
Vitamin B12 Attenuates Changes in Phospholipid Levels Related to Oxidative Stress in SH-SY5Y Cells

 , , , , and
, , , , and 
Abstract
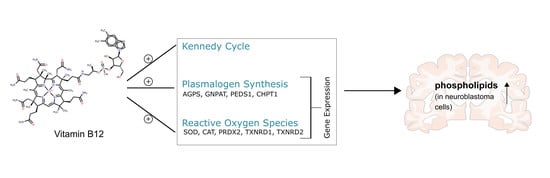
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Standards
2.2. Cell Culture and Vitamin B12 or Hydrogen Peroxide Treatment
2.3. Sample Preparation
2.4. Lipid Extraction
2.5. Targeted Shotgun Mass Spectrometry
2.6. Gene Expression Analysis
2.7. Analysis of β- and γ-Secretase Activity
2.8. Data and Statistical Analysis
3. Results
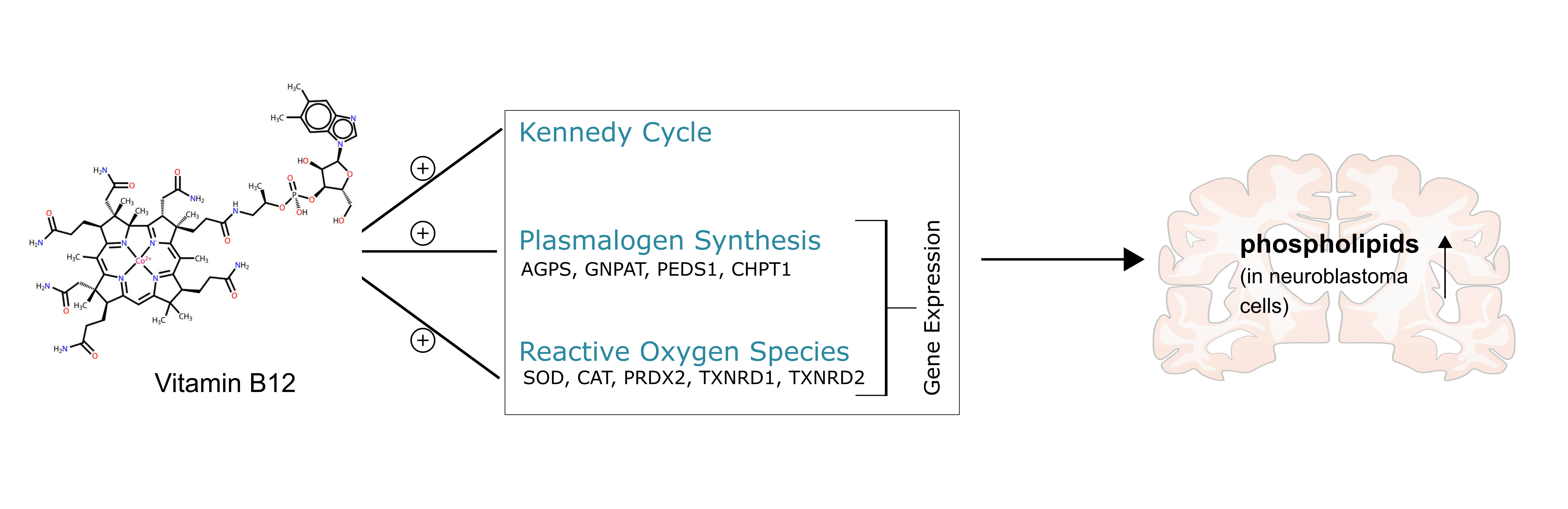
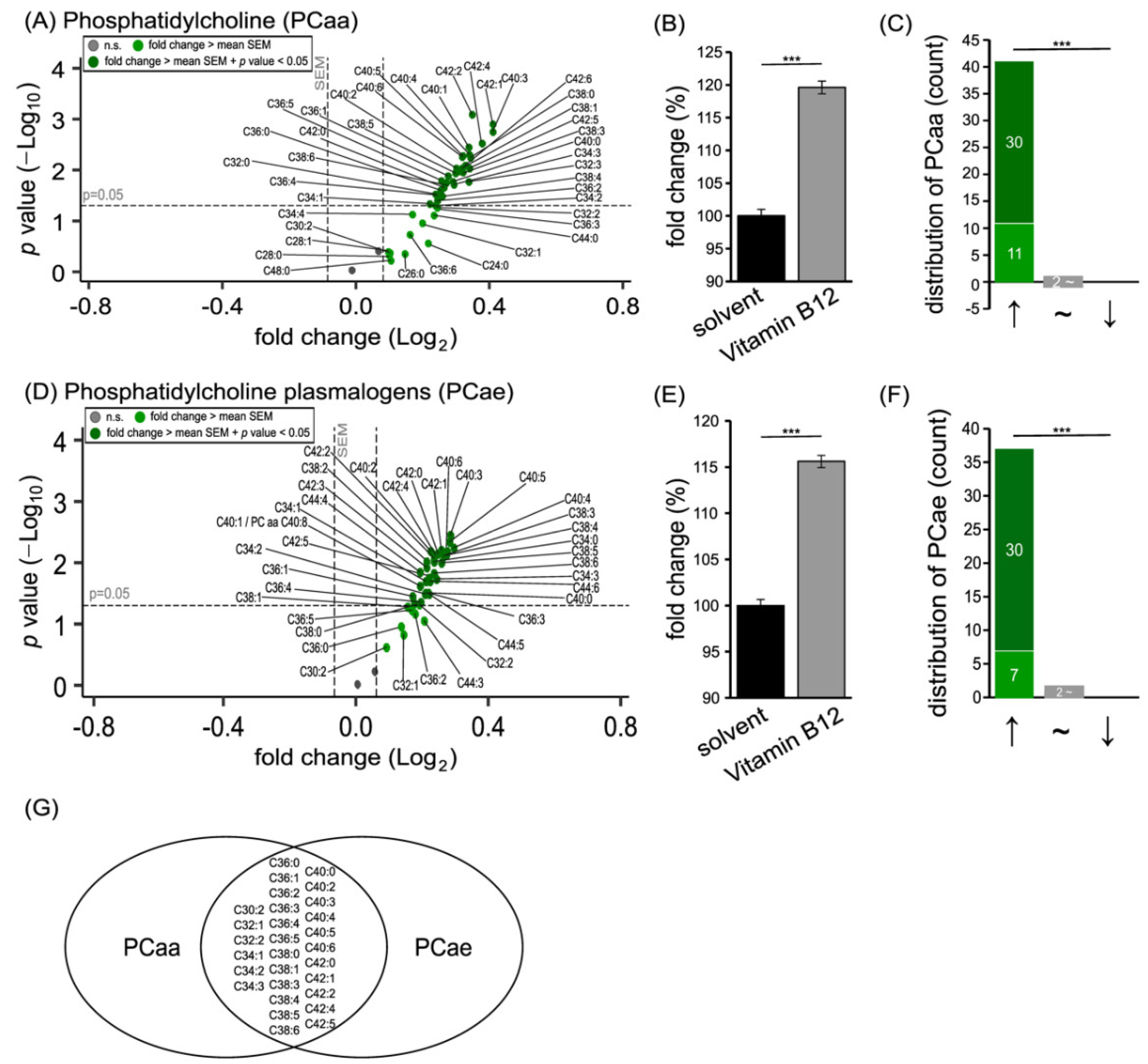
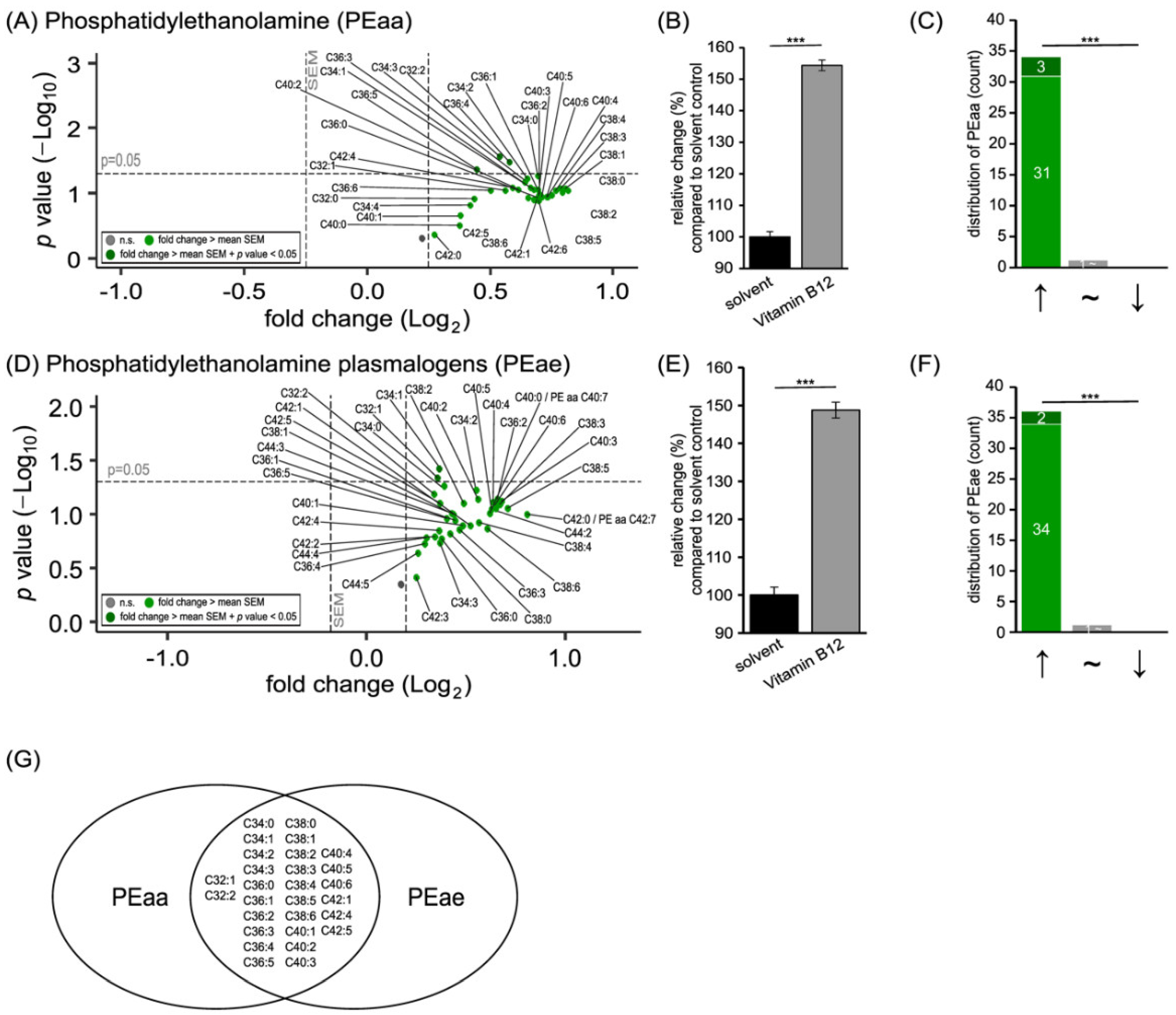
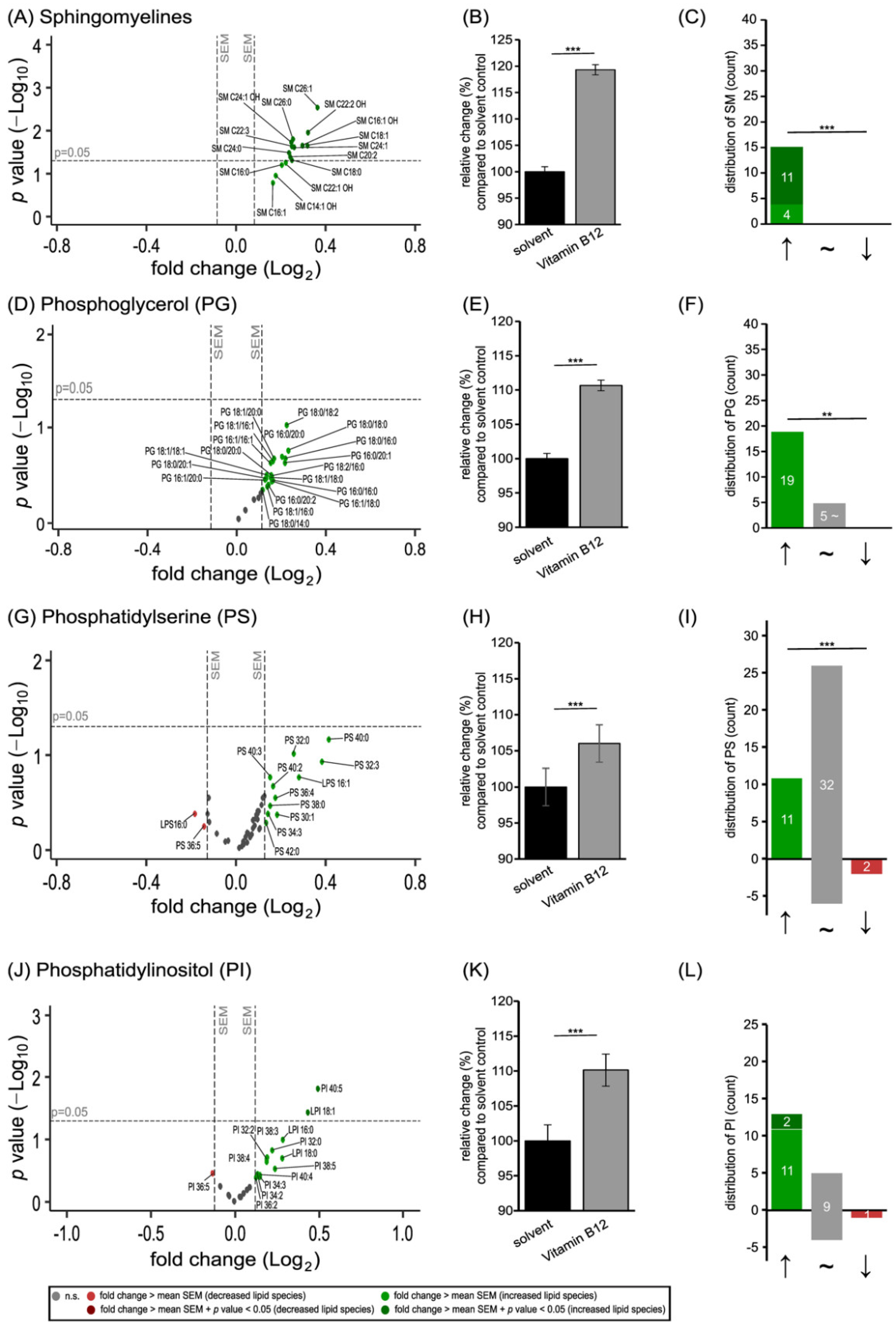
3.1. Phospholipid Species in SH-SY5Y Cells Treated with Vitamin B12
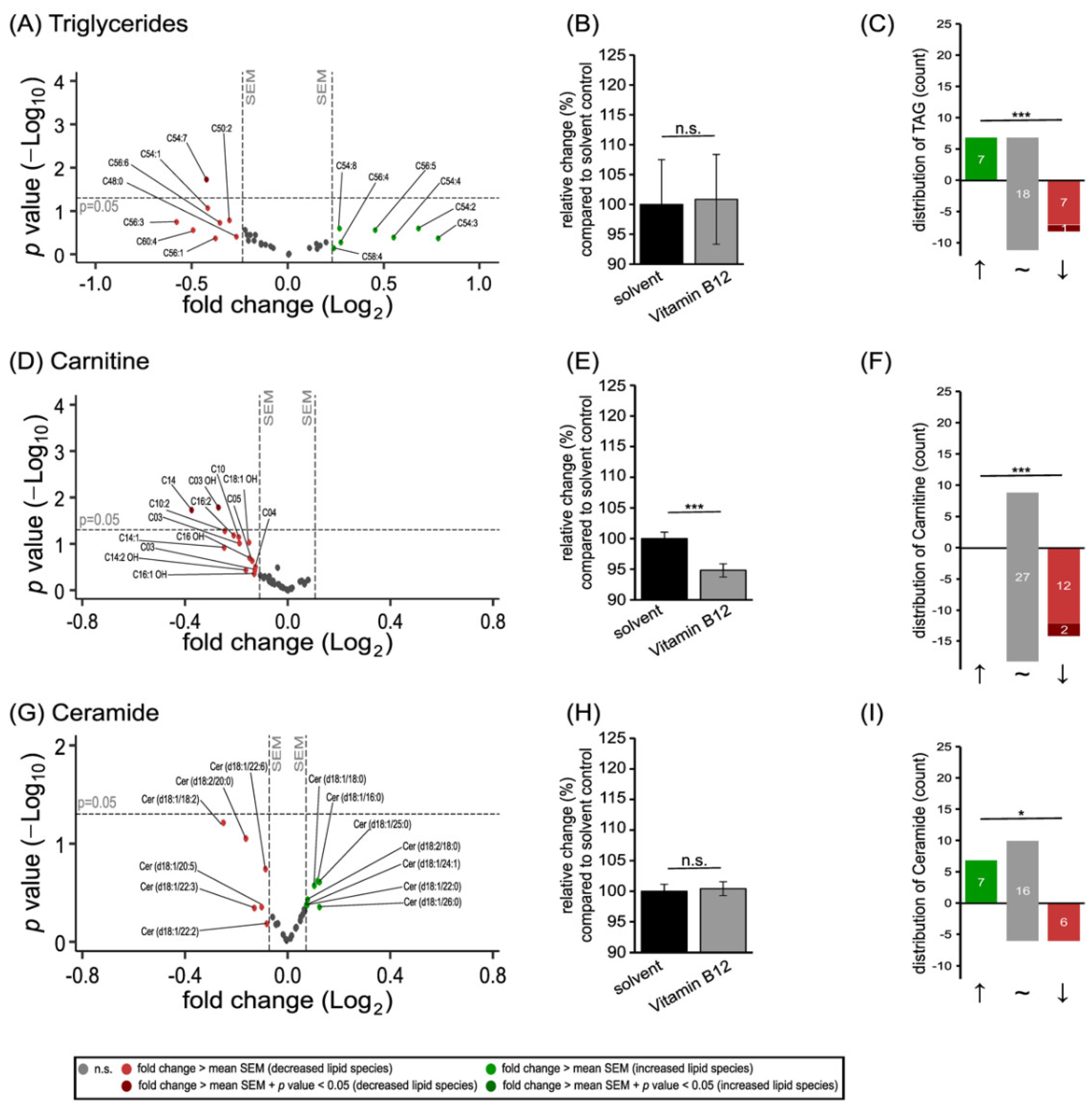
3.2. Further Lipid Species in SH-SY5Y Cells Treated with Vitamin B12
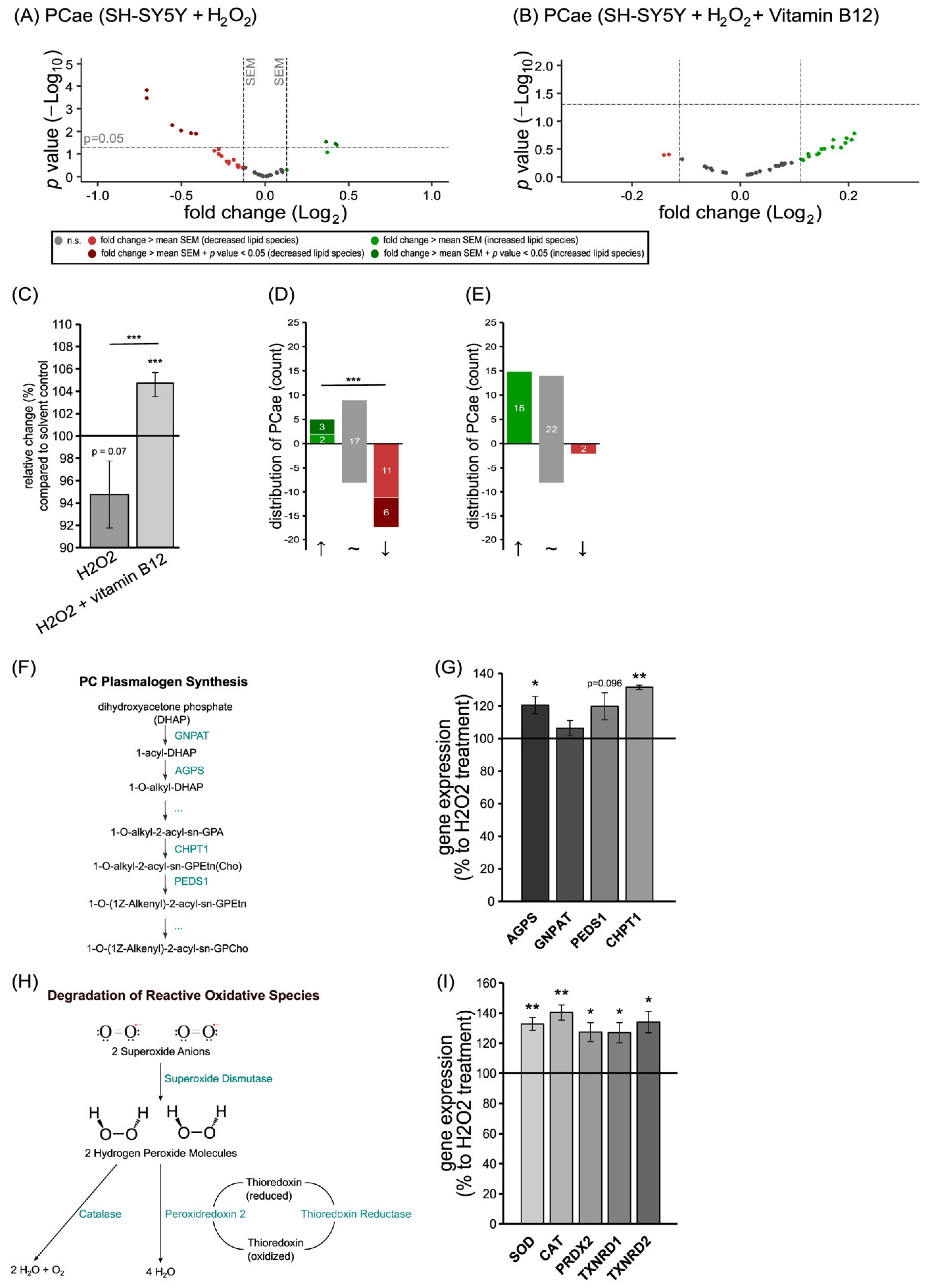
3.3. The Anti-Oxidative Properties of Vitamin B12 in the Presence of Hydrogen Peroxide in Relation to Phosphatidylcholine Plasmalogen Species
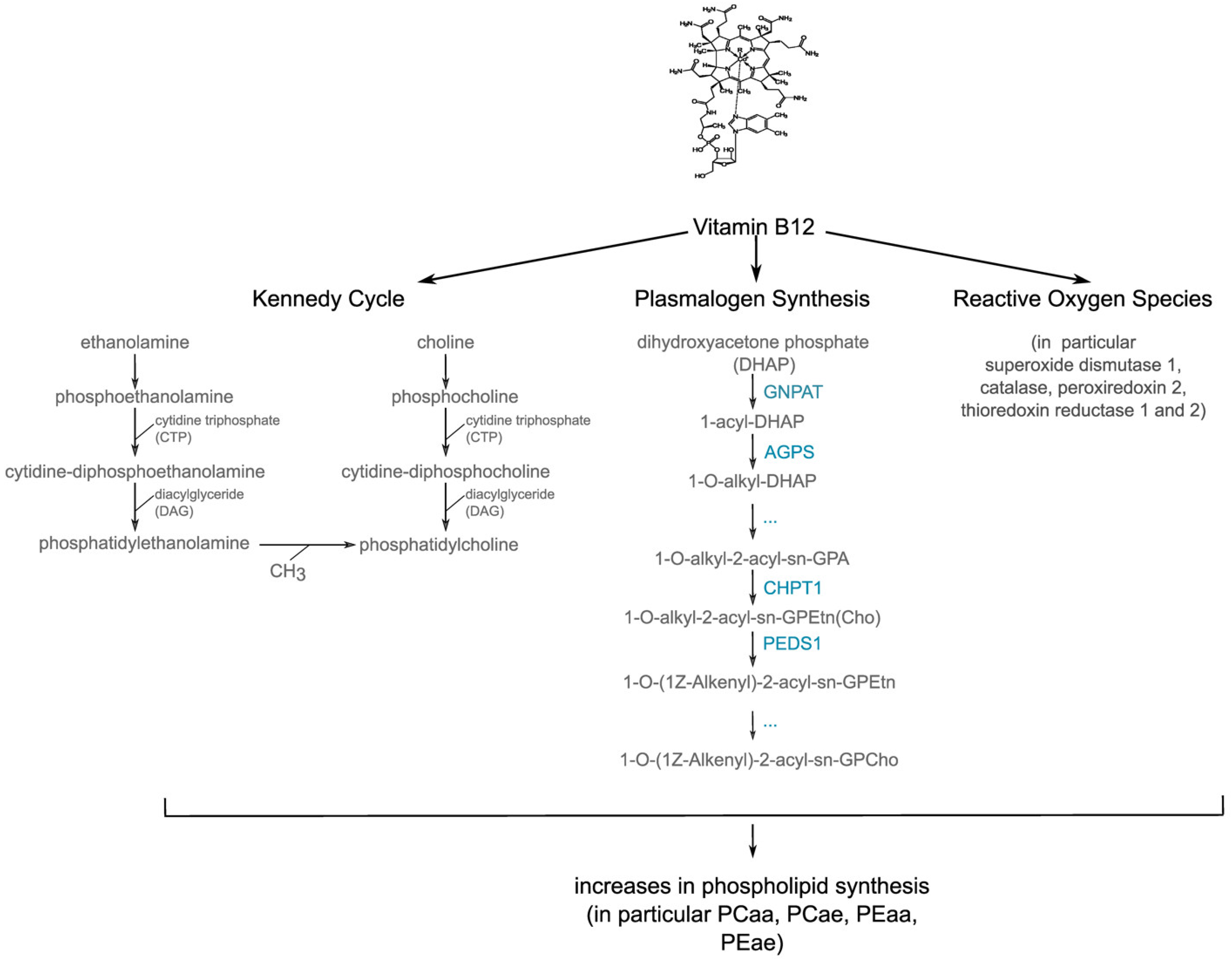
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gauthier, S.; Rosa-Neto, P.; Morais, J.; Webster, C. World Alzheimer Report 2021: Journey through the Diagnosis of Dementzia; Alzheimer’s Disease International: London, UK, 2021. [Google Scholar]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, S.; Bobo-Jimenez, V.; Requejo-Aguilar, R.; Gonzalez-Fernandez, S.; Resch, M.; Carabias-Carrasco, M.; Ros, J.; Almeida, A.; Bolanos, J.P. Hippocampal neurons require a large pool of glutathione to sustain dendrite integrity and cognitive function. Redox Biol. 2018, 19, 52–61. [Google Scholar] [CrossRef]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Zaloga, G.P. Narrative review of n-3 polyunsaturated fatty acid supplementation upon immune functions, resolution molecules and lipid peroxidation. Nutrients 2021, 13, 662. [Google Scholar] [CrossRef]
- Grimm, M.O.; Haupenthal, V.J.; Mett, J.; Stahlmann, C.P.; Blumel, T.; Mylonas, N.T.; Endres, K.; Grimm, H.S.; Hartmann, T. Oxidized docosahexaenoic acid species and lipid peroxidation products increase amyloidogenic amyloid precursor protein processing. Neurodegener. Dis. 2016, 16, 44–54. [Google Scholar] [CrossRef]
