
Maresin1 Suppresses High-Glucose-Induced Ferroptosis in Osteoblasts via NRF2 Activation in Type 2 Diabetic Osteoporosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Osteoblast Culture and Treatment
2.2. NRF2-siRNA Knockdown of Osteoblasts
2.3. Cell Proliferation Assessments
2.3.1. 5′-ethynyl-2′-deoxyuridine (EdU) Assay
2.3.2. Cell Counting Kit-8 (CCK-8) Assay
2.4. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.5. Protein Isolation and Western Blotting
2.6. Osteogenic Differentiation Assessments
2.6.1. Alkaline Phosphatase (ALP) Staining
2.6.2. Alizarin Red S (ARS) Staining
2.7. Cells Immunofluorescence Staining
2.8. Measurement of Malondialdehyde (MDA) Levels
2.9. Determination of Reduced Glutathione (GSH) Levels
2.10. Intracellular Iron Ion Content Detection
2.11. Determination of Lipid ROS Levels
2.12. Determination of Intracellular Reactive Oxygen Species (ROS) Levels
2.13. Transmission Electron Microscopy (TEM)
2.14. Ethics Statement
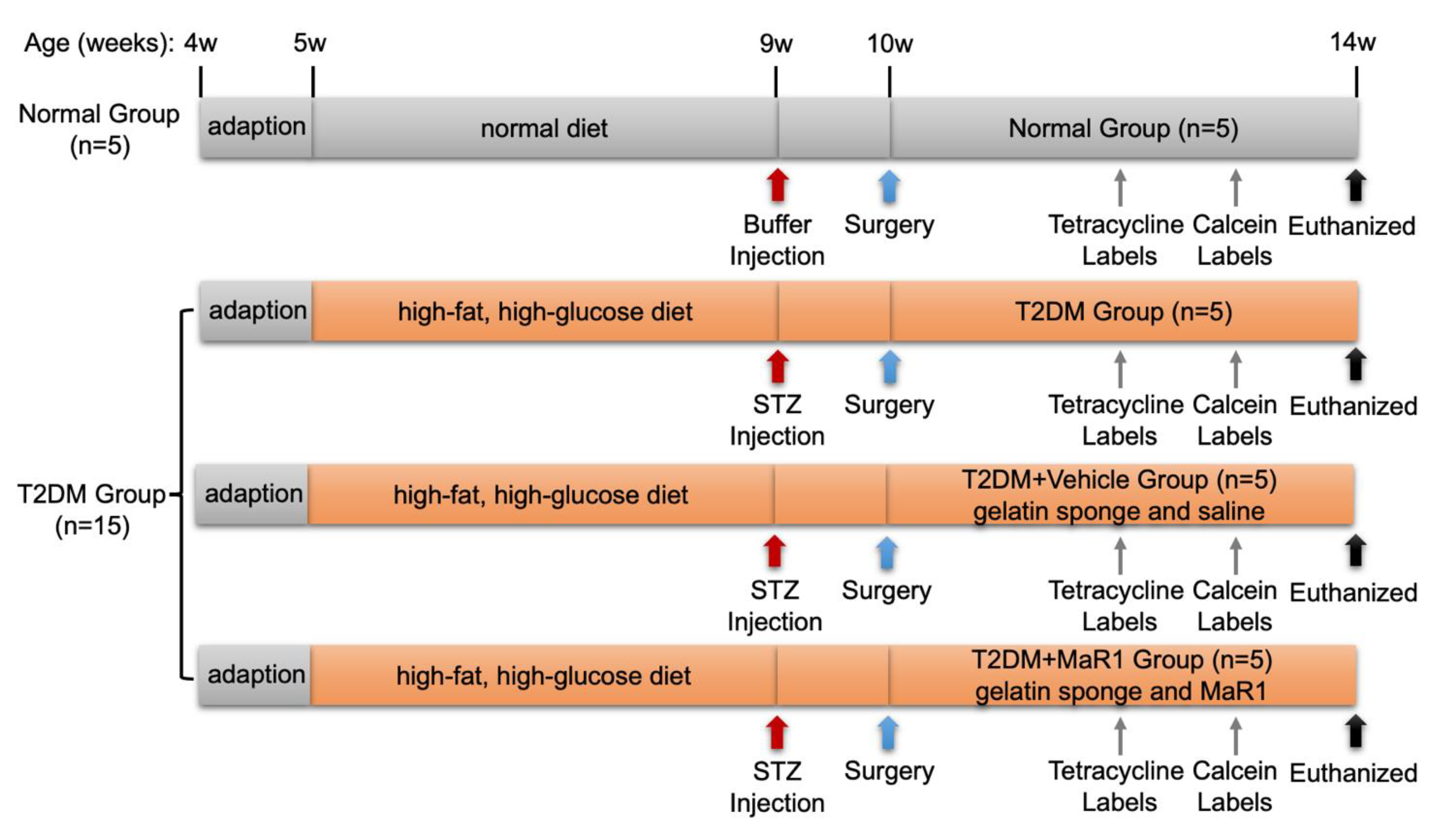
2.15. Experimental Animals and Models
2.16. Sequential Fluorescence Labeling
2.17. Microscopic Computed Tomography (MicroCT)
2.18. Immunohistochemistry (IHC)
2.19. Statistical Analysis
3. Results
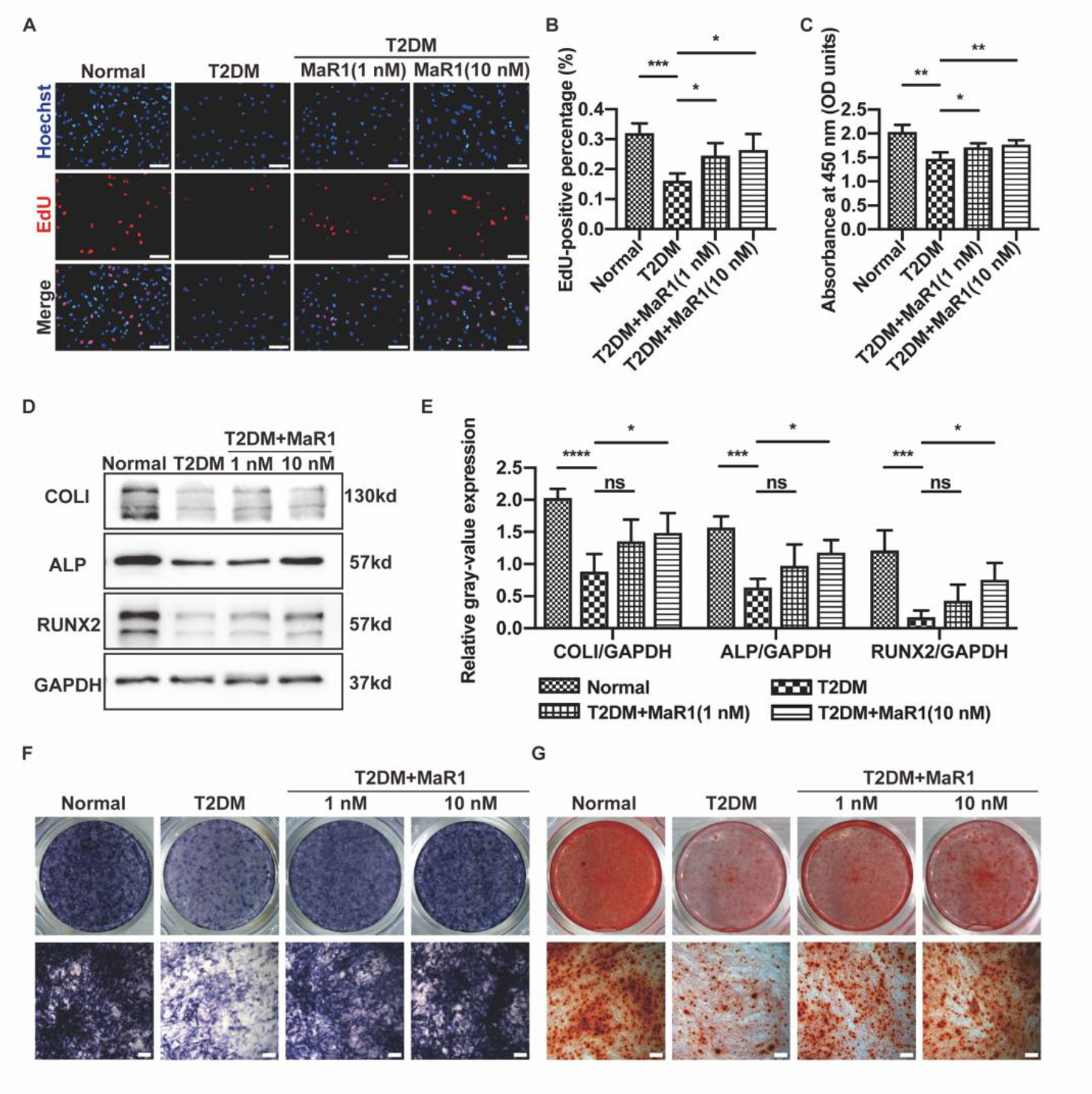
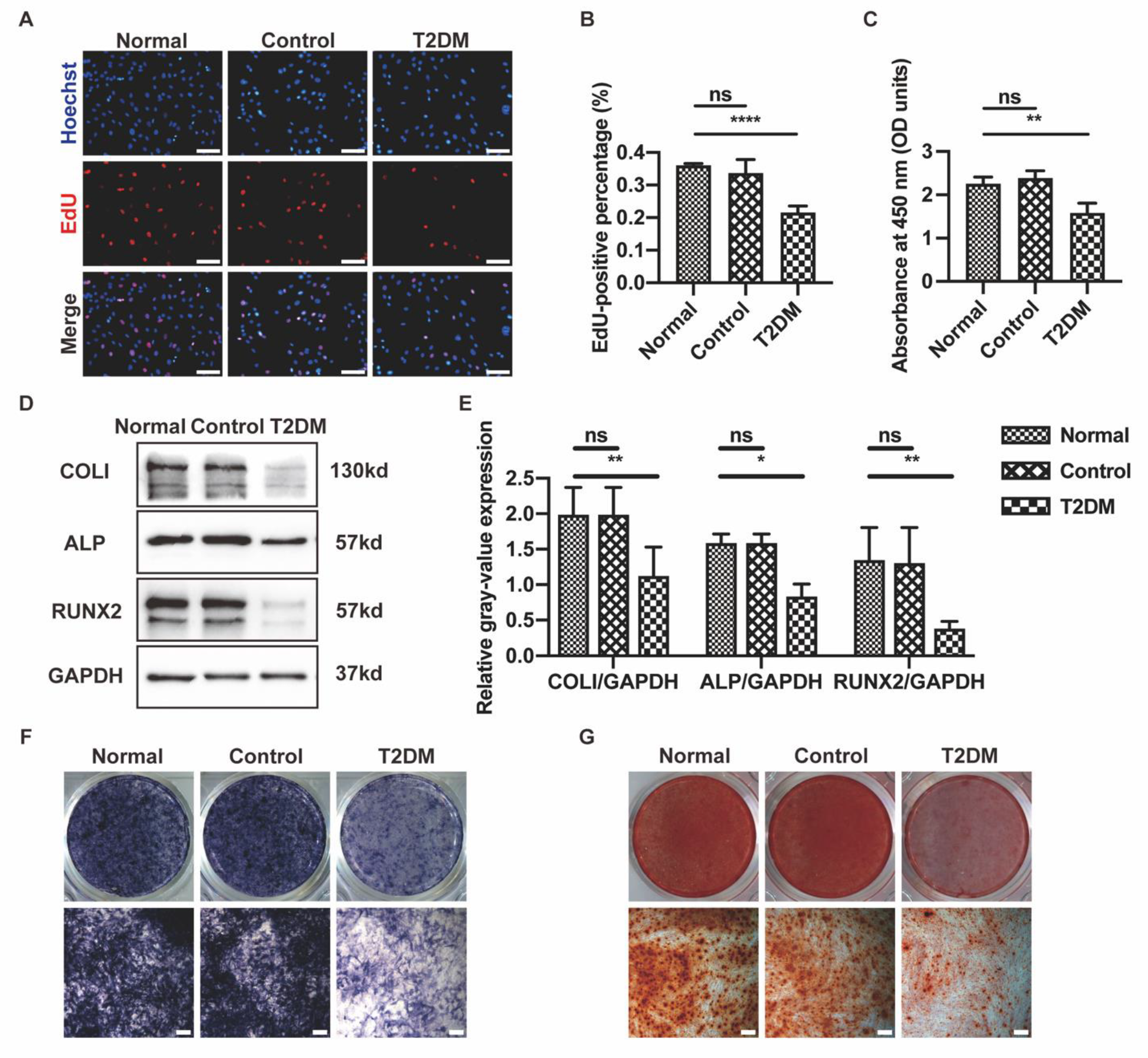
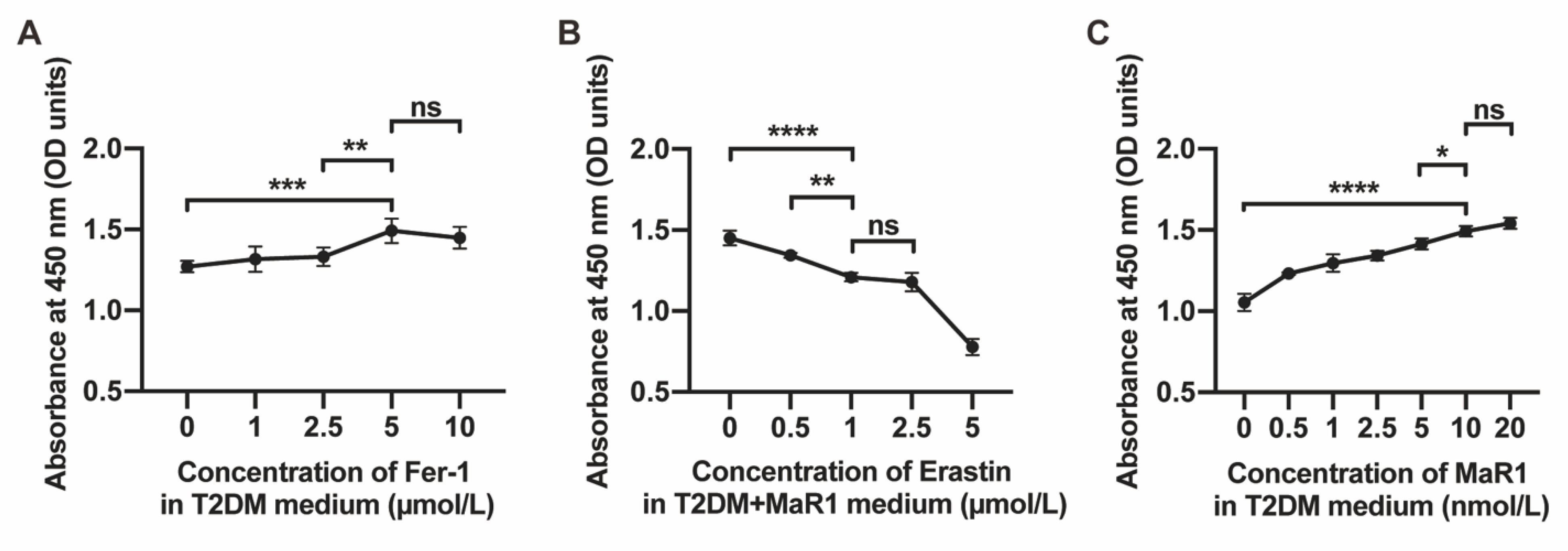
3.1. MaR1 Improved Osteogenesis Inhibited by High Glucose in MC3T3-E1
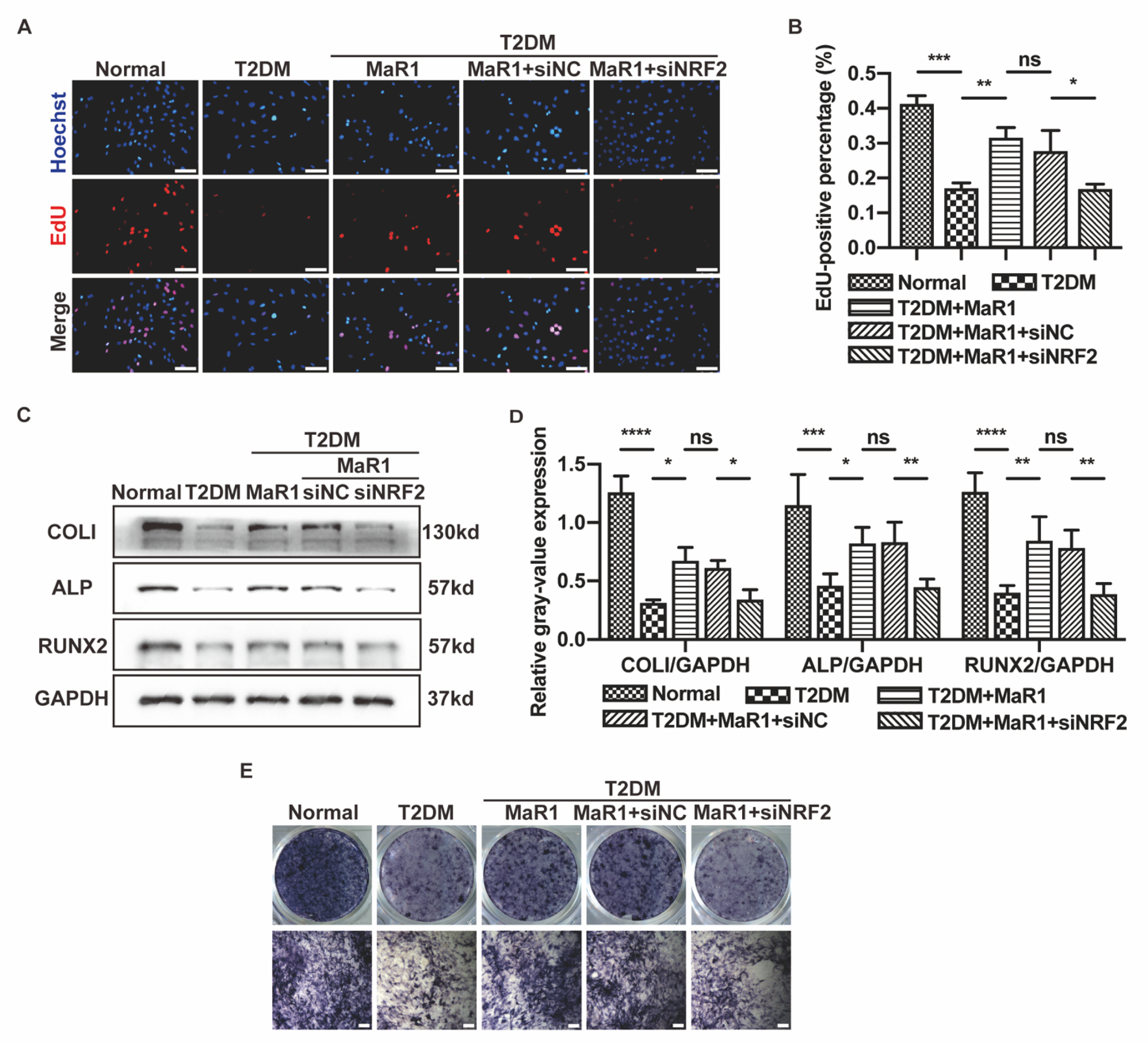
3.2. MaR1 Improved the Osteogenic Function of Osteoblasts in High-Glucose Medium by Activating NRF2 Signaling
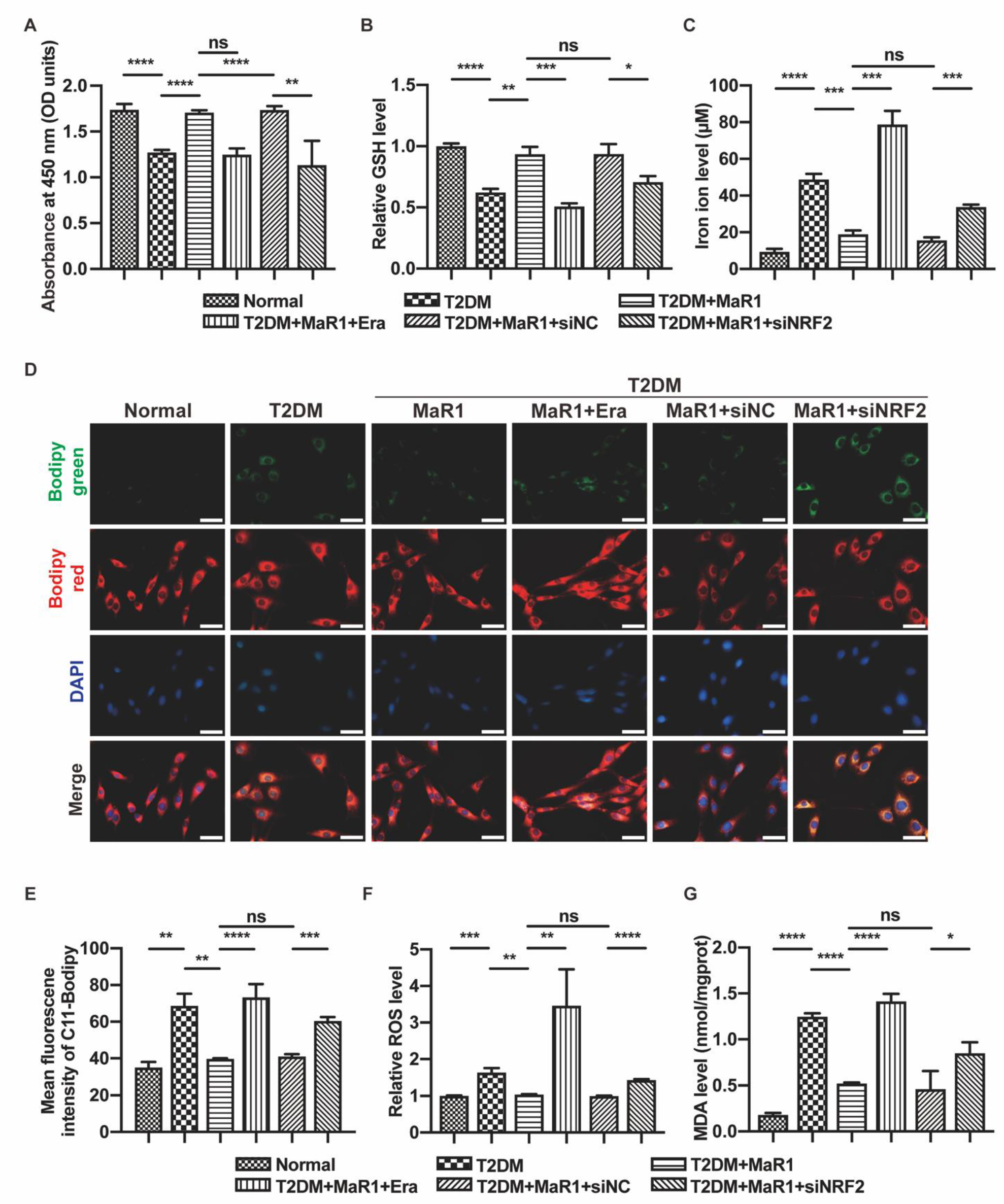
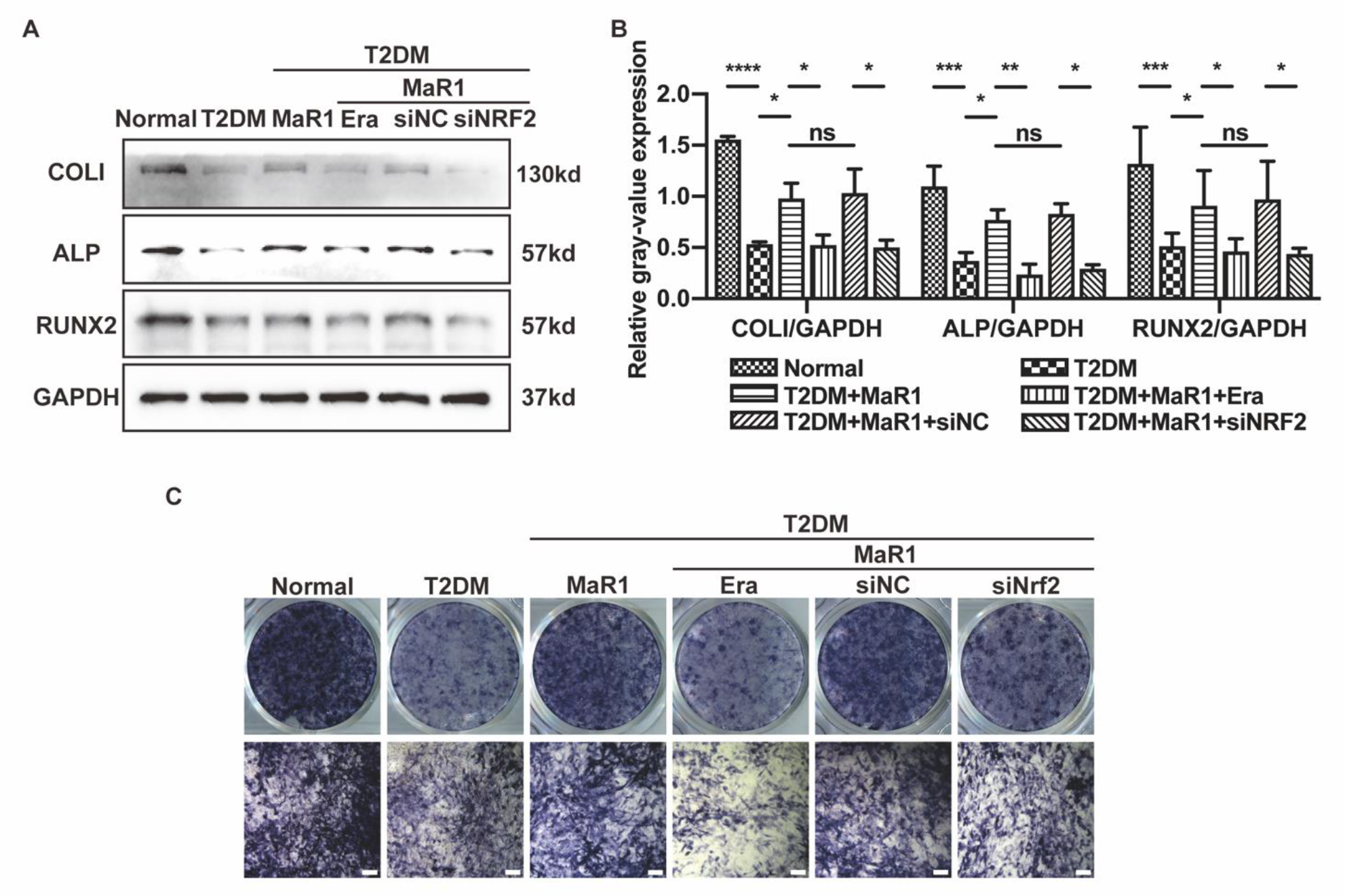
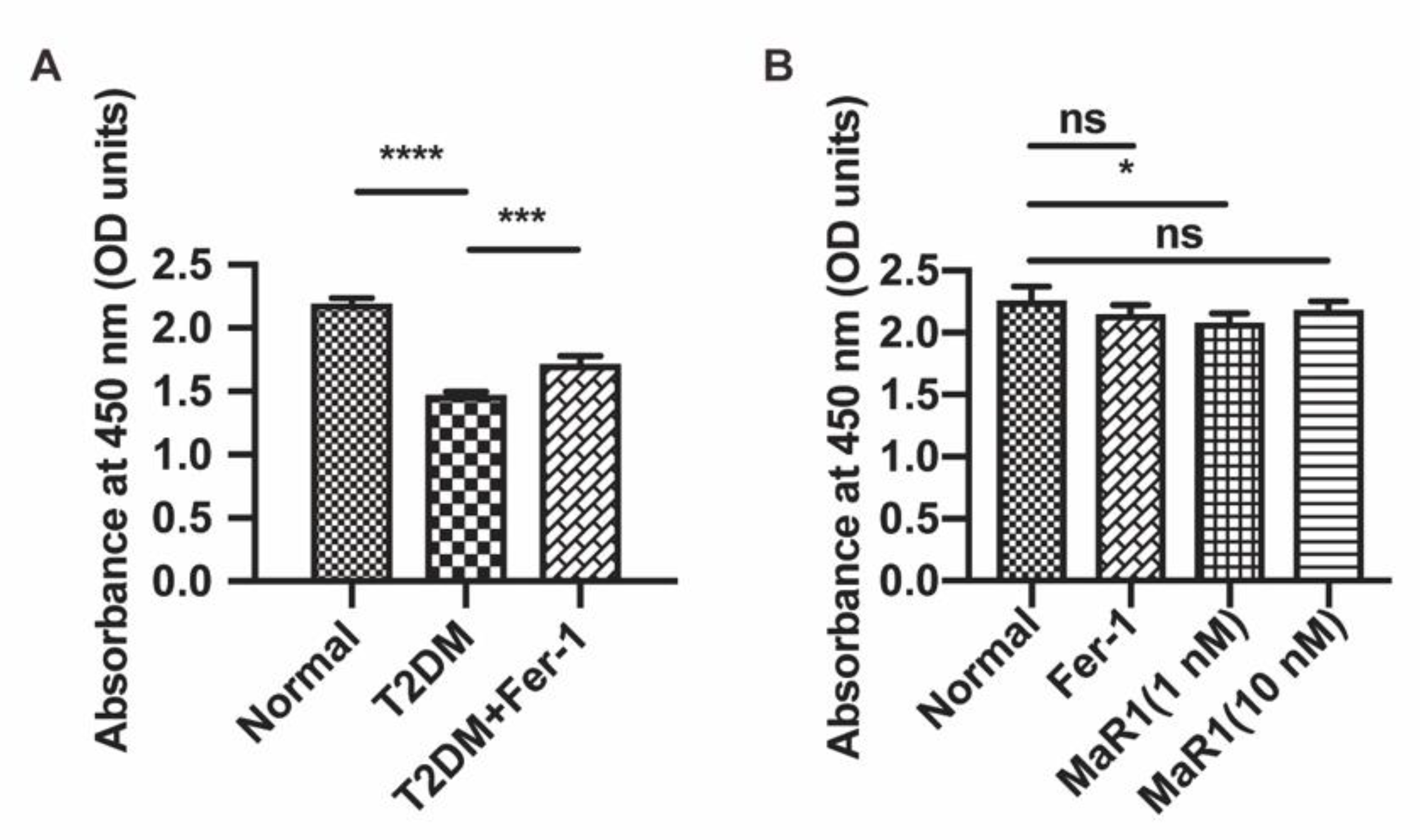
3.3. MaR1 Ameliorated High-Glucose-Induced Osteoblast Ferroptosis via NRF2 Activation
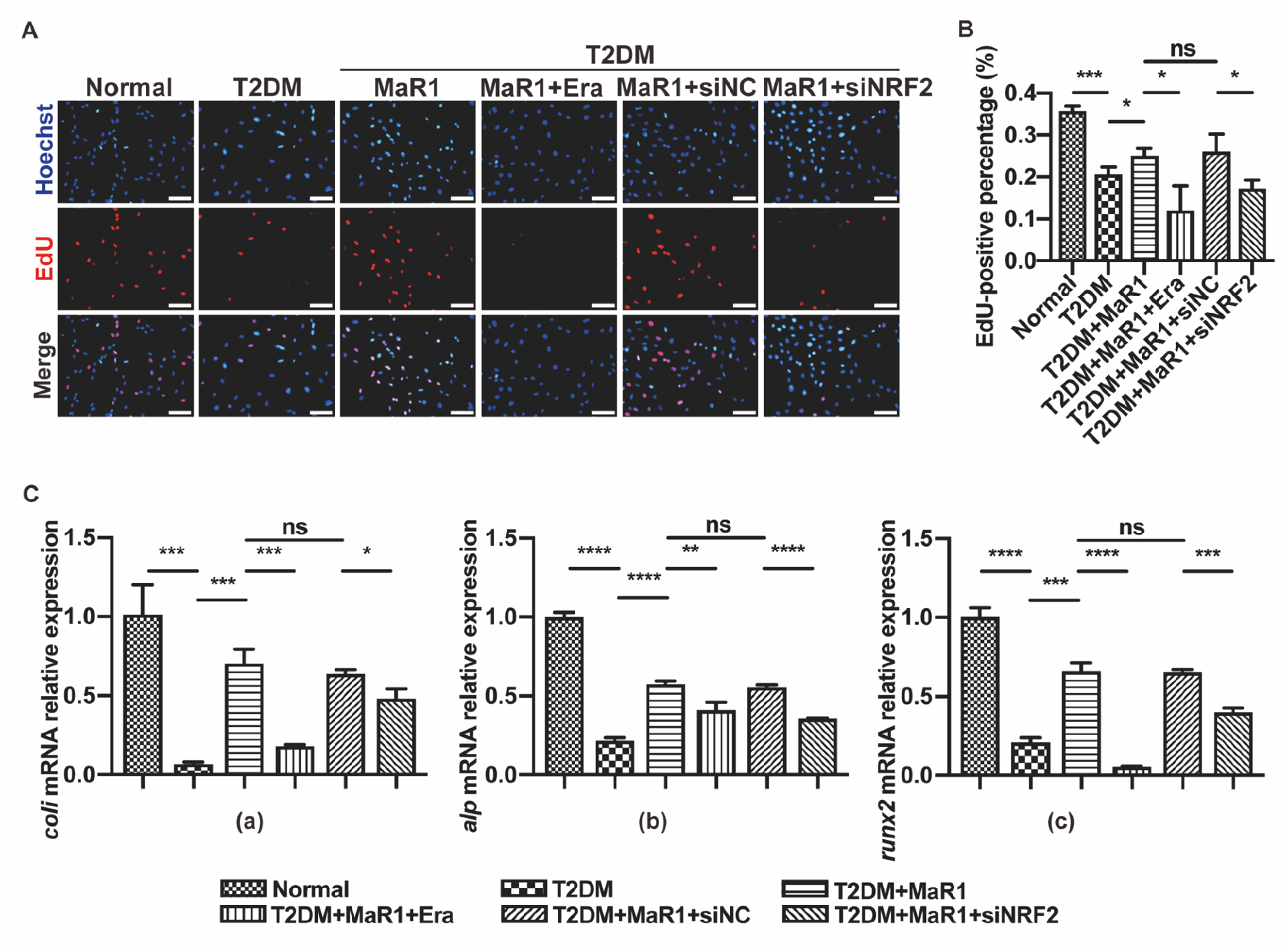
3.4. MaR1 Protected Osteoblasts from Ferroptosis-Mediated Impairment of Osteogenic Differentiation by Modulating NRF2 Signaling in T2DOP
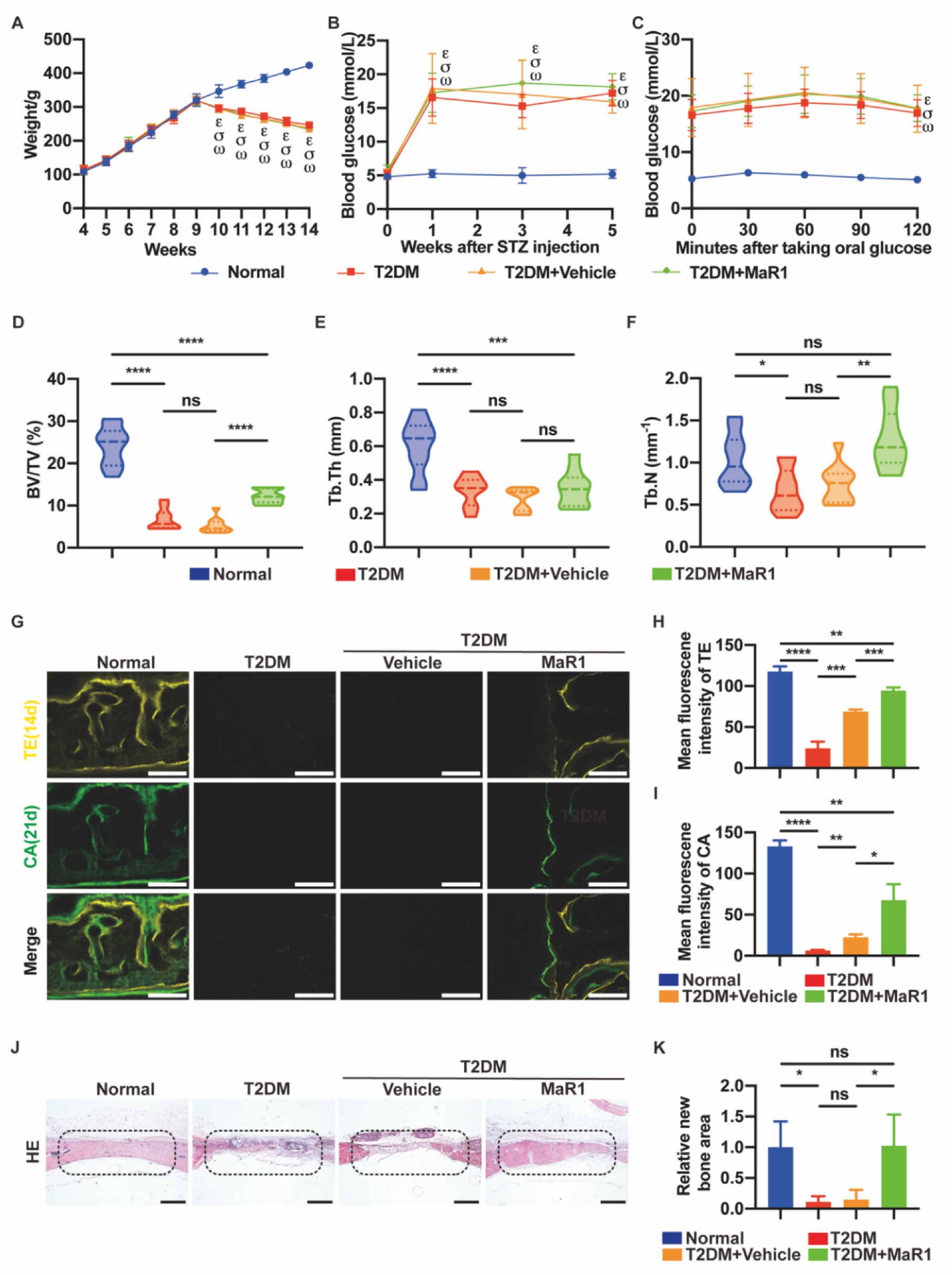
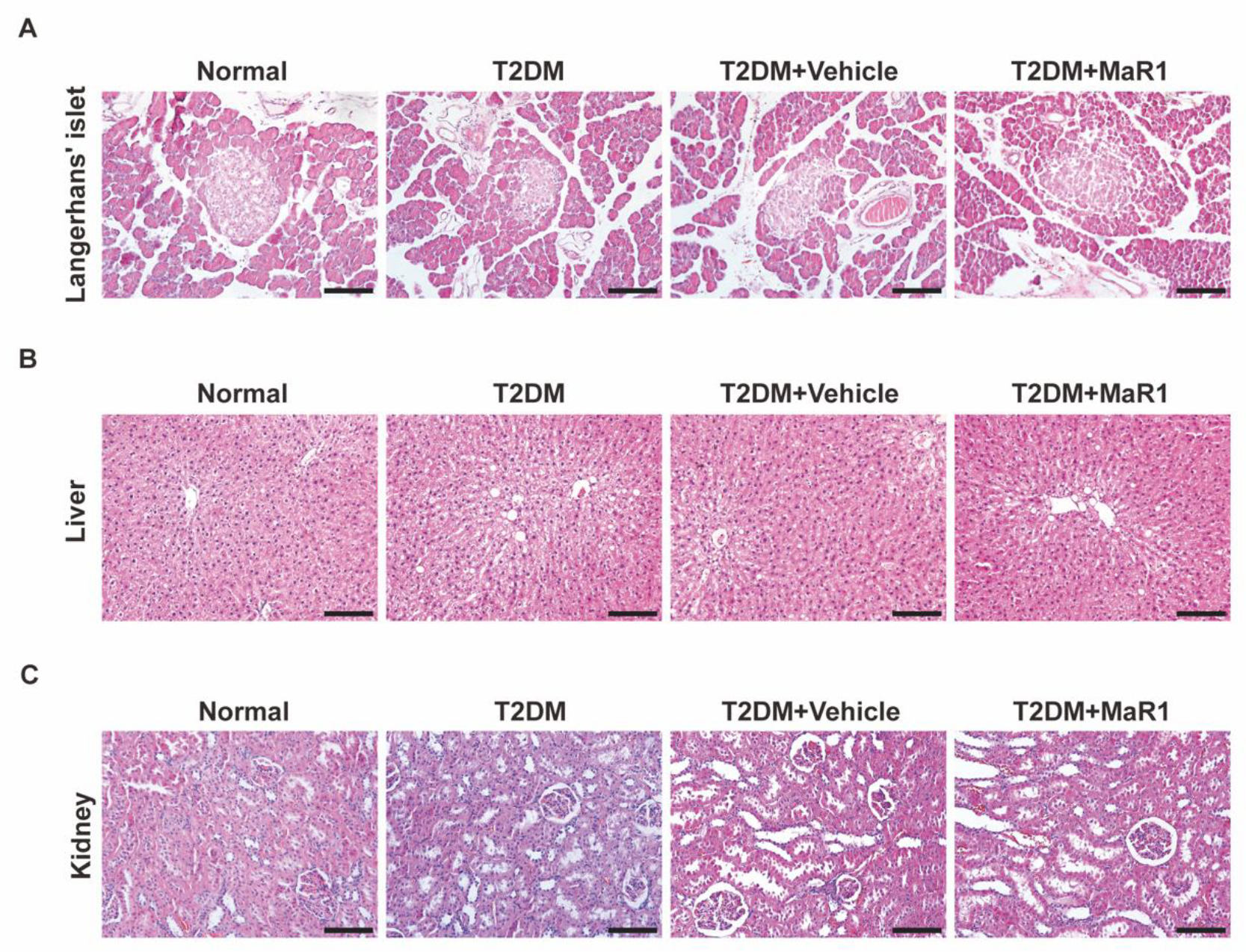
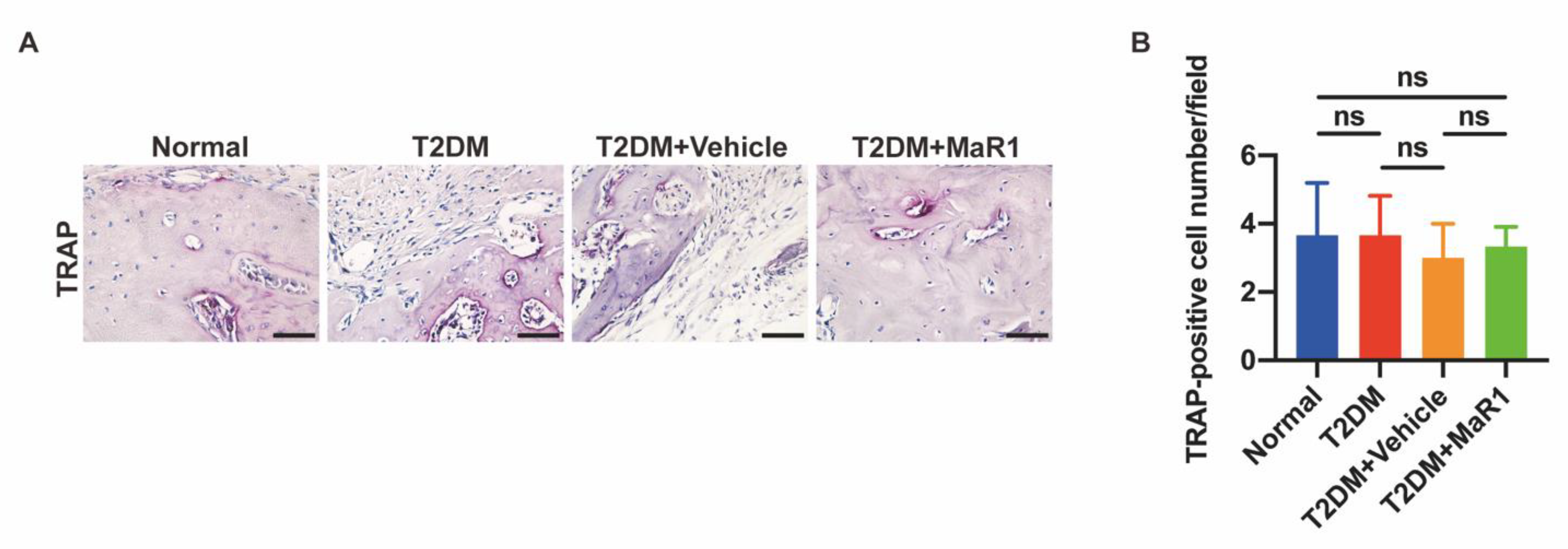
3.5. MaR1 Promotes In Situ Bone Regeneration in a Calvarial Defect Model in Diabetic Rats
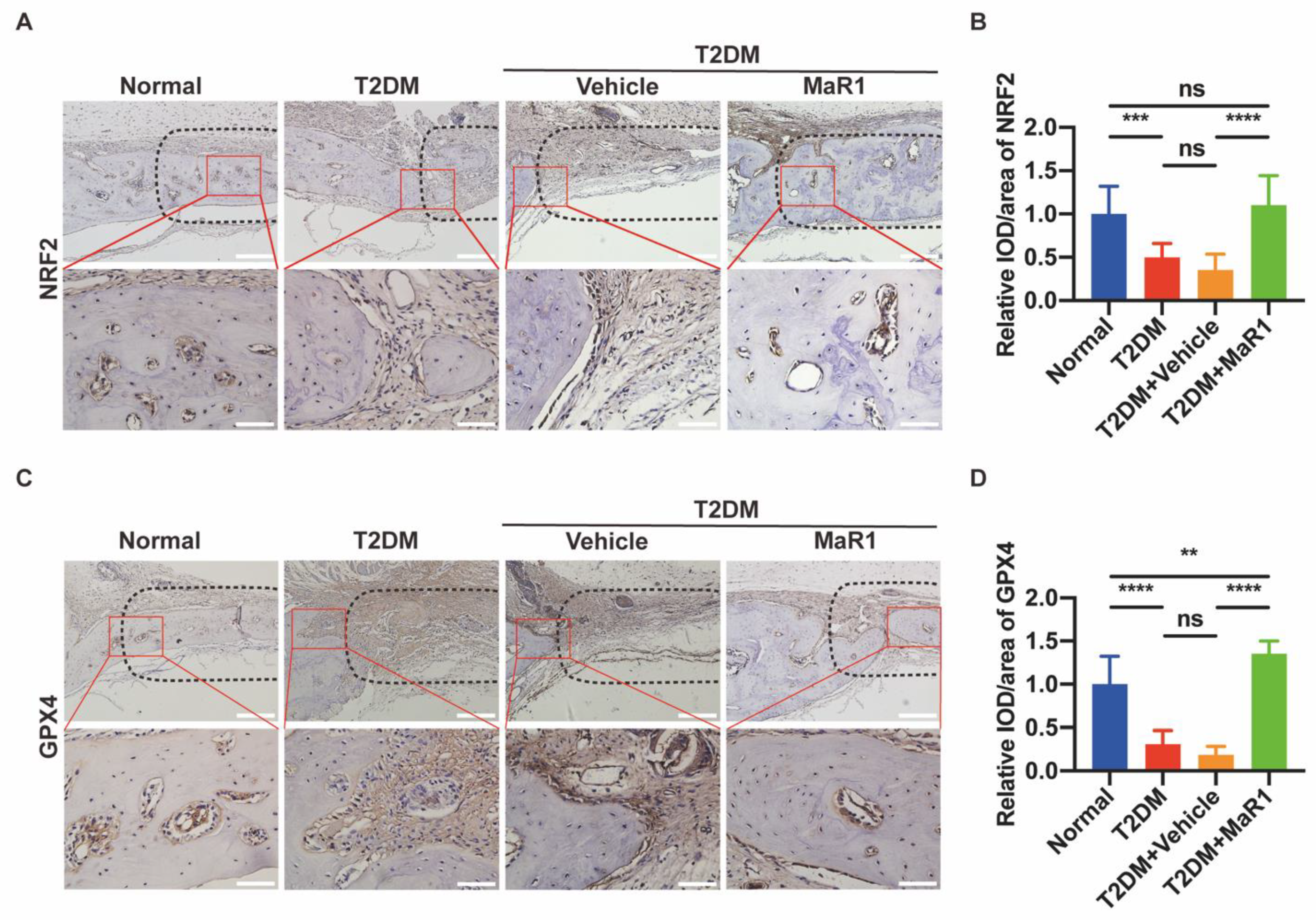
3.5.1. MaR1 Enhances In Situ Bone Regeneration
3.5.2. MaR1 Enhances the Expression of NRF2 and GPX4
