
Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer



Abstract
:1. General Introduction
2. Mechanism of CMA
3. Targeting of Proteins for Degradation by CMA
4. Hsc70 and Its Interaction with Target Proteins
5. Assembly of the LAMP-2A/Hsc70/Protein Complexes on the Lysosomal Surface and Translocation into the Lysosomal Lumen
6. LAMP-2A—The Lysosomal Receptor for the Hsc70/Protein Complex and a Central Player in CMA
7. Visualising CMA
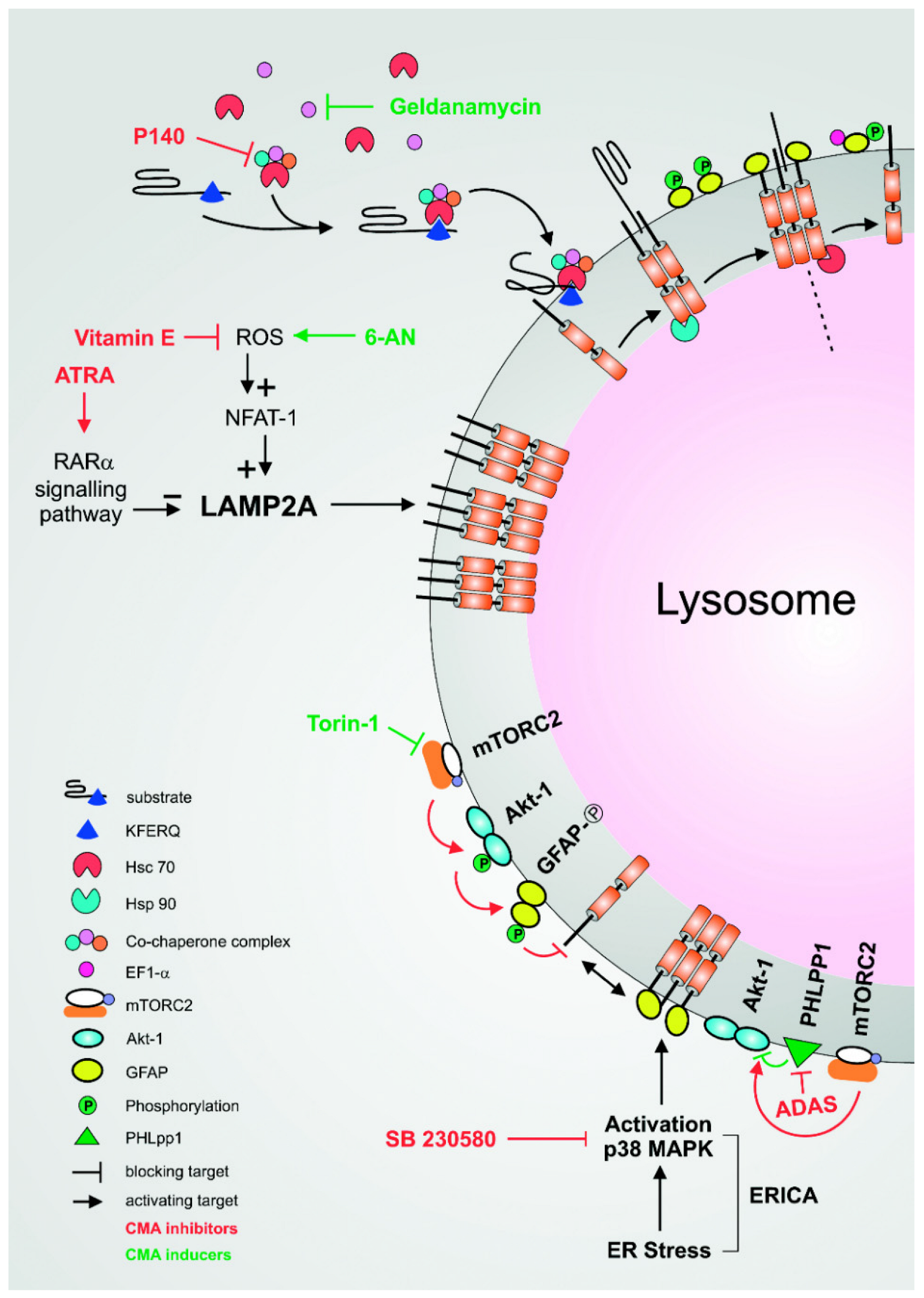
8. CMA Pathways and Their Pharmacological Modulation
8.1. The mTORC2/PHLPP1/AKT Pathway
8.2. The RARα Signalling Pathway and ATRA
8.3. The ERICA Pathway
8.4. Hsp90
8.5. Hsc70
8.6. LAMP-2A
8.7. Oxidative Stress
8.8. Cross-Talk between Macroautophagy and CMA
9. CMA and Cancer
9.1. Anti-Oncogenic Roles of CMA in Healthy Cells
9.2. The Roles of CMA in Malignant Neoplasias
9.2.1. Pro-Oncogenic Role of CMA in Cancer Cells
9.2.2. Glycolytic Capability
9.2.3. Cell Cycle and Proliferation
9.2.4. DNA Damage Response
10. CMA Modulation in Cancer
11. CMA, a New Target in Cancer Therapy?
12. Conclusions and Perspectives
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Klionsky, D.J. Autophagy Revisited: A Conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef]
- Clark, S.L. Cellular Differentiation in the Kidneys of Newborn Mice Studies with the Electron Microscope. J. Biophys. Biochem. Cytol. 1957, 3, 349–362. [Google Scholar] [CrossRef]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in Yeast Demonstrated with Proteinase-Deficient Mutants and Conditions for Its Induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in Mammalian Cells: Revisiting a 40-Year-Old Conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef]
- Lescat, L.; Herpin, A.; Mourot, B.; Véron, V.; Guiguen, Y.; Bobe, J.; Seiliez, I. CMA Restricted to Mammals and Birds: Myth or Reality? Autophagy 2018, 14, 1267–1270. [Google Scholar] [CrossRef]
- Lescat, L.; Véeron, V.; Mourot, B.; Péron, S.; Chenais, N.; Dias, K.; Riera-Heredia, N.; Beaumatin, F.; Pinel, K.; Priault, M.; et al. Chaperone-Mediated Autophagy in the Light of Evolution: Insight from Fish. Mol. Biol. Evol. 2020, 37, 2887–2899. [Google Scholar] [CrossRef]
- Reddy Bonam, S.; Wang, F.; Muller, S. Autophagy: A New Concept in Autoimmunity Regulation and a Novel Therpeutic Option. J. Autoimmun. 2018, 94, 16–32. [Google Scholar] [CrossRef]
- Rothaug, M.; Stroobants, S.; Schweizer, M.; Peters, J.; Zunke, F.; Allerding, M.; D’Hooge, R.; Saftig, P.; Blanz, J. LAMP-2 Deficiency Leads to Hippocampal Dysfunction but Normal Clearance of Neuronal Substrates of Chaperone-Mediated Autophagy in a Mouse Model for Danon Disease. Acta Neuropathol. Commun. 2015, 3, 6. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, B.; Wang, J.; Wu, H.; Xu, S.; Zhang, J.; Wang, L. Discovery of LAMP-2A as Potential Biomarkers for Glioblastoma Development by Modulating Apoptosis through N-CoR Degradation. Cell Commun. Signal. 2021, 19, 40. [Google Scholar] [CrossRef]