- Stadelmann-Ingrand, S.; Favreliere, S.; Fauconneau, B.; Mauco, G.; Tallineau, C. Plasmalogen degradation by oxidative stress: Production and disappearance of specific fatty aldehydes and fatty alpha-hydroxyaldehydes. Free Radic. Biol. Med. 2001, 31, 1263–1271. [Google Scholar] [CrossRef]
- Senanayake, V.; Goodenowe, D.B. Plasmalogen deficiency and neuropathology in Alzheimer’s disease: Causation or coincidence? Alzheimer’s Dement. 2019, 5, 524–532. [Google Scholar] [CrossRef]
- Moreira, E.S.; Brasch, N.E.; Yun, J. Vitamin b12 protects against superoxide-induced cell injury in human aortic endothelial cells. Free Radic. Biol. Med. 2011, 51, 876–883. [Google Scholar] [CrossRef]
- Chan, W.; Almasieh, M.; Catrinescu, M.M.; Levin, L.A. Cobalamin-associated superoxide scavenging in neuronal cells is a potential mechanism for vitamin b12-deprivation optic neuropathy. Am. J. Pathol. 2018, 188, 160–172. [Google Scholar] [CrossRef]
- Politis, A.; Olgiati, P.; Malitas, P.; Albani, D.; Signorini, A.; Polito, L.; De Mauro, S.; Zisaki, A.; Piperi, C.; Stamouli, E.; et al. Vitamin b12 levels in Alzheimer’s disease: Association with clinical features and cytokine production. J. Alzheimer’s Dis. 2010, 19, 481–488. [Google Scholar] [CrossRef]
- Birch, C.S.; Brasch, N.E.; McCaddon, A.; Williams, J.H. A novel role for vitamin b(12): Cobalamins are intracellular antioxidants in vitro. Free Radic. Biol. Med. 2009, 47, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N. Mechanisms of increased vascular oxidant stress in hyperhomocys-teinemia and its impact on endothelial function. Curr. Drug Metab. 2005, 6, 27–36. [Google Scholar] [CrossRef]
- Allen, L.H. Vitamin b-12. Adv. Nutr. 2012, 3, 54–55. [Google Scholar] [CrossRef]
- Vincenti, A.; Bertuzzo, L.; Limitone, A.; D’Antona, G.; Cena, H. Perspective: Practical approach to preventing subclinical b12 deficiency in elderly population. Nutrients 2021, 13, 1913. [Google Scholar] [CrossRef] [PubMed]
- Feldman, M.; Cryer, B.; McArthur, K.E.; Huet, B.A.; Lee, E. Effects of aging and gastritis on gastric acid and pepsin secretion in humans: A prospective study. Gastroenterology 1996, 110, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Lopes da Silva, S.; Vellas, B.; Elemans, S.; Luchsinger, J.; Kamphuis, P.; Yaffe, K.; Sijben, J.; Groenendijk, M.; Stijnen, T. Plasma nutrient status of patients with Alzheimer’s disease: Systematic review and meta-analysis. Alzheimer’s Dement. 2014, 10, 485–502. [Google Scholar] [CrossRef]