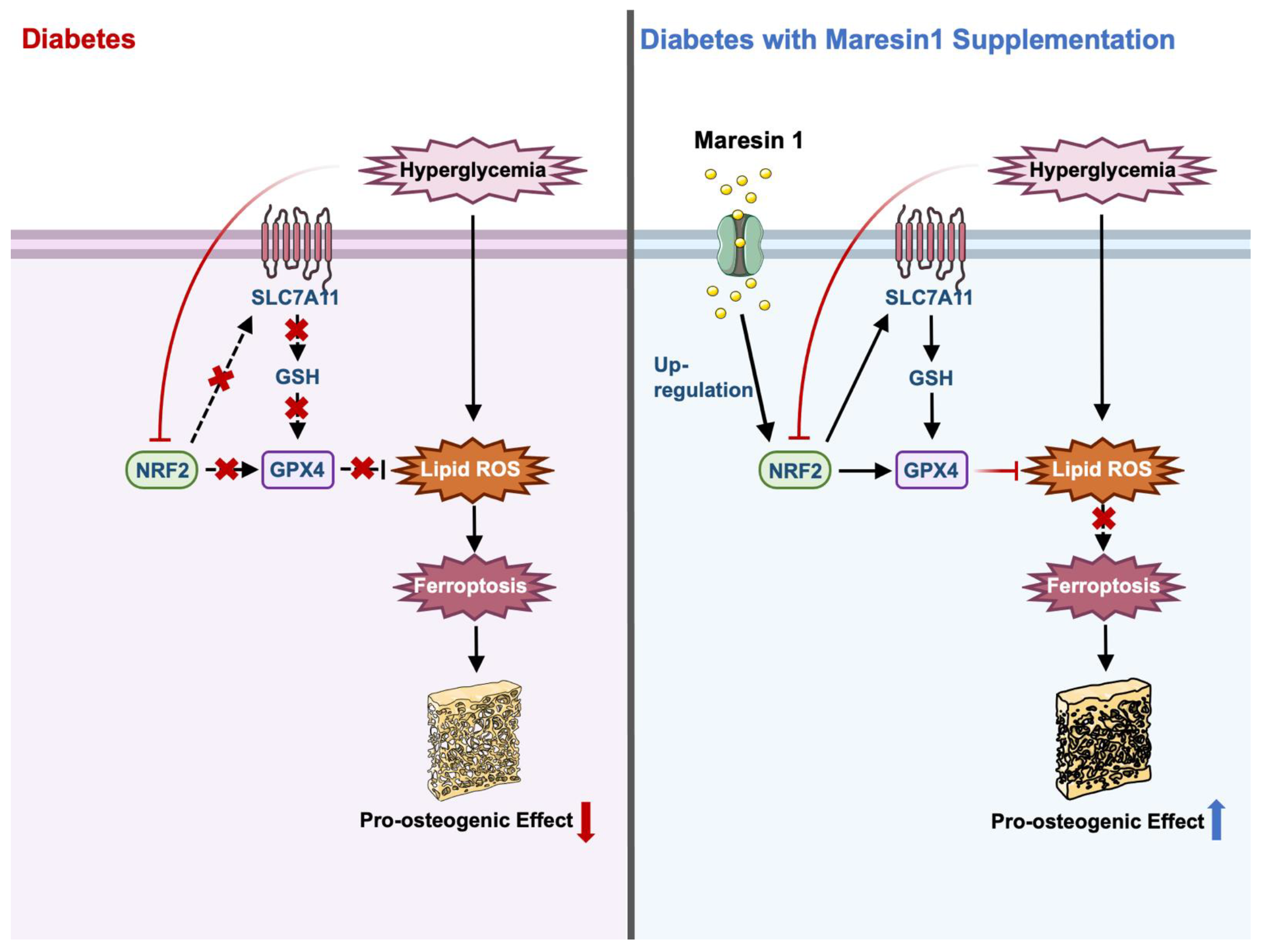
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A









References
- Compston, J. Type 2 diabetes mellitus and bone. J. Intern. Med. 2018, 283, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Khosla, S.; Samakkarnthai, P.; Monroe, D.G.; Farr, J.N. Update on the pathogenesis and treatment of skeletal fragility in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2021, 17, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Veronese, N.; Cooper, C.; Reginster, J.-Y.; Hochberg, M.; Branco, J.; Bruyère, O.; Chapurlat, R.; Al-Daghri, N.; Dennison, E.; Herrero-Beaumont, G.; et al. Type 2 diabetes mellitus and osteoarthritis. Semin. Arthritis Rheum. 2019, 49, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-C.; Guntur, A.R.; Long, F.; Rosen, C.J. Energy Metabolism of the Osteoblast: Implications for Osteoporosis. Endocr. Rev. 2017, 38, 255–266. [Google Scholar] [CrossRef]
- Ferrari, S.; Abrahamsen, B.; Napoli, N.; Akesson, K.; Chandran, M.; Eastell, R.; Fuleihan, G.E.-H.; Josse, R.; Kendler, D.; Kraenzlin, M.; et al. Diagnosis and management of bone fragility in diabetes: An emerging challenge. Osteoporos. Int. 2018, 29, 2585–2596. [Google Scholar] [CrossRef]
- Sheu, A.; Greenfield, J.R.; White, C.P.; Center, J.R. Assessment and treatment of osteoporosis and fractures in type 2 diabetes. Trends Endocrinol. Metab. 2022, 33, 333–344. [Google Scholar] [CrossRef]
- Saito, M.; Marumo, K. Collagen cross-links as a determinant of bone quality: A possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos. Int. 2010, 21, 195–214. [Google Scholar] [CrossRef]
- Javed, F.; Romanos, G.E. Chronic hyperglycemia as a risk factor in implant therapy. Periodontol. 2000 2019, 81, 57–63. [Google Scholar] [CrossRef]
- De Oliveira, P.G.F.P.; Bonfante, E.A.; Bergamo, E.T.; de Souza, S.L.S.; Riella, L.; Torroni, A.; Jalkh, E.B.B.; Witek, L.; Lopez, C.D.; Zambuzzi, W.F.; et al. Obesity/Metabolic Syndrome and Diabetes Mellitus on Peri-implantitis. Trends Endocrinol. Metab. 2020, 31, 596–610. [Google Scholar] [CrossRef]
- Mohsin, S.; Baniyas, M.M.; Aldarmaki, R.S.; Tekes, K.; Kalász, H.; Adeghate, E.A. An update on therapies for the treatment of diabetes-induced osteoporosis. Expert Opin. Biol. Ther. 2019, 19, 937–948. [Google Scholar] [CrossRef]
