate MicroRNA Biogenesis. Mol. Cell 2017, 66, 270–284.e13. [Google Scholar] [CrossRef] [PubMed]
- Tsvetanova, N.G.; Klass, D.M.; Salzman, J.; Brown, P.O. Proteome-Wide Search Reveals Unexpected RNA-Binding Proteins in Saccharomyces cerevisiae. PLoS ONE 2010, 5, e12671. [Google Scholar] [CrossRef]
- Chu, E.; Allegra, C.J. The role of thymidylate synthase in cellular regulation. Adv. Enzym. Regul. 1996, 36, 143–163. [Google Scholar] [CrossRef]
- Scherrer, T.; Mittal, N.; Janga, S.C.; Gerber, A.P. A Screen for RNA-Binding Proteins in Yeast Indicates Dual Functions for Many Enzymes. PLoS ONE 2010, 5, e15499. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Horos, R.; Strein, C.; Fischer, B.; Eichelbaum, K.; Steinmetz, L.M.; Krijgsveld, J.; Hentze, M.W. System-wide identification of RNA-binding proteins by interactome capture. Nat. Protoc. 2013, 8, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Han, M.; Meng, L.; Chen, X. Transcriptome-wide discovery of coding and noncoding RNA-binding proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E3879–E3887. [Google Scholar] [CrossRef]
- Van Nostrand, E.L.; Pratt, G.A.; Shishkin, A.A.; Gelboin-Burkhart, C.; Fang, M.Y.; Sundararaman, B.; Blue, S.M.; Nguyen, T.B.; Surka, C.; Elkins, K.; et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 2016, 13, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Xiao, R.; Blue, S.M.; Chen, J.-Y.; Cody, N.A.L.; Dominguez, D.; et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 2020, 583, 711–719. [Google Scholar] [CrossRef]
- Cook, K.B.; Kazan, H.; Zuberi, K.; Morris, Q.; Hughes, T.R. RBPDB: A database of RNA-binding specificities. Nucleic Acids Res. 2011, 39, D301–D308. [Google Scholar] [CrossRef]
- Paz, I.; Kosti, I.; Ares, M., Jr.; Cline, M.; Mandel-Gutfreund, Y. RBPmap: A web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014, 42, W361–W367. [Google Scholar] [CrossRef]
- Giudice, G.; Sánchez-Cabo, F.; Torroja, C.; Lara-Pezzi, E. ATtRACT-a database of RNA-binding proteins and associated motifs. Database 2016, 2016, baw035. [Google Scholar] [CrossRef]
- Ghosh, P.; Murugavel, P.; Sowdhamini, R. hRBPome: A central repository of all known human RNA-binding proteins. bioRxiv 2018, 269043. [Google Scholar] [CrossRef]
- Bouvrette, L.P.B.; Bovaird, S.; Blanchette, M.; Lécuyer, E. oRNAment: A database of putative RNA binding protein target sites in the transcriptomes of model species. Nucleic Acids Res. 2019, 48, D166–D173. [Google Scholar] [CrossRef] [PubMed]
- Stepniewski, J.; Kachamakova-Trojanowska, N.; Ogrocki, D.; Szopa, M.; Matlok, M.; Beilharz, M.; Dyduch, G.; Malecki, M.T.; Jozkowicz, A.; Dulak, J. Induced pluripotent stem cells as a model for diabetes investigation. Sci. Rep. 2015, 5, 8597. [Google Scholar] [CrossRef] [PubMed]