- Shen, L.; Ji, H.F. Associations between homocysteine, folic acid, vitamin b12 and Alzheimer’s disease: Insights from meta-analyses. J. Alzheimer’s Dis. 2015, 46, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, C.E.; Lutjohann, D.; von Bergmann, K.; Verhey, F.; Vreeling, F.; Wauters, A.; Bosmans, E.; Bosma, H.; van Boxtel, M.P.; Maes, M.; et al. Combination of serum markers related to several mechanisms in Alzheimer’s disease. Neurobiol. Aging 2003, 24, 893–902. [Google Scholar] [CrossRef]
- Chen, H.; Liu, S.; Ge, B.; Zhou, D.; Li, M.; Li, W.; Ma, F.; Liu, Z.; Ji, Y.; Huang, G. Effects of folic acid and vitamin b12 supplementation on cognitive impairment and inflammation in patients with Alzheimer’s disease: A randomized, single-blinded, placebo-controlled trial. J. Prev. Alzheimer’s Dis. 2021, 8, 249–256. [Google Scholar] [CrossRef]
- Remington, R.; Bechtel, C.; Larsen, D.; Samar, A.; Doshanjh, L.; Fishman, P.; Luo, Y.; Smyers, K.; Page, R.; Morrell, C.; et al. A phase ii randomized clinical trial of a nutritional formulation for cognition and mood in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 395–405. [Google Scholar] [CrossRef]
- Kurokin, I.; Lauer, A.A.; Janitschke, D.; Winkler, J.; Theiss, E.L.; Griebsch, L.V.; Pilz, S.M.; Matschke, V.; van der Laan, M.; Grimm, H.S.; et al. Targeted lipidomics of mitochondria in a cellular Alzheimer’s disease model. Biomedicines 2021, 9, 1062. [Google Scholar] [CrossRef]
- Zhong, L.; Zhou, J.; Chen, X.; Lou, Y.; Liu, D.; Zou, X.; Yang, B.; Yin, Y.; Pan, Y. Quantitative proteomics study of the neuroprotective effects of b12 on hydrogen peroxide-induced apoptosis in sh-sy5y cells. Sci. Rep. 2016, 6, 22635. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Lauer, A.A.; Griebsch, L.V.; Pilz, S.M.; Janitschke, D.; Theiss, E.L.; Reichrath, J.; Herr, C.; Beisswenger, C.; Bals, R.; Valencak, T.G.; et al. Impact of vitamin d3 deficiency on phosphatidylcholine-/ethanolamine, plasmalogen-, lyso-phosphatidylcholine-/ethanolamine, carnitine- and triacyl glyceride-homeostasis in neuroblastoma cells and murine brain. Biomolecules 2021, 11, 1699. [Google Scholar] [CrossRef]
- Grimm, M.O.; Grosgen, S.; Riemenschneider, M.; Tanila, H.; Grimm, H.S.; Hartmann, T. From brain to food: Analysis of phosphatidylcholins, lyso-phosphatidylcholins and phosphatidylcholin-plasmalogens derivates in Alzheimer’s disease human post mortem brains and mice model via mass spectrometry. J. Chromatogr. A 2011, 1218, 7713–7722. [Google Scholar] [CrossRef]