- Vestergaard, P. Bone metabolism in type 2 diabetes and role of thiazolidinediones. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Napoli, N.; Chandran, M.; Pierroz, D.D.; Abrahamsen, B.; Schwartz, A.V.; Ferrari, S.L. Mechanisms of diabetes mellitus-induced bone fragility. Nat. Rev. Endocrinol. 2017, 13, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Dalli, J.; Karamnov, S.; Choi, A.; Park, C.-K.; Xu, Z.-Z.; Ji, R.-R.; Zhu, M.; Petasis, N.A. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012, 26, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Saito-Sasaki, N.; Sawada, Y.; Nakamura, M. Maresin-1 and Inflammatory Disease. Int. J. Mol. Sci. 2022, 23, 1367. [Google Scholar] [CrossRef]
- Barden, A.E.; Moghaddami, M.; Mas, E.; Phillips, M.; Cleland, L.G.; Mori, T.A. Specialised pro-resolving mediators of inflammation in inflammatory arthritis. Prostaglandins Leukot. Essent. Fat. Acids 2016, 107, 24–29. [Google Scholar] [CrossRef]
- Krishnamoorthy, N.; Burkett, P.R.; Dalli, J.; Abdulnour, R.-E.E.; Colas, R.; Ramon, S.; Phipps, R.P.; Petasis, N.; Kuchroo, V.K.; Serhan, C.N.; et al. Cutting Edge: Maresin-1 Engages Regulatory T Cells to Limit Type 2 Innate Lymphoid Cell Activation and Promote Resolution of Lung Inflammation. J. Immunol. 2015, 194, 863–867. [Google Scholar] [CrossRef]
- Francos-Quijorna, I.; Santos-Nogueira, E.; Gronert, K.; Sullivan, A.B.; Kopp, M.A.; Brommer, B.; David, S.; Schwab, J.M.; López-Vales, R. Maresin 1 promotes inflammatory resolution, neuroprotection, and functional neurological recovery after spinal cord injury. J. Neurosci. 2017, 37, 11731–11743. [Google Scholar] [CrossRef]
- Li, D.; Wang, M.; Ye, J.; Zhang, J.; Xu, Y.; Wang, Z.; Zhao, M.; Ye, D.; Wan, J. Maresin 1 alleviates the inflammatory response, reduces oxidative stress and protects against cardiac injury in LPS-induced mice. Life Sci. 2021, 277, 119467. [Google Scholar] [CrossRef]
- Qiu, S.; Li, P.; Zhao, H.; Li, X. Maresin 1 alleviates dextran sulfate sodium-induced ulcerative colitis by regulating NRF2 and TLR4/NF-kB signaling pathway. Int. Immunopharmacol. 2020, 78, 106018. [Google Scholar] [CrossRef]