- Leite, N.C.; Sintov, E.; Meissner, T.B.; Brehm, M.A.; Greiner, D.L.; Harlan, D.M.; Melton, D.A. Modeling Type 1 Diabetes In Vitro Using Human Pluripotent Stem Cells. Cell Rep. 2020, 32, 107894. [Google Scholar] [CrossRef] [PubMed]
- Sa, S.; Gu, M.; Chappell, J.; Shao, N.-Y.; Ameen, M.; Elliott, K.A.T.; Li, D.; Grubert, F.; Li, C.G.; Taylor, S.; et al. Induced Pluripotent Stem Cell Model of Pulmonary Arterial Hypertension Reveals Novel Gene Expression and Patient Specificity. Am. J. Respir. Crit. Care Med. 2017, 195, 930–941. [Google Scholar] [CrossRef]
- Gu, M.; Shao, N.Y.; Sa, S.; Li, D.; Termglinchan, V.; Ameen, M.; Karakikes, I.; Sosa, G.; Grubert, F.; Lee, J.; et al. Patient-Specific iPSC-Derived Endothelial Cells Uncover Pathways that Protect against Pulmonary Hypertension in BMPR2 Mutation Carriers. Cell Stem Cell 2017, 20, 490–504.e495. [Google Scholar] [CrossRef]
- Theodoris, C.V.; Li, M.; White, M.P.; Liu, L.; He, D.; Pollard, K.S.; Bruneau, B.G.; Srivastava, D. Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell 2015, 160, 1072–1086. [Google Scholar] [CrossRef]
- Ge, X.; Ren, Y.; Bartulos, O.; Lee, M.Y.; Yue, Z.; Kim, K.Y.; Li, W.; Amos, P.J.; Bozkulak, E.C.; Iyer, A.; et al. Modeling supravalvular aortic stenosis syndrome with human induced pluripotent stem cells. Circulation 2012, 126, 1695–1704. [Google Scholar] [CrossRef]
- Kinnear, C.; Chang, W.Y.; Khattak, S.; Hinek, A.; Thompson, T.; Rodrigues, D.D.C.; Kennedy, K.; Mahmut, N.; Pasceri, P.; Stanford, W.L.; et al. Modeling and Rescue of the Vascular Phenotype of Williams-Beuren Syndrome in Patient Induced Pluripotent Stem Cells. STEM CELLS Transl. Med. 2013, 2, 2–15. [Google Scholar] [CrossRef]
- Ong, S.-B.; Lee, W.H.; Shao, N.-Y.; Ismail, N.I.; Katwadi, K.; Lim, M.-M.; Kwek, X.-Y.; Michel, N.A.; Li, J.; Newson, J.; et al. Calpain Inhibition Restores Autophagy and Prevents Mitochondrial Fragmentation in a Human iPSC Model of Diabetic Endotheliopathy. Stem Cell Rep. 2019, 12, 597–610. [Google Scholar] [CrossRef]
- Wimmer, R.A.; Leopoldi, A.; Aichinger, M.; Kerjaschki, D.; Penninger, J.M. Generation of blood vessel organoids from human pluripotent stem cells. Nat. Protoc. 2019, 14, 3082–3100. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, R.A.; Leopoldi, A.; Aichinger, M.; Wick, N.; Hantusch, B.; Novatchkova, M.; Taubenschmid, J.; Hämmerle, M.; Esk, C.; Bagley, J.A.; et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature 2019, 565, 505–510. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, R.G.; van der Veer, E.P.; Prins, J.; Lee, D.H.; Dane, M.J.; Zhang, H.; Roeten, M.K.; Bijkerk, R.; de Boer, H.C.; Rabelink, T.J.; et al. The RNA-binding protein quaking maintains endothelial barrier function and affects VE-cadherin and beta-catenin protein expression. Sci. Rep. 2016, 6, 21643. [Google Scholar] [CrossRef]