- Lauer, A.A.; Janitschke, D.; Dos Santos Guilherme, M.; Nguyen, V.T.T.; Bachmann, C.M.; Qiao, S.; Schrul, B.; Boehm, U.; Grimm, H.S.; Hartmann, T.; et al. Shotgun lipidomics of liver and brain tissue of Alzheimer’s disease model mice treated with acitretin. Sci. Rep. 2021, 11, 15301. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-pcr data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Thiel, A.; Lauer, A.A.; Winkler, J.; Lehmann, J.; Regner, L.; Nelke, C.; Janitschke, D.; Benoist, C.; Streidenberger, O.; et al. Vitamin d and its analogues decrease amyloid-beta (abeta) formation and increase abeta-degradation. Int. J. Mol. Sci. 2017, 18, 2764. [Google Scholar] [CrossRef]
- Jakubec, M.; Barias, E.; Kryuchkov, F.; Hjornevik, L.V.; Halskau, O. Fast and quantitative phospholipidomic analysis of sh-sy5y neuroblastoma cell cultures using liquid chromatography-tandem mass spectrometry and (31)p nuclear magnetic resonance. ACS Omega 2019, 4, 21596–21603. [Google Scholar] [CrossRef]
- Morgado, I.; Garvey, M. Lipids in amyloid-beta processing, aggregation, and toxicity. Adv. Exp. Med. Biol 2015, 855, 67–94. [Google Scholar]
- Chew, H.; Solomon, V.A.; Fonteh, A.N. Involvement of lipids in Alzheimer’s disease pathology and potential therapies. Front. Physiol. 2020, 11, 598. [Google Scholar] [CrossRef]
- Mett, J.; Lauer, A.A.; Janitschke, D.; Griebsch, L.V.; Theiss, E.L.; Grimm, H.S.; Koivisto, H.; Tanila, H.; Hartmann, T.; Grimm, M.O.W. Medium-chain length fatty acids enhance abeta degradation by affecting insulin-degrading enzyme. Cells 2021, 10, 2941. [Google Scholar] [CrossRef]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Haupenthal, V.J.; Blumel, T.; Stotzel, H.; Grimm, H.S.; Hartmann, T. Eicosapentaenoic acid and docosahexaenoic acid increase the degradation of amyloid-beta by affecting insulin-degrading enzyme. Biochem. Cell Biol. 2016, 94, 534–542. [Google Scholar] [CrossRef]
- Grimm, M.O.; Stahlmann, C.P.; Mett, J.; Haupenthal, V.J.; Zimmer, V.C.; Lehmann, J.; Hundsdorfer, B.; Endres, K.; Grimm, H.S.; Hartmann, T. Vitamin e: Curse or benefit in Alzheimer’s disease? A systematic investigation of the impact of alpha-, gamma- and delta-tocopherol on ass generation and degradation in neuroblastoma cells. J. Nutr. Health Aging 2015, 19, 646–656. [Google Scholar] [CrossRef]