- Soto, G.; Rodríguez, M.J.; Fuentealba, R.; Treuer, A.V.; Castillo, I.; González, D.R.; Zúñiga-Hernández, J. Maresin 1, a Proresolving Lipid Mediator, Ameliorates Liver Ischemia-Reperfusion Injury and Stimulates Hepatocyte Proliferation in Sprague-Dawley Rats. Int. J. Mol. Sci. 2020, 21, 540. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Wu, Y.; Zhao, H.; Sun, H.; Gao, S. Maresin 1 mitigates renal ischemia/reperfusion injury in mice via inhibition of the TLR4/MAPK/NF-κB pathways and activation of the Nrf2 pathway. Drug Des. Dev. Ther. 2019, 13, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wang, Y.; Zhang, C.; Huang, Y.; Yu, J.; Shi, L.; Zhang, P.; Yin, Y.; Li, R.; Tao, K. Maresin1 Protect Against Ferroptosis-Induced Liver Injury Through ROS Inhibition and Nrf2/HO-1/GPX4 Activation. Front. Pharmacol. 2022, 13, 865689. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yu, S.; Fretwurst, T.; Larsson, L.; Sugai, J.; Oh, J.; Lehner, K.; Jin, Q.; Giannobile, W. Maresin 1 Promotes Wound Healing and Socket Bone Regeneration for Alveolar Ridge Preservation. J. Dent. Res. 2020, 99, 930–937. [Google Scholar] [CrossRef]
- Huang, R.; Vi, L.; Zong, X.; Baht, G.S. Maresin 1 resolves aged-associated macrophage inflammation to improve bone regeneration. FASEB J. 2020, 34, 13521–13532. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1–Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637–651. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, L.; Shen, Y.; He, Y.; Cheng, G.; Yin, M.; Zhang, Q.; Qin, L. Curculigoside Protects against Excess-Iron-Induced Bone Loss by Attenuating Akt-FoxO1-Dependent Oxidative Damage to Mice and Osteoblastic MC3T3-E1 Cells. Oxidative Med. Cell. Longev. 2019, 2019, 9281481. [Google Scholar] [CrossRef] [PubMed]
- Tsay, J.; Yang, Z.; Ross, F.P.; Cunningham-Rundles, S.; Lin, H.; Coleman, R.; Mayer-Kuckuk, P.; Doty, S.B.; Grady, R.W.; Giardina, P.J.; et al. Bone loss caused by iron overload in a murine model: Importance of oxidative stress. Blood 2010, 116, 2582–2589. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Du, T.; Chen, K.; Guo, J.; Xiang, W.; Yao, X.; Sun, K.; Ye, Y.; Guo, F. Icariin protects against iron overload-induced bone loss via suppressing oxidative stress. J. Cell. Physiol. 2019, 234, 10123–10137. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Yamagishi, S.-I. Role of advanced glycation end products (AGEs) in osteoporosis in diabetes. Curr. Drug Targets 2011, 12, 2096–2102. [Google Scholar] [CrossRef]