- van der Veer, E.P.; de Bruin, R.G.; Kraaijeveld, A.O.; de Vries, M.R.; Bot, I.; Pera, T.; Segers, F.M.; Trompet, S.; van Gils, J.M.; Roeten, M.K.; et al. Quaking, an RNA-binding protein, is a critical regulator of vascular smooth muscle cell phenotype. Circ. Res. 2013, 113, 1065–1075. [Google Scholar] [CrossRef]
- Yang, C.; Eleftheriadou, M.; Kelaini, S.; Morrison, T.; González, M.V.; Caines, R.; Edwards, N.; Yacoub, A.; Edgar, K.; Moez, A.; et al. Targeting QKI-7 in vivo restores endothelial cell function in diabetes. Nat. Commun. 2020, 11, 3812. [Google Scholar] [CrossRef]
- Amadio, M.; Scapagnini, G.; Lupo, G.; Drago, F.; Govoni, S.; Pascale, A. PKCbetaII/HuR/VEGF: A new molecular cascade in retinal pericytes for the regulation of VEGF gene expression. Pharm. Res. 2008, 57, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Castello, A.; Fischer, B.; Leicht, S.; Föehr, S.; Frese, C.K.; Ragan, C.; Kurscheid, S.; Pagler, E.; Yang, H.; et al. The Cardiomyocyte RNA-Binding Proteome: Links to Intermediary Metabolism and Heart Disease. Cell Rep. 2016, 16, 1456–1469. [Google Scholar] [CrossRef]
- de Bruin, R.G.; Shiue, L.; Prins, J.; de Boer, H.C.; Singh, A.; Fagg, W.S.; van Gils, J.M.; Duijs, J.M.G.J.; Katzman, S.; Kraaijeveld, A.O.; et al. Quaking promotes monocyte differentiation into pro-atherogenic macrophages by controlling pre-mRNA splicing and gene expression. Nat. Commun. 2016, 7, 10846. [Google Scholar] [CrossRef]
- Kelaini, S.; Chan, C.; Cornelius, V.; Margariti, A. RNA-Binding Proteins Hold Key Roles in Function, Dysfunction, and Disease. Biology 2021, 10, 366. [Google Scholar] [CrossRef]
- Hou, S.C.; Chan, L.W.; Chou, Y.C.; Su, C.Y.; Chen, X.; Shih, Y.L.; Tsai, P.C.; Shen, C.K.; Yan, Y.T. Ankrd17, an ubiquitously expressed ankyrin factor, is essential for the vascular integrity during embryogenesis. FEBS Lett. 2009, 583, 2765–2771. [Google Scholar] [CrossRef]
- Green, L.C.; Anthony, S.R.; Slone, S.; Lanzillotta, L.; Nieman, M.L.; Wu, X.; Robbins, N.; Jones, S.M.; Roy, S.; Owens, A.P., III; et al. Human antigen R as a therapeutic target in pathological cardiac hypertrophy. JCI Insight. 2019, 4, e121541. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, T.; Ohga, N.; Hida, Y.; Maishi, N.; Akiyama, K.; Kakuguchi, W.; Kuroshima, T.; Kondo, M.; Akino, T.; Totsuka, Y.; et al. HuR keeps an angiogenic switch on by stabilising mRNA of VEGF and COX-2 in tumour endothelium. Br. J. Cancer 2011, 104, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.-I.; Li, W.-M.; Wang, Y.-H.; Wu, T.-F.; Wu, W.-R.; Liao, A.C.; Shen, K.-H.; Wei, Y.-C.; Hsing, C.-H.; Shiue, Y.-L.; et al. HuR cytoplasmic expression is associated with increased cyclin A expression and poor outcome with upper urinary tract urothelial carcinoma. BMC Cancer 2012, 12, 611. [Google Scholar] [CrossRef]