- Notomi, S.; Ishihara, K.; Efstathiou, N.E.; Lee, J.J.; Hisatomi, T.; Tachibana, T.; Konstantinou, E.K.; Ueta, T.; Murakami, Y.; Maidana, D.E.; et al. Genetic LAMP2 Deficiency Accelerates the Age-Associated Formation of Basal Laminar Deposits in the Retina. Proc. Natl. Acad. Sci. USA 2019, 116, 23724–23734. [Google Scholar] [CrossRef]
- Dong, S.; Wang, Q.; Kao, Y.R.; Diaz, A.; Tasset, I.; Kaushik, S.; Thiruthuvanathan, V.; Zintiridou, A.; Nieves, E.; Dzieciatkowska, M.; et al. Chaperone-Mediated Autophagy Sustains Haematopoietic Stem-Cell Function. Nature 2021, 591, 117–123. [Google Scholar] [CrossRef]
- Dice, J.F. Altered Degradation of Proteins Microinjected into Senescent Human Fibroblasts. J. Biol. Chem. 1982, 257, 14624–14627. [Google Scholar] [CrossRef]
- Dice, J.F.; Chiang, H.L.; Spencer, E.P.; Backer, J.M. Regulation of Catabolism of Microinjected Ribonuclease A. Identification of Residues 7-11 as the Essential Pentapeptide. J. Biol. Chem. 1986, 261, 6853–6859. [Google Scholar] [CrossRef]
- Chiang, H.-L.; Terlecky, S.; Plant, C.; Dice, J.F. A Role for a 70-Kilodaton Heat Shock Protein in Lysosomal Degradation of Intracellular Proteins. Science 1989, 246, 382–384. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. A Receptor for the Selective Uptake and Degradation of Proteins by Lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef]
- Dice, J.F. Peptide Sequences That Target Cytosolic Proteins for Lysosomal Proteolysis. Trends Biochem. Sci. 1990, 8, 305–309. [Google Scholar] [CrossRef]
- Kirchner, P.; Bourdenx, M.; Madrigal-Matute, J.; Tiano, S.; Diaz, A.; Bartholdy, B.A.; Will, B.; Cuervo, A.M. Proteome-Wide Analysis of Chaperone-Mediated Autophagy Targeting Motifs. PLoS Biol. 2019, 17, e3000301. [Google Scholar] [CrossRef]
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation Targets the M2 Isoform of Pyruvate Kinase for Degradation through Chaperone-Mediated Autophagy and Promotes Tumor Growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef]
- Thompson, L.M.; Aiken, C.T.; Kaltenbach, L.S.; Agrawal, N.; Illes, K.; Khoshnan, A.; Martinez-Vincente, M.; Arrasate, M.; O’Rourke, J.G.; Khashwji, H.; et al. IKK Phosphorylates Huntingtin and Targets It for Degradation by the Proteasome and Lysosome. J. Cell Biol. 2009, 187, 1083–1099. [Google Scholar] [CrossRef]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of Chaperone-Mediated Autophagy during Oxidative Stress. Mol. Biol. Cell 2004, 15, 5318–5328. [Google Scholar] [CrossRef]
- Agarraberes, F.A.; Dice, J.F. A Molecular Chaperone Complex at the Lysosomal Membrane Is Required for Protein Translocation. J. Cell Sci. 2001, 114, 2491–2499. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, P.; Lovchik, M.A.; Li, Y.; Tang, L.; Chen, Z.; Zeng, R.; Ma, D.; Yuan, J.; Yu, Q. Cyclodepsipeptide Toxin Promotes the Degradation of Hsp90 Client Proteins through Chaperone-Mediated Autophagy. J. Cell Biol. 2009, 185, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The Chaperone-Mediated Autophagy Receptor Organizes in Dynamic Protein Complexes at the Lysosomal Membrane. Mol. Cell. Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef]