- He, Y.-F.; Ma, Y.; Gao, C.; Zhao, G.-Y.; Zhang, L.-L.; Li, G.; Pan, Y.-Z.; Li, K.; Xu, Y.-J. Iron Overload Inhibits Osteoblast Biological Activity Through Oxidative Stress. Biol. Trace Elem. Res. 2013, 152, 292–296. [Google Scholar] [CrossRef]
- Ma, H.; Wang, X.; Zhang, W.; Li, H.; Zhao, W.; Sun, J.; Yang, M. Melatonin Suppresses Ferroptosis Induced by High Glucose via Activation of the Nrf2/HO-1 Signaling Pathway in Type 2 Diabetic Osteoporosis. Oxidative Med. Cell. Longev. 2020, 2020, 9067610. [Google Scholar] [CrossRef]
- Wang, X.; Ma, H.; Sun, J.; Zheng, T.; Zhao, P.; Li, H.; Yang, M. Mitochondrial Ferritin Deficiency Promotes Osteoblastic Ferroptosis via Mitophagy in Type 2 Diabetic Osteoporosis. Biol. Trace Elem. Res. 2022, 200, 298–307. [Google Scholar] [CrossRef]
- Lin, Y.; Shen, X.; Ke, Y.; Lan, C.; Chen, X.; Liang, B.; Zhang, Y.; Yan, S. Activation of osteoblast ferroptosis via the METTL3/ASK1-p38 signaling pathway in high glucose and high fat (HGHF)-induced diabetic bone loss. FASEB J. 2022, 36, e22147. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shrestha, A.; Zhang, H.; Li, L.; Li, D.; Fu, T.; Song, J.; Ji, P.; Huang, Y.; Chen, T. Impact of diabetes mellitus simulations on bone cell behavior through in vitro models. J. Bone Miner. Metab. 2020, 38, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ma, C.; Rong, X.; Zou, S.; Liu, X. Immunomodulatory ECM-like Microspheres for Accelerated Bone Regeneration in Diabetes Mellitus. ACS Appl. Mater. Interfaces 2018, 10, 2377–2390. [Google Scholar] [CrossRef]
- Jia, T.; Wang, Y.-N.; Zhang, J.; Hao, X.; Zhang, D.; Xu, X. Cinaciguat in combination with insulin induces a favorable effect on implant osseointegration in type 2 diabetic rats. Biomed. Pharmacother. 2019, 118, 109216. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Wang, Y.-N.; Feng, Y.; Wang, C.; Zhang, D.; Xu, X. Pharmic Activation of PKG2 Alleviates Diabetes-Induced Osteoblast Dysfunction by Suppressing PLCβ1-Ca2+-Mediated Endoplasmic Reticulum Stress. Oxidative Med. Cell. Longev. 2021, 2021, 5552530. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, T.; Feng, Y.; Liu, S.; Zhang, W.; Zhang, D.; Xu, X. Hyperlipidemia Impairs Osseointegration via the ROS/Wnt/β-Catenin Pathway. J. Dent. Res. 2021, 100, 658–665. [Google Scholar] [CrossRef]