- Pullmann, R.; Juhaszova, M.; de Silanes, I.L.; Kawai, T.; Mazan-Mamczarz, K.; Halushka, M.; Gorospe, M. Enhanced Proliferation of Cultured Human Vascular Smooth Muscle Cells Linked to Increased Function of RNA-binding Protein HuR. J. Biol. Chem. 2005, 280, 22819–22826. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Lu, J.-G.; Wang, Q.; He, X.-L.; Chu, Y.-K.; Ma, Q.-J. Stabilization of Snail by HuR in the process of hydrogen peroxide induced cell migration. Biochem. Biophys. Res. Commun. 2007, 356, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Hur, J.; Yoon, C.H.; Kim, J.H.; Lee, C.S.; Youn, S.W.; Oh, I.Y.; Skurk, C.; Murohara, T.; Park, Y.B.; et al. Augmentation of therapeutic angiogenesis using genetically modified human endothelial progenitor cells with altered glycogen synthase kinase-3beta activity. J. Biol. Chem. 2004, 279, 49430–49438. [Google Scholar] [CrossRef]
- Caines, R.; Cochrane, A.; Kelaini, S.; Vila-Gonzalez, M.; Yang, C.; Eleftheriadou, M.; Moez, A.; Stitt, A.W.; Zeng, L.; Grieve, D.J.; et al. The RNA-binding protein QKI controls alternative splicing in vascular cells, producing an effective model for therapy. J. Cell Sci. 2019, 132, jcs230276. [Google Scholar] [CrossRef]
- Cochrane, A.; Kelaini, S.; Tsifaki, M.; Bojdo, J.; Vilà-González, M.; Drehmer, D.; Caines, R.; Magee, C.; Eleftheriadou, M.; Hu, Y.; et al. Quaking Is a Key Regulator of Endothelial Cell Differentiation, Neovascularization, and Angiogenesis. Stem Cells 2017, 35, 952–966. [Google Scholar] [CrossRef]
- Kasirer-Friede, A.; Peuhu, E.; Ivaska, J.; Shattil, S.J. Platelet SHARPIN regulates platelet adhesion and inflammatory responses through associations with αIIbβ3 and LUBAC. Blood Adv. 2022, 6, 2595–2607. [Google Scholar] [CrossRef]
- HogenEsch, H.; Sola, M.; Stearns, T.M.; Silva, K.A.; Kennedy, V.E.; Sundberg, J.P. Angiogenesis in the skin of SHARPIN-deficient mice with chronic proliferative dermatitis. Exp. Mol. Pathol. 2016, 101, 303–307. [Google Scholar] [CrossRef]
- Zhang, H.; Taylor, W.R.; Joseph, G.; Caracciolo, V.; Gonzales, D.M.; Sidell, N.; Seli, E.; Blackshear, P.J.; Kallen, C.B. mRNA-binding protein ZFP36 is expressed in atherosclerotic lesions and reduces inflammation in aortic endothelial cells. Arter. Thromb. Vasc. Biol. 2013, 33, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Bollmann, F.; Wu, Z.; Oelze, M.; Siuda, D.; Xia, N.; Henke, J.; Daiber, A.; Li, H.; Stumpo, D.J.; Blackshear, P.; et al. Endothelial Dysfunction in Tristetraprolin-deficient Mice Is Not Caused by Enhanced Tumor Necrosis Factor-α Expression. J. Biol. Chem. 2014, 289, 15653–15665. [Google Scholar] [CrossRef]