- Gong, Z.; Tasset, I.; Diaz, A.; Anguiano, J.; Tas, E.; Cui, L.; Kuliawat, R.; Liu, H.; Kühn, B.; Cuervo, A.M.; et al. Humanin Is an Endogenous Activator of Chaperone-mediated Autophagy. J. Cell Biol. 2018, 217, 635–647. [Google Scholar] [CrossRef]
- Gough, N.R.; Fambrough, D.M. Different Steady State Subcellular Distributions of the Three Splice Variants of Lysosome-Associated Membrane Protein LAMP-2 Are Determined Largely by the COOH-Terminal Amino Acid Residue. J. Cell Biol. 1997, 137, 1161–1169. [Google Scholar] [CrossRef]
- Bandyopad, U.; Cuervo, A.M. Entering the Lysosome. Autophagy 2010, 4, 1101–1103. [Google Scholar]
- Rout, A.K.; Strub, M.P.; Piszczek, G.; Tjandra, N. Structure of Transmembrane Domain of Lysosome-Associated Membrane Protein Type 2a (LAMP-2A) Reveals Key Features for Substrate Specificity in Chaperone-Mediated Autophagy. J. Biol. Chem. 2014, 289, 35111–35123. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. Unique Properties of Lamp2a Compared to Other Lamp2 Isoforms. J. Cell Sci. 2000, 113, 4441–4450. [Google Scholar] [CrossRef]
- Arias, E.; Koga, H.; Diaz, A.; Mocholi, E.; Patel, B.; Cuervo, A.M. Lysosomal MTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol. Cell 2015, 59, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Hall, M.N. Regulation of MTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Methods Mol. Biol. 2018, 1691, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, J.; Dou, J.; She, H.; Tao, K.; Xu, H.; Yang, Q.; Mao, Z. Phosphorylation of LAMP2A by P38 MAPK Couples ER Stress to Chaperone-Mediated Autophagy. Nat. Commun. 2017, 8, 1763. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Massey, A.C.; Cuervo, A.M. Lysosome Membrane Lipid Microdomains: Novel Regulators of Chaperone-Mediated Autophagy. EMBO J. 2006, 25, 3921–3933. [Google Scholar] [CrossRef]
- Kiffin, R.; Kaushik, S.; Zeng, M.; Bandyopadhyay, U.; Zhang, C.; Massey, A.C.; Martinez-Vicente, M.; Cuervo, A.M. Altered Dynamics of the Lysosomal Receptor for Chaperone-Mediated Autophagy with Age. J. Cell Sci. 2007, 120, 782–791. [Google Scholar] [CrossRef]
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F.; Valdor, R.; et al. Chaperone-Mediated Autophagy Regulates T Cell Responses through Targeted Degradation of Negative Regulators of T Cell Activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical Modulation of Chaperone-Mediated Autophagy by Retinoic Acid Derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef]
- Gomez-Sintes, R.; Xin, Q.; Jimenez-Loygorri, J.I.; McCabe, M.; Diaz, A.; Garner, T.P.; Cotto-Rios, X.M.; Wu, Y.; Dong, S.; Reynolds, C.A.; et al. Targeting Retinoic Acid Receptor Alpha-Corepressor Interaction Activates Chaperone-Mediated Autophagy and Protects against Retinal Degeneration. Nat. Commun. 2022, 13, 4220. [Google Scholar] [CrossRef]
- Tang, F.L.; Erion, J.R.; Tian, Y.; Liu, W.; Yin, D.M.; Ye, J.; Tang, B.; Mei, L.; Xiong, W.C. VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for α-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. 2015, 35, 10613–10628. [Google Scholar] [CrossRef]