- Yang, T.; Xu, G.; Newton, P.; Chagin, A.; Mkrtchian, S.; Carlström, M.; Zhang, X.-M.; Harris, R.; Cooter, M.; Berger, M.; et al. Maresin 1 attenuates neuroinflammation in a mouse model of perioperative neurocognitive disorders. Br. J. Anaesth. 2019, 122, 350–360. [Google Scholar] [CrossRef]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef]
- Gao, Y.; Patil, S.; Jia, J. The Development of Molecular Biology of Osteoporosis. Int. J. Mol. Sci. 2021, 22, 8182. [Google Scholar] [CrossRef]
- Hamann, C.; Kirschner, S.; Günther, K.-P.; Hofbauer, L.C. Bone, sweet bone—Osteoporotic fractures in diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Gheibi, S.; Kashfi, K.; Ghasemi, A. A practical guide for induction of type-2 diabetes in rat: Incorporating a high-fat diet and streptozotocin. Biomed. Pharmacother. 2017, 95, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Skovsø, S. Modeling type 2 diabetes in rats using high fat diet and streptozotocin. J. Diabetes Investig. 2014, 5, 349–358. [Google Scholar] [CrossRef]
- Vajgel, A.; Mardas, N.; Farias, B.C.; Petrie, A.; Cimões, R.; Donos, N. A systematic review on the critical size defect model. Clin. Oral Implant. Res. 2014, 25, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.S.; Fernandes, M.H. Rodent models in bone-related research: The relevance of calvarial defects in the assessment of bone regeneration strategies. Lab. Anim. 2011, 45, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Czekanska, E.; Stoddart, M.; Richards, R.; Hayes, J. In search of an osteoblast cell model for in vitro research. Eur. Cells Mater. 2012, 24, 1–17. [Google Scholar] [CrossRef]
- Levy, B.D.; Serhan, C.N. Resolution of Acute Inflammation in the Lung. Annu. Rev. Physiol. 2014, 76, 467–492. [Google Scholar] [CrossRef]
- Li, Q.-F.; Hao, H.; Tu, W.-S.; Guo, N.; Zhou, X.-Y. Maresins: Anti-inflammatory pro-resolving mediators with therapeutic potential. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7442–7453. [Google Scholar] [CrossRef]
- Tang, S.; Gao, C.; Long, Y.; Huang, W.; Chen, J.; Fan, F.; Jiang, C.; Xu, Y. Maresin 1 Mitigates High Glucose-Induced Mouse Glomerular Mesangial Cell Injury by Inhibiting Inflammation and Fibrosis. Mediat. Inflamm. 2017, 2017, 2438247. [Google Scholar] [CrossRef]