- Carballo, E.; Blackshear, P. Roles of tumor necrosis factor-α receptor subtypes in the pathogenesis of the tristetraprolin-deficiency syndrome. Blood 2001, 98, 2389–2395. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Lei, T.; Song, Y.; Yanes, N.; Qi, Y.; Fu, M. RNA-destabilizing factor tristetraprolin negatively regulates NF-kappaB signaling. J. Biol. Chem. 2009, 284, 29383–29390. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.-Y.; Cai, Y.; Sun, W.; Ding, Y.; Wang, W.; Kong, W.; Tang, C.; Zhu, Y.; Xu, M.-J.; Wang, X. Intermedin inhibits macrophage foam-cell formation via tristetraprolin-mediated decay of CD36 mRNA. Cardiovasc. Res. 2013, 101, 297–305. [Google Scholar] [CrossRef]
- Teplova, M.; Hafner, M.; Teplov, D.; Essig, K.; Tuschl, T.; Patel, D.J. Structure–function studies of STAR family Quaking proteins bound to their in vivo RNA target sites. Genes Dev. 2013, 27, 928–940. [Google Scholar] [CrossRef]
- Kondo, T.; Furuta, T.; Mitsunaga, K.; Ebersole, T.A.; Shichiri, M.; Wu, J.; Artzt, K.; Yamamura, K.-i.; Abe, K. Genomic organization and expression analysis of the mouse qkI locus. Mamm. Genome 1999, 10, 662–669. [Google Scholar]
- Sidman, R.L.; Dickie, M.M.; Appel, S.H. Mutant Mice (Quaking and Jimpy) with Deficient Myelination in the Central Nervous System. Science 1964, 144, 309–311. [Google Scholar] [CrossRef]
- Galarneau, A.; Richard, S. Target RNA motif and target mRNAs of the Quaking STAR protein. Nat. Struct. Mol. Biol. 2005, 12, 691–698. [Google Scholar] [CrossRef]
- Zaffran, S.; Astier, M.; Gratecos, D.; Semeriva, M. The held out wings (how) Drosophila gene encodes a putative RNA-binding protein involved in the control of muscular and cardiac activity. Development 1997, 124, 2087–2098. [Google Scholar] [CrossRef]
- Noveroske, J.K.; Lai, L.; Gaussin, V.; Northrop, J.L.; Nakamura, H.; Hirschi, K.K.; Justice, M.J. Quaking is essential for blood vessel development. Genesis 2002, 32, 218–230. [Google Scholar] [CrossRef]
- Li, Z.; Takakura, N.; Oike, Y.; Imanaka, T.; Araki, K.; Suda, T.; Kaname, T.; Kondo, T.; Abe, K.; Yamamura, K. Defective smooth muscle development in qkI-deficient mice. Dev. Growth Differ. 2003, 45, 449–462. [Google Scholar] [CrossRef] [PubMed]
- van Mil, A.; Grundmann, S.; Goumans, M.-J.; Lei, Z.; Oerlemans, M.I.; Jaksani, S.; Doevendans, P.A.; Sluijter, J.P.G. MicroRNA-214 inhibits angiogenesis by targeting Quaking and reducing angiogenic growth factor release. Cardiovasc. Res. 2012, 93, 655–665. [Google Scholar] [CrossRef]