- Zhang, J.; Johnson, J.L.; He, J.; Napolitano, G.; Ramadass, M.; Rocca, C.; Kiosses, W.B.; Bucci, C.; Xin, Q.; Gavathiotis, E.; et al. Cystinosin, the Small GTPase Rab11, and the Rab7 Effector RILP Regulate Intracellular Trafficking of the Chaperone-Mediated Autophagy Receptor LAMP2A. J. Biol. Chem. 2017, 292, 10328–10346. [Google Scholar] [CrossRef]
- Arias, E. Methods to Study Chaperone-Mediated Autophagy. Methods Enzymol. 2017, 588, 283–305. [Google Scholar] [CrossRef]
- Wen, X.; Yang, Y.; Klionsky, D.J. Moments in Autophagy and Disease: Past and Present. Mol. Asp. Med. 2021, 82, 100966. [Google Scholar] [CrossRef]
- Koga, H.; Martinez-Vicente, M.; Macian, F.; Verkhusha, V.V.; Cuervo, A.M. A Photoconvertible Fluorescent Reporter to Track Chaperone-Mediated Autophagy. Nat. Commun. 2011, 2, 386. [Google Scholar] [CrossRef]
- Sato, M.; Seki, T.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H. Fluorescent-Based Evaluation of Chaperone-Mediated Autophagy and Microautophagy Activities in Cultured Cells. Genes Cells 2016, 21, 861–873. [Google Scholar] [CrossRef]
- Seki, T.; Yoshino, K.-i.; Tanaka, S.; Dohi, E.; Onji, T.; Yamamoto, K.; Hide, I.; Paulson, H.L.; Saito, N.; Sakai, N. Establishment of a Novel Fluorescence-Based Method to Evaluate Chaperone-Mediated Autophagy in a Single Neuron. PLoS ONE 2012, 7, e31232. [Google Scholar] [CrossRef]
- Dong, S.; Aguirre-Hernandez, C.; Scrivo, A.; Eliscovich, C.; Arias, E.; Bravo-Cordero, J.J.; Cuervo, A.M. Monitoring Spatiotemporal Changes in Chaperone-Mediated Autophagy In Vivo. Nat. Commun. 2020, 11, 645. [Google Scholar] [CrossRef]
- Liu, Q.; Chang, J.W.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Markhard, A.; Hur, W.; Zhang, J.; Sim, T.; Sabatini, D.M.; et al. Discovery of 1-(4-(4-Propionylpiperazin-1-Yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-Yl)benzo[h][1,6]naphthyridin-2(1 H)-one as a Highly Potent, Selective Mammalian Target of Rapamycin (MTOR) Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 7146–7155. [Google Scholar] [CrossRef]
- Bradley, E.W.; Taylor, E.L.; Becerra, C.C.; Lorang, I.M.; Burhow, S.A.; Reid, J.M.; Westendorf, J.J. Inhibitors of Phlpp Phosphatases Are Highly Stable and Suppress Pain Following Joint Injury. Osteoarthr. Cartil. 2018, 26, S356–S357. [Google Scholar] [CrossRef]
- Hu, Y.; Chan, E.; Wang, S.X.; Li, B. Activation of P38 Mitogen-Activated Protein Kinase Is Required for Osteoblast Differentiation. Endocrinology 2003, 144, 2068–2074. [Google Scholar] [CrossRef]
- Brown, K.K.; Heitmeyer, S.A.; Hookfin, E.B.; Hsieh, L.; Buchalova, M.; Taiwo, Y.O.; Janusz, M.J. P38 MAP Kinase Inhibitors as Potential Therapeutics for the Treatment of Joint Degeneration and Pain Associated with Osteoarthritis. J. Inflamm. 2008, 5, 22. [Google Scholar] [CrossRef]
- Kankaanranta, H.; De Souza, P.M.; Barnes, P.J.; Salmon, M.; Giembycz, M.A.; Lindsay, M.A. SB 203580, an Inhibitor of P38 Mitogen-Activated Protein Kinase, Enhances Constitutive Apoptosis of Cytokine-Deprived Human Eosinophils. J. Pharmacol. Exp. Ther. 1999, 290, 621–628. [Google Scholar]
- Finn, P.F.; Mesires, N.T.; Vine, M.; Dice, J.F. Effects of Small Molecules on Chaperone-Mediated Autophagy. Autophagy 2005, 1, 141–145. [Google Scholar] [CrossRef]