- Grimm, M.O.; Regner, L.; Mett, J.; Stahlmann, C.P.; Schorr, P.; Nelke, C.; Streidenberger, O.; Stoetzel, H.; Winkler, J.; Zaidan, S.R.; et al. Tocotrienol affects oxidative stress, cholesterol homeostasis and the amyloidogenic pathway in neuroblastoma cells: Consequences for Alzheimer’s disease. Int. J. Mol. Sci. 2016, 17, 1809. [Google Scholar] [CrossRef]
- Grimm, M.O.; Haupenthal, V.J.; Rothhaar, T.L.; Zimmer, V.C.; Grosgen, S.; Hundsdorfer, B.; Lehmann, J.; Grimm, H.S.; Hartmann, T. Effect of different phospholipids on alpha-secretase activity in the non-amyloidogenic pathway of Alzheimer’s disease. Int. J. Mol. Sci. 2013, 14, 5879–5898. [Google Scholar] [CrossRef]
- Rothhaar, T.L.; Grosgen, S.; Haupenthal, V.J.; Burg, V.K.; Hundsdorfer, B.; Mett, J.; Riemenschneider, M.; Grimm, H.S.; Hartmann, T.; Grimm, M.O. Plasmalogens inhibit app processing by directly affecting gamma-secretase activity in Alzheimer’s disease. Sci. World J. 2012, 2012, 141240. [Google Scholar] [CrossRef]
- Grimm, M.O.; Kuchenbecker, J.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef]
- Simons, M.; Keller, P.; De Strooper, B.; Beyreuther, K.; Dotti, C.G.; Simons, K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar] [CrossRef]
- Grimm, M.O.; Grimm, H.S.; Tomic, I.; Beyreuther, K.; Hartmann, T.; Bergmann, C. Independent inhibition of Alzheimer disease beta- and gamma-secretase cleavage by lowered cholesterol levels. J. Biol. Chem. 2008, 283, 11302–11311. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L.; Ellis, B.C.; Saunders, A.J.; Kovacs, D.M. Ceramide stabilizes beta-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid beta-peptide biogenesis. J. Biol. Chem. 2003, 278, 19777–19783. [Google Scholar] [CrossRef]
- Jazvinscak Jembrek, M.; Hof, P.R.; Simic, G. Ceramides in Alzheimer’s disease: Key mediators of neuronal apoptosis induced by oxidative stress and abeta accumulation. Oxid. Med. Cell Longev. 2015, 2015, 346783. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; He, Q.; Kong, J.N.; Bieberich, E. The 5xfad mouse model of Alzheimer’s disease exhibits an age-dependent increase in anti-ceramide igg and exogenous administration of ceramide further increases anti-ceramide titers and amyloid plaque burden. J. Alzheimer’s Dis. 2015, 46, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Mett, J.; Grimm, H.S.; Hartmann, T. App function and lipids: A bidirectional link. Front. Mol. Neurosci. 2017, 10, 63. [Google Scholar] [CrossRef]