- Das, U.N. Influence of polyunsaturated fatty acids and their metabolites on stem cell biology. Nutrition 2011, 27, 21–25. [Google Scholar] [CrossRef]
- Pillai, R.; Hayashi, M.; Zavitsanou, A.-M.; Papagiannakopoulos, T. NRF2: KEAPing Tumors Protected. Cancer Discov. 2022, 12, 625–643. [Google Scholar] [CrossRef]
- Ulasov, A.V.; Rosenkranz, A.A.; Georgiev, G.P.; Sobolev, A.S. Nrf2/Keap1/ARE signaling: Towards specific regulation. Life Sci. 2022, 291, 120111. [Google Scholar] [CrossRef]
- Tao, W.; Wang, N.; Ruan, J.; Cheng, X.; Fan, L.; Zhang, P.; Lu, C.; Hu, Y.; Che, C.; Sun, D.; et al. Enhanced ROS-Boosted Phototherapy against Pancreatic Cancer via Nrf2-Mediated Stress-Defense Pathway Suppression and Ferroptosis Induction. ACS Appl. Mater. Interfaces 2022, 14, 6404–6416. [Google Scholar] [CrossRef]
- Pasupuleti, V.R.; Arigela, C.S.; Gan, S.H.; Salam, S.K.N.; Krishnan, K.T.; Rahman, N.A.; Jeffree, M.S. A Review on Oxidative Stress, Diabetic Complications, and the Roles of Honey Polyphenols. Oxidative Med. Cell. Longev. 2020, 2020, 8878172. [Google Scholar] [CrossRef]
- Liu, P.; Wang, W.; Li, Z.; Li, Y.; Yu, X.; Tu, J.; Zhang, Z. Ferroptosis: A New Regulatory Mechanism in Osteoporosis. Oxidative Med. Cell. Longev. 2022, 2022, 2634431. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef]
- Gan, B. Mitochondrial regulation of ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef]
- Bebber, C.M.; Thomas, E.S.; Stroh, J.; Chen, Z.; Androulidaki, A.; Schmitt, A.; Höhne, M.N.; Stüker, L.; de Pádua Alves, C.; Khonsari, A.; et al. Ferroptosis response segregates small cell lung cancer (SCLC) neuroendocrine subtypes. Nat. Commun. 2021, 12, 2048. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Jang, S.; Chapa-Dubocq, X.R.; Tyurina, Y.Y.; St Croix, C.M.; Kapralov, A.A.; Tyurin, V.A.; Bayır, H.; Kagan, V.E.; Javadov, S. Elucidating the contribution of mitochondrial glutathione to ferroptosis in cardiomyocytes. Redox Biol. 2021, 45, 102021. [Google Scholar] [CrossRef]