- Xu, Y.; Wallace, M.A.G.; Fitzgerald, M.C. Thermodynamic Analysis of the Geldanamycin–Hsp90 Interaction in a Whole Cell Lysate Using a Mass Spectrometry-Based Proteomics Approach. J. Am. Soc. Mass Spectrom. 2016, 27, 1670–1676. [Google Scholar] [CrossRef]
- Sõti, C.; Nagy, E.; Giricz, Z.; Vígh, L.; Csermely, P.; Ferdinandy, P. Heat Shock Proteins as Emerging Therapeutic Targets. Br. J. Pharmacol. 2005, 146, 769–780. [Google Scholar] [CrossRef]
- Wang, F.; Bonam, S.R.; Schall, N.; Kuhn, L.; Hammann, P.; Chaloin, O.; Madinier, J.B.; Briand, J.P.; Page, N.; Muller, S. Blocking Nuclear Export of HSPA8 after Heat Shock Stress Severely Alters Cell Survival. Sci. Rep. 2018, 8, 16820. [Google Scholar] [CrossRef]
- Muller, S.; Monneaux, F.; Schal, N.; Rashkov, R.K.; Oparanov, B.A.; Wiesel, P.; Geiger, J.M.; Zimmer, R. Spliceosomal Peptide P140 for Immunotherapy of Systemic Lupus Erythematosus: Results of an Early Phase II Clinical Trial. Arthritis Rheum. 2008, 58, 3873–3883. [Google Scholar] [CrossRef]
- Page, N.; Gros, F.; Schall, N.; Décossas, M.; Bagnard, D.; Briand, J.-P.P.; Muller, S. HSC70 Blockade by the Therapeutic Peptide P140 Affects Autophagic Processes and Endogenous MHCII Presentation in Murine Lupus. Ann. Rheum. Dis. 2011, 70, 837–843. [Google Scholar] [CrossRef]
- Macri, C.; Wang, F.; Tasset, I.; Schall, N.; Page, N.; Briand, J.P.; Cuervo, A.M.; Muller, S.; Briand, P.; Cuervo, A.M.; et al. Modulation of Deregulated Chaperone-Mediated Autophagy by a Phosphopeptide. Autophagy 2015, 11, 472–486. [Google Scholar] [CrossRef]
- Zhou, D.; Li, P.; Lin, Y.; Lott, J.M.; Hislop, A.D.; Canaday, D.H.; Brutkiewicz, R.R.; Blum, J.S. Lamp-2a Facilitates MHC Class II Presentation of Cytoplasmic Antigens. Immunity 2005, 22, 571–581. [Google Scholar] [CrossRef]
- Wang, F.; Tasset, I.; Cuervo, A.M.; Muller, S. In Vivo Remodeling of Altered Autophagy-Lysosomal Pathway by a Phosphopeptide in Lupus. Cells 2020, 9, 2328. [Google Scholar] [CrossRef]
- Zhang, C.; Cuervo, A.M. Restoration of Chaperone-Mediated Autophagy in Aging Liver Improves Cellular Maintenance and Hepatic Function. Nat. Med. 2008, 14, 959–965. [Google Scholar] [CrossRef]
- Hubert, V.; Peschel, A.; Langer, B.; Groeger, M.; Rees, A.J.; Kain, R.; Gröger, M.; Rees, A.J.; Kain, R.; Gro, M.; et al. LAMP-2 Is Required for Incorporating Syntaxin-17 into Autophagosomes and for Their Fusion with Lysosomes. Biol. Open 2016, 5, 1516–1529. [Google Scholar] [CrossRef]
- Tanak, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lullmann-Rauch, R.; Janssen, P.M.; Blanz, J.; von Figura, K.; Saftig, P. Accumulation of Autophagic Vacuoles Anc Cardiomyopathy in LAMP-2 Deficient Mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 Deficiency Causes X-Linked Vacuolar Cardiomyopathy and Myopathy (Danon Disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef]