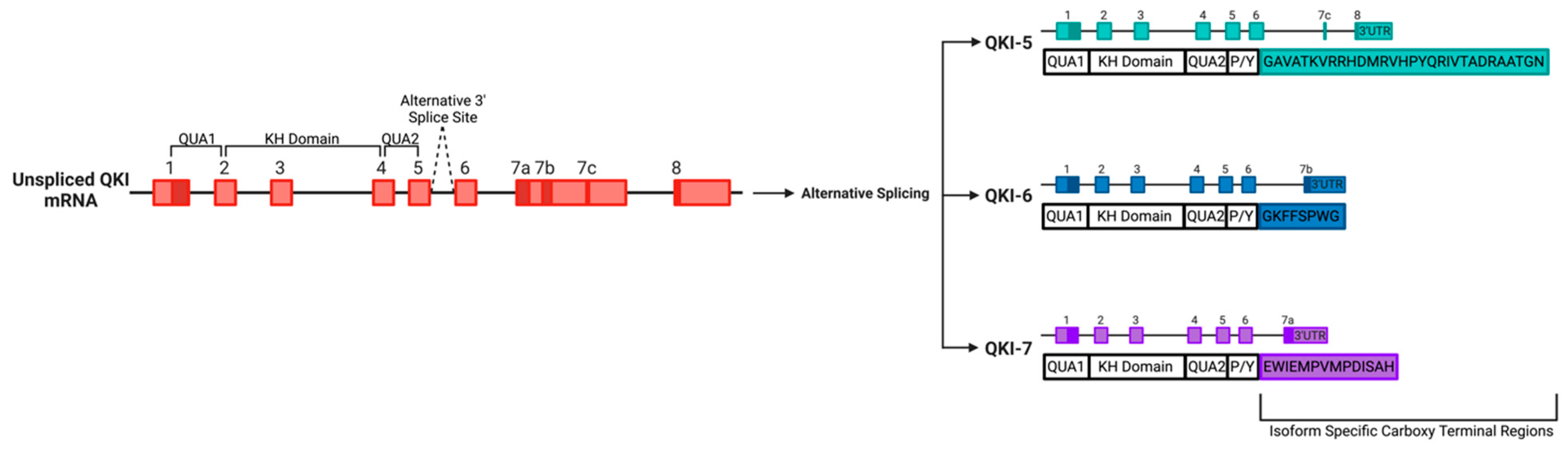
- Wu, J.; Zhou, L.; Tonissen, K.; Tee, R.; Artzt, K. The Quaking I-5 Protein (QKI-5) Has a Novel Nuclear Localization Signal and Shuttles between the Nucleus and the Cytoplasm. J. Biol. Chem. 1999, 274, 29202–29210. [Google Scholar] [CrossRef] [PubMed]
- Pilotte, J.; Larocque, D.; Richard, S. Nuclear translocation controlled by alternatively spliced isoforms inactivates the QUAKING apoptotic inducer. Genes Dev. 2001, 15, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Jiang, T.; Lian, C.; Wang, H.; Zheng, Q.; Ma, H. QKI deficiency promotes FoxO1 mediated nitrosative stress and endoplasmic reticulum stress contributing to increased vulnerability to ischemic injury in diabetic heart. J. Mol. Cell. Cardiol. 2014, 75, 131–140. [Google Scholar] [CrossRef]
- Wang, F.; Yuan, Y.; Yang, P.; Li, X. Extracellular vesicles-mediated transfer of miR-208a/b exaggerate hypoxia/reoxygenation injury in cardiomyocytes by reducing QKI expression. Mol. Cell. Biochem. 2017, 431, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Garg, A.; Bär, C.; Chatterjee, S.; Foinquinos, A.; Milting, H.; Streckfuß-Bömeke, K.; Fiedler, J.; Thum, T. Quaking Inhibits Doxorubicin-Mediated Cardiotoxicity Through Regulation of Cardiac Circular RNA Expression. Circ. Res. 2018, 122, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, Z.; Yang, M.; Dai, B.; Hu, F.; Yang, F.; Zhu, J.; Chen, T.; Zhang, L. MicroRNA-214 regulates smooth muscle cell differentiation from stem cells by targeting RNA-binding protein QKI. Oncotarget 2017, 8, 19866–19878. [Google Scholar] [CrossRef]
- Sun, Z.; Zhou, D.; Xie, X.; Wang, S.; Wang, Z.; Zhao, W.; Xu, H.; Zheng, L. Cross-talk between macrophages and atrial myocytes in atrial fibrillation. Basic Res. Cardiol. 2016, 111, 63. [Google Scholar] [CrossRef]
- Yamagishi, R.; Tsusaka, T.; Mitsunaga, H.; Maehata, T.; Hoshino, S.-I. The STAR protein QKI-7 recruits PAPD4 to regulate post-transcriptional polyadenylation of target mRNAs. Nucleic Acids Res. 2016, 44, 2475–2490. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; McGee, D.L. Diabetes and Glucose Tolerance as Risk Factors for Cardiovascular Disease: The Framingham Study. Diabetes Care 1979, 2, 120–126. [Google Scholar] [CrossRef]
- Lieberman, J. Tapping the RNA world for therapeutics. Nat. Struct. Mol. Biol. 2018, 25, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Setten, R.L.; Rossi, J.J.; Han, S.-P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef]
- Kashima, T.; Manley, J. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003, 34, 460–463. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RBPs | Clinical Outcome | References |