- Sparvero, L.J.; Tian, H.; Amoscato, A.A.; Sun, W.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Kapralov, O.; Javadov, S.; He, R.; Watkins, S.C.; et al. Direct Mapping of Phospholipid Ferroptotic Death Signals in Cells and Tissues by Gas Cluster Ion Beam Secondary Ion Mass Spectrometry (GCIB-SIMS). Angew. Chem. Int. Ed. Engl. 2021, 60, 11784–11788. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, M.I.J.; Aichler, M.; Walch, M.A.A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Çolakoğlu, M.; Tunçer, S.; Banerjee, S. Emerging cellular functions of the lipid metabolizing enzyme 15-Lipoxygenase-1. Cell Prolif. 2018, 51, e12472. [Google Scholar] [CrossRef]
- Sha, W.; Hu, F.; Xi, Y.; Chu, Y.; Bu, S. Mechanism of Ferroptosis and Its Role in Type 2 Diabetes Mellitus. J. Diabetes Res. 2021, 2021, 9999612. [Google Scholar] [CrossRef]
- Liu, Y.-P.; Shao, S.-J.; Guo, H.-D. Schwann cells apoptosis is induced by high glucose in diabetic peripheral neuropathy. Life Sci. 2020, 248, 117459. [Google Scholar] [CrossRef]
- Cao, Z.; Huang, D.; Tang, C.; Lu, Y.; Huang, S.; Peng, C.; Hu, X. Pyroptosis in diabetes and diabetic nephropathy. Clin. Chim. Acta 2022, 531, 188–196. [Google Scholar] [CrossRef]
- Gao, S.; Zhang, Y.; Zhang, M. Targeting Novel Regulated Cell Death: Pyroptosis, Necroptosis, and Ferroptosis in Diabetic Retinopathy. Front. Cell Dev. Biol. 2022, 10, 932886. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Ji, C.; Wang, Y.-N.; Liu, S.; Wang, M.; Xu, X.; Zhang, D. Maresin1 Suppresses High-Glucose-Induced Ferroptosis in Osteoblasts via NRF2 Activation in Type 2 Diabetic Osteoporosis. Cells 2022, 11, 2560. https://doi.org/10.3390/cells11162560
Zhang Z, Ji C, Wang Y-N, Liu S, Wang M, Xu X, Zhang D. Maresin1 Suppresses High-Glucose-Induced Ferroptosis in Osteoblasts via NRF2 Activation in Type 2 Diabetic Osteoporosis. Cells. 2022; 11(16):2560. https://doi.org/10.3390/cells11162560
Chicago/Turabian StyleZhang, Zhanwei, Chonghao Ji, Ya-Nan Wang, Shiyue Liu, Maoshan Wang, Xin Xu, and Dongjiao Zhang. 2022. "Maresin1 Suppresses High-Glucose-Induced Ferroptosis in Osteoblasts via NRF2 Activation in Type 2 Diabetic Osteoporosis" Cells 11, no. 16: 2560. https://doi.org/10.3390/cells11162560
APA StyleZhang, Z., Ji, C., Wang, Y. -N., Liu, S., Wang, M., Xu, X., & Zhang, D. (2022). Maresin1 Suppresses High-Glucose-Induced Ferroptosis in Osteoblasts via NRF2 Activation in Type 2 Diabetic Osteoporosis. Cells, 11(16), 2560. https://doi.org/10.3390/cells11162560