- Pérez, L.; McLetchie, S.; Gardiner, G.J.; Sarah, N.; Zhou, D.; Blum, J.S.; Deffit, S.N.; Zhou, D.; Blum, J.S. LAMP-2C Inhibits MHC Class II Presentation of Cytoplasmic Antigens by Disrupting Chaperone-Mediated Autophagy. J. Immunol. 2016, 196, 2457–2465. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a Selective Pathway of Lysosomal Proteolysis in Rat Liver by Prolonged Starvation. Am. J. Physiol. Cell Physiol. 1995, 269, 1200–1208. [Google Scholar] [CrossRef]
- Dohi, E.; Tanaka, S.; Seki, T.; Miyagi, T.; Hide, I.; Takahashi, T.; Matsumoto, M.; Sakai, N. Hypoxic Stress Activates Chaperone-Mediated Autophagy and Modulates Neuronal Cell Survival. Neurochem. Int. 2012, 60, 431–442. [Google Scholar] [CrossRef]
- Tasset, I.; Cuervo, A.M. Role of Chaperone-Mediated Autophagy in Metabolism. FEBS J. 2016, 283, 2403–2413. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, J.A.; Kaushik, S.; Koga, H.; Dall’Armi, C.; Shui, G.; Wenk, M.R.; Di Paolo, G.; Cuervo, A.M. Inhibitory Effect of Dietary Lipids on Chaperone-Mediated Autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, E705–E714. [Google Scholar] [CrossRef]
- Aw, T.Y.; Rhoads, C.A. Glucose Regulation of Hydroperoxide Metabolism in Rat Intestinal Cells. Stimulation of Reduced Nicotinamide Adenine Dinucleotide Phosphate Supply. J. Clin. Investig. 1994, 94, 2426–2434. [Google Scholar] [CrossRef]
- Yu, Y.; Hou, L.; Song, H.; Xu, P.; Sun, Y.; Wu, K. Akt/AMPK/MTOR Pathway Was Involved in the Autophagy Induced by Vitamin E Succinate in Human Gastric Cancer SGC-7901 Cells. Mol. Cell. Biochem. 2017, 424, 173–183. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. Age-Related Decline in Chaperone-Mediated Autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef]
- Okada, A.A.; Dice, J.F. Altered Degradation of Intracellular Proteins in Aging Human Fibroblast. Mech. Aging Dev. 1984, 26, 341–356. [Google Scholar] [CrossRef]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the Selective Blockage of Chaperone-Mediated Autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; De Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo Recognition Failure Is Responsible for Inefficient Autophagy in Huntington’s Disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef]
- Wu, H.; Chen, S.; Ammar, A.B.; Xu, J.; Wu, Q.; Pan, K.; Zhang, J.; Hong, Y. Crosstalk Between Macroautophagy and Chaperone-Mediated Autophagy: Implications for the Treatment of Neurological Diseases. Mol. Neurobiol. 2015, 52, 1284–1296. [Google Scholar] [CrossRef]
- Gomes, L.R.; Menck, C.F.M.; Cuervo, A.M. Chaperone-Mediated Autophagy Prevents Cellular Transformation by Regulating MYC Proteasomal Degradation. Autophagy 2017, 13, 928–940. [Google Scholar] [CrossRef]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-Mediated Autophagy Is Required for Tumor Growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef]
- Saha, T. LAMP2A Overexpression in Breast Tumors Promotes Cancer Cell Survival via Chaperone-Mediated Autophagy. Autophagy 2012, 8, 1643–1656. [Google Scholar] [CrossRef]
- Schneider, J.L.; Villarroya, J.; Diaz-Carretero, A.; Patel, B.; Urbanska, A.M.; Thi, M.M.; Villarroya, F.; Santambrogio, L.; Cuervo, A.M. Loss of Hepatic Chaperone-Mediated Autophagy Accelerates Proteostasis Failure in Aging. Aging Cell 2015, 14, 249–264. [Google Scholar] [CrossRef]