- Grimm, M.O.; Zimmer, V.C.; Lehmann, J.; Grimm, H.S.; Hartmann, T. The impact of cholesterol, dha, and sphingolipids on Alzheimer’s disease. Biomed. Res. Int. 2013, 2013, 814390. [Google Scholar] [CrossRef]
- Guan, Z.; Wang, Y.; Cairns, N.J.; Lantos, P.L.; Dallner, G.; Sindelar, P.J. Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with Alzheimer disease. J. Neuropathol. Exp. Neurol. 1999, 58, 740–747. [Google Scholar] [CrossRef]
- Svennerholm, L.; Gottfries, C.G. Membrane lipids, selectively diminished in Alzheimer brains, suggest synapse loss as a primary event in early-onset form (type i) and demyelination in late-onset form (type ii). J. Neurochem. 1994, 62, 1039–1047. [Google Scholar] [CrossRef]
- Agranoff, B.W.; Benjamins, J.A.; Hajra, A.K. Properties of brain lipids. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Siegel, G.J., Agranoff, B.W., Albers, R.W., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1999. [Google Scholar]
- Nitsch, R.M.; Blusztajn, J.K.; Pittas, A.G.; Slack, B.E.; Growdon, J.H.; Wurtman, R.J. Evidence for a membrane defect in Alzheimer disease brain. Proc. Natl. Acad. Sci. USA 1992, 89, 1671–1675. [Google Scholar] [CrossRef]
- Fonteh, A.N.; Chiang, J.; Cipolla, M.; Hale, J.; Diallo, F.; Chirino, A.; Arakaki, X.; Harrington, M.G. Alterations in cerebrospinal fluid glycerophospholipids and phospholipase a2 activity in Alzheimer’s disease. J. Lipid Res. 2013, 54, 2884–2897. [Google Scholar] [CrossRef]
- Prasad, M.R.; Lovell, M.A.; Yatin, M.; Dhillon, H.; Markesbery, W.R. Regional membrane phospholipid alterations in Alzheimer’s disease. Neurochem. Res. 1998, 23, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Wells, K.; Farooqui, A.A.; Liss, L.; Horrocks, L.A. Neural membrane phospholipids in Alzheimer disease. Neurochem. Res. 1995, 20, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Carrera, I.; Martinez-Iglesias, O.; Cacabelos, N.; Naidoo, V. What is the gold standard model for Alzheimer’s disease drug discovery and development? Expert Opin. Drug Discov. 2021, 16, 1415–1440. [Google Scholar] [CrossRef]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Grosgen, S.; Haupenthal, V.J.; Blumel, T.; Hundsdorfer, B.; Zimmer, V.C.; Mylonas, N.T.; Tanila, H.; et al. App intracellular domain derived from amyloidogenic beta- and gamma-secretase cleavage regulates neprilysin expression. Front. Aging Neurosci. 2015, 7, 77. [Google Scholar] [CrossRef]