|---|---|---|
| ANKRD17 | ANKRD17 deficient mice are embryonic lethal due to cardiovascular defects as a result of incomplete vascular maturation. | [71] |
| HuR | HuR has a range of roles across the vasculature. For example, HuR has critical roles in vascular smooth muscle contraction and maintains blood pressure regulation. In addition, HuR regulates multiple angiogenic factors and promotes cell proliferation and migration of endothelial cells. Deficiency has been associated with decreased angiogenesis as well as abnormal vasculature. HuR has also arisen as a viable therapeutic target for pathological cardiac hypertrophy and heart failure and expression to correlate with poor prognosis in coronary artery disease. | [19,72,73,74,75,76,77] |
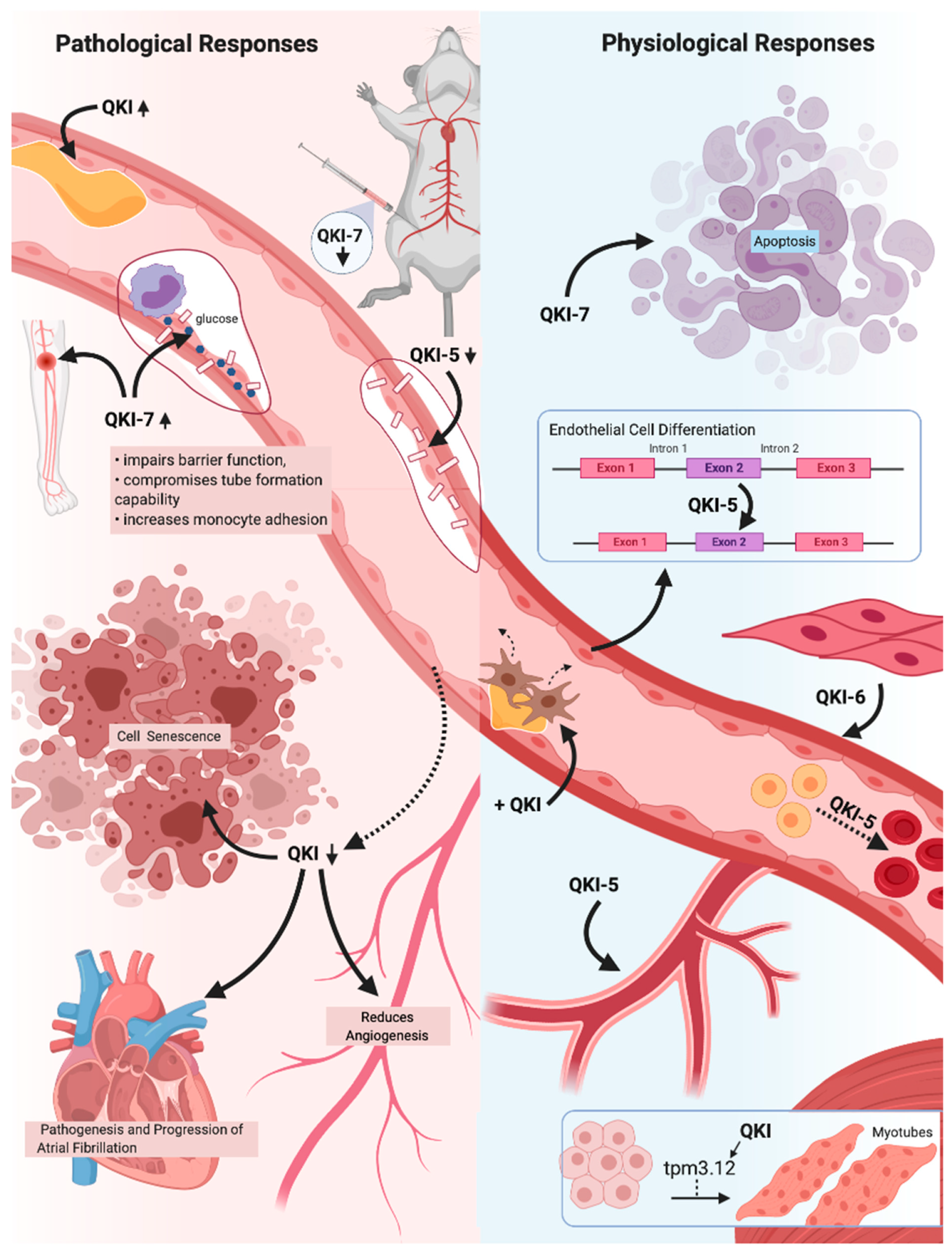
| QKI | A lack of QKI-5 in endothelial cells impairs cell-to-cell adhesions and barrier function and is associated with multiple vascular diseases, including ischemia. QKI-5 and QKI-6 expression improves vascular cell health as well as encourages neovascularisation and blood flow recovery in a hind limb ischemia model. QKI-7 upregulation disrupts endothelial cell–cell barrier, compromises angiogenesis and enhances monocyte adhesion. | [64,66,69,78,79] |
| SHARPIN | A role for SHARPIN has emerged in blood vessel health as well as angiogenesis. Moreover, SHARPIN has been shown to have differential and context-dependent roles in platelets to regulate inflammatory and integrin adhesive functions. | [80,81] |
| ZFP36/TTP | ZFP36 expression can suppress atherosclerosis. Deficiency reduces blood pressure, triggers vascular inflammation, reduces relaxation upon acetylcholine and orchestrates endothelial cell dysfunction. | [82,83,84,85,86] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornelius, V.A.; Naderi-Meshkin, H.; Kelaini, S.; Margariti, A. RNA-Binding Proteins: Emerging Therapeutics for Vascular Dysfunction. Cells 2022, 11, 2494. https://doi.org/10.3390/cells11162494
Cornelius VA, Naderi-Meshkin H, Kelaini S, Margariti A. RNA-Binding Proteins: Emerging Therapeutics for Vascular Dysfunction. Cells. 2022; 11(16):2494. https://doi.org/10.3390/cells11162494
Chicago/Turabian StyleCornelius, Victoria A., Hojjat Naderi-Meshkin, Sophia Kelaini, and Andriana Margariti. 2022. "RNA-Binding Proteins: Emerging Therapeutics for Vascular Dysfunction" Cells 11, no. 16: 2494. https://doi.org/10.3390/cells11162494
APA StyleCornelius, V. A., Naderi-Meshkin, H., Kelaini, S., & Margariti, A. (2022). RNA-Binding Proteins: Emerging Therapeutics for Vascular Dysfunction. Cells, 11(16), 2494. https://doi.org/10.3390/cells11162494