- Lu, T.J.L.; Huang, G.J.; Wang, H.J.; Chen, J.L.; Hsu, H.P.; Lu, T.J.L. Hispolon Promotes MDM2 Downregulation through Chaperone-Mediated Autophagy. Biochem. Biophys. Res. Commun. 2010, 398, 26–31. [Google Scholar] [CrossRef]
- Kielbik, M.; Szulc-Kielbik, I.; Klink, M. Calreticulin—Multifunctional Chaperone in Immunogenic Cell Death: Potential Significance as a Prognostic Biomarker in Ovarian Cancer Patients. Cells 2021, 10, 130. [Google Scholar] [CrossRef]
- Serrano-del Valle, A.; Anel, A.; Naval, J.; Marzo, I. Immunogenic Cell Death and Immunotherapy of Multiple Myeloma. Front. Cell Dev. Biol. 2019, 7, 50. [Google Scholar] [CrossRef]
- Garg, A.D.; Dudek, A.M.; Agostinis, P. Calreticulin Surface Exposure Is Abrogated in Cells Lacking, Chaperone-Mediated Autophagy-Essential Gene, LAMP2A. Cell Death Dis. 2013, 4, e826. [Google Scholar] [CrossRef]
- Franch, H.A.; Sooparb, S.; Du, J. A Mechanism Regulating Proteolysis of Specific Proteins during Renal Tubular Cell Growth. J. Biol. Chem. 2001, 276, 19126–19131. [Google Scholar] [CrossRef]
- Valdor, R.; García-Bernal, D.; Riquelme, D.; Martinez, C.M.; Moraleda, J.M.; Cuervo, A.M.; Macian, F.; Martinez, S. Glioblastoma Ablates Pericytes Antitumor Immune Function through Aberrant Up-Regulation of Chaperone-Mediated Autophagy. Proc. Natl. Acad. Sci. USA 2019, 116, 20655–20665. [Google Scholar] [CrossRef]
- Arias, E.; Cuervo, A.M. Pros and Cons of Chaperone-Mediated Autophagy in Cancer Biology. Trends Endocrinol. Metab. 2020, 31, 53–66. [Google Scholar] [CrossRef]
- Ding, Z.-B.; Fu, X.T.; Shi, Y.H.; Zhou, J.; Peng, Y.F.; Liu, W.R.; Shi, G.M.; Gao, Q.; Wang, X.Y.; Song, K.; et al. Lamp2a Is Required for Tumor Growth and Promotes Tumor Recurrence of Hepatocellular Carcinoma. Int. J. Oncol. 2016, 49, 2367–2376. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, J.; Fan, X.; Hu, S.; Zhou, F.; Dong, J.; Zhang, S.; Shang, Y.; Jiang, X.; Guo, H.; et al. Chaperone-Mediated Autophagy Regulates Proliferation by Targeting RND3 in Gastric Cancer. Autophagy 2016, 12, 515–528. [Google Scholar] [CrossRef]
- Rios, J.; Sequeida, A.; Albornoz, A.; Budini, M. Chaperone Mediated Autophagy Substrates and Components in Cancer. Front. Oncol. 2021, 10, 614677. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.-G.; Najafov, A.; Geng, J.; Galan-Acosta, L.; Han, X.; Guo, Y.; Shan, B.; Zhang, Y.; Norberg, E.; Zhang, T.; et al. Degradation of HK2 by Chaperone-Mediated Autophagy Promotes Metabolic Catastrophe and Cell Death. J. Cell Biol. 2015, 210, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Ren, C.; Qiao, P.; Han, X.; Wang, L.; Lv, S.; Sun, Y.; Liu, Z.; Du, Y.; Yu, Z. PIM2-Mediated Phosphorylation of Hexokinase 2 Is Critical for Tumor Growth and Paclitaxel Resistance in Breast Cancer. Oncogene 2018, 37, 5997–6009. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Hu, W.; Lim, B.; Dice, J.F. IκB Is a Substrate for a Selective Pathway of Lysosomal Proteolysis. Mol. Biol. Cell 1998, 9, 1995–2010. [Google Scholar] [CrossRef]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, Death, and Autophagy in Cancer: NF-ΚB Turns up Everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef]