- Cai, X.D.; Golde, T.E.; Younkin, S.G. Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science 1993, 259, 514–516. [Google Scholar] [CrossRef]
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef]
- Belyaev, N.D.; Kellett, K.A.; Beckett, C.; Makova, N.Z.; Revett, T.J.; Nalivaeva, N.N.; Hooper, N.M.; Turner, A.J. The transcriptionally active amyloid precursor protein (app) intracellular domain is preferentially produced from the 695 isoform of app in a {beta}-secretase-dependent pathway. J. Biol. Chem. 2010, 285, 41443–41454. [Google Scholar] [CrossRef]
- Song, X.J.; Zhou, H.Y.; Sun, Y.X.; Huang, H.C. Inhibitory effects of curcumin on h2o2-induced cell damage and app expression and processing in sh-sy5y cells transfected with app gene with swedish mutation. Mol. Biol. Rep. 2020, 47, 2047–2059. [Google Scholar] [CrossRef]
- Pahrudin Arrozi, A.; Shukri, S.N.S.; Wan Ngah, W.Z.; Mohd Yusof, Y.A.; Ahmad Damanhuri, M.H.; Makpol, S. Evaluation of the expression of amyloid precursor protein and the ratio of secreted amyloid beta 42 to amyloid beta 40 in sh-sy5y cells stably transfected with wild-type, single-mutant and double-mutant forms of the app gene for the study of Alzheimer’s disease pathology. Appl. Biochem. Biotechnol. 2017, 183, 853–866. [Google Scholar]
- Di, X.; Yan, J.; Zhao, Y.; Zhang, J.; Shi, Z.; Chang, Y.; Zhao, B. L-theanine protects the app (swedish mutation) transgenic sh-sy5y cell against glutamate-induced excitotoxicity via inhibition of the nmda receptor pathway. Neuroscience 2010, 168, 778–786. [Google Scholar] [CrossRef]
- Wang, X.; Yu, S.; Gao, S.J.; Hu, J.P.; Wang, Y.; Liu, H.X. Insulin inhibits abeta production through modulation of app processing in a cellular model of Alzheimer’s disease. Neuro. Endocrinol. Lett. 2014, 35, 224–229. [Google Scholar] [PubMed]
- Kong, J.J.; Zhang, D.D.; Li, P.; Wei, C.Y.; Yu, H.J.; Zhang, H.; Zhang, W.; Wang, Y.F.; Cao, Y.P. Nicorandil inhibits oxidative stress and amyloid-beta precursor protein processing in sh-sy5y cells overexpressing appsw. Int. J. Clin. Exp. Med. 2015, 8, 1966–1975. [Google Scholar] [PubMed]