- Jiang, L.; Huang, S.; Wang, J.; Zhang, Y.; Xiong, Y.; Zeng, S.X.; Lu, H. Inactivating P53 Is Essential for Nerve Growth Factor Receptor to Promote Melanoma-Initiating Cell-Stemmed Tumorigenesis. Cell Death Dis. 2020, 11, 550. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Gilkes, D.M.; Hu, H.; Kshitiz, K.; Ahmed, I.; Semenza, G.L. Cyclin-Dependent Kinases Regulate Lysosomal Degradation of Hypoxia-Inducible Factor 1α to Promote Cell-Cycle Progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3325–E3334. [Google Scholar] [CrossRef]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated Degradation of Chk1 by Chaperone-Mediated Autophagy in Response to DNA Damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef]
- Galan-Acosta, L.; Xia, H.; Yuan, J.; Vakifahmetoglu-Norberg, H. Activation of Chaperone-Mediated Autophagy as a Potential Anticancer Therapy. Autophagy 2015, 11, 2370–2371. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-Mediated Autophagy Degrades Mutant P53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Method | Pros | Cons |
|---|---|---|
| Immunoblot of LAMP-2A | - Technically simple | - Overall estimate of CMA changes |
| Quantification of CMA active lysosomes | - Technically simple | - Overall estimate of CMA changes - Methanol fixation |
| Monitoring the degradation of radiolabelled long-lived protein | - Measures CMA flux | - Involves use of radioactivity - Requires inhibition of other autophagic pathways - Measures CMA and microautophagy |
| Measurement of the uptake of CMA substrate by isolated lysosomes | - Reconstitution of CMA in vitro - Measures functional CMA definitively | - Requires lysosome isolation - Requires large volume of cells or tissues |
| Photoswitchable/photoactivable CMA reporter | - Measures CMA flux | - Overall estimate of CMA changes - Must be complemented by other methods - Difficult to distinguish surface bound from translocated substrate |
| GAPDH-Halo tag fluorescence-based method | - Measures CMA flux | - Overall estimate of CMA changes - Must be complemented by other methods - Measures CMA and microautophagy |
| KFERQ Dendra reporter | - Measures CMA flux in vitro and in vivo | - Must be complemented by other methods |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hubert, V.; Weiss, S.; Rees, A.J.; Kain, R. Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer. Cells 2022, 11, 2562. https://doi.org/10.3390/cells11162562
Hubert V, Weiss S, Rees AJ, Kain R. Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer. Cells. 2022; 11(16):2562. https://doi.org/10.3390/cells11162562
Chicago/Turabian StyleHubert, Virginie, Sebastian Weiss, Andrew Jackson Rees, and Renate Kain. 2022. "Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer" Cells 11, no. 16: 2562. https://doi.org/10.3390/cells11162562
APA StyleHubert, V., Weiss, S., Rees, A. J., & Kain, R. (2022). Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer. Cells, 11(16), 2562. https://doi.org/10.3390/cells11162562