- Zheng, L.; Terman, A.; Hallbeck, M.; Dehvari, N.; Cowburn, R.F.; Benedikz, E.; Kagedal, K.; Cedazo-Minguez, A.; Marcusson, J. Macroautophagy-generated increase of lysosomal amyloid beta-protein mediates oxidant-induced apoptosis of cultured neuroblastoma cells. Autophagy 2011, 7, 1528–1545. [Google Scholar] [CrossRef]
- Chan, K.H.; Lam, K.S.; Cheng, O.Y.; Kwan, J.S.; Ho, P.W.; Cheng, K.K.; Chung, S.K.; Ho, J.W.; Guo, V.Y.; Xu, A. Adiponectin is protective against oxidative stress induced cytotoxicity in amyloid-beta neurotoxicity. PLoS ONE 2012, 7, e52354. [Google Scholar] [CrossRef]
- Lopez Sanchez, M.I.G.; Waugh, H.S.; Tsatsanis, A.; Wong, B.X.; Crowston, J.G.; Duce, J.A.; Trounce, I.A. Amyloid precursor protein drives down-regulation of mitochondrial oxidative phosphorylation independent of amyloid beta. Sci. Rep. 2017, 7, 9835. [Google Scholar] [CrossRef] [PubMed]
- Kling, M.A.; Goodenowe, D.B.; Senanayake, V.; MahmoudianDehkordi, S.; Arnold, M.; Massaro, T.J.; Baillie, R.; Han, X.; Leung, Y.Y.; Saykin, A.J.; et al. Circulating ethanolamine plasmalogen indices in Alzheimer’s disease: Relation to diagnosis, cognition, and csf tau. Alzheimer’s Dement. 2020, 16, 1234–1247. [Google Scholar] [CrossRef]
- Otoki, Y.; Kato, S.; Nakagawa, K.; Harvey, D.J.; Jin, L.W.; Dugger, B.N.; Taha, A.Y. Lipidomic analysis of postmortem prefrontal cortex phospholipids reveals changes in choline plasmalogen containing docosahexaenoic acid and stearic acid between cases with and without Alzheimer’s disease. Neuromol. Med. 2021, 23, 161–175. [Google Scholar] [CrossRef]
- Wood, P.L.; Barnette, B.L.; Kaye, J.A.; Quinn, J.F.; Woltjer, R.L. Non-targeted lipidomics of csf and frontal cortex grey and white matter in control, mild cognitive impairment, and Alzheimer’s disease subjects. Acta Neuropsychiatr. 2015, 27, 270–278. [Google Scholar] [CrossRef]
- Wood, P.L.; Locke, V.A.; Herling, P.; Passaro, A.; Vigna, G.B.; Volpato, S.; Valacchi, G.; Cervellati, C.; Zuliani, G. Targeted lipidomics distinguishes patient subgroups in mild cognitive impairment (mci) and late onset Alzheimer’s disease (load). BBA Clin. 2016, 5, 25–28. [Google Scholar] [CrossRef]
- Han, X.; Holtzman, D.M.; McKeel, D.W., Jr. Plasmalogen deficiency in early Alzheimer’s disease subjects and in animal models: Molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 2001, 77, 1168–1180. [Google Scholar] [CrossRef]
- Igarashi, M.; Ma, K.; Gao, F.; Kim, H.W.; Rapoport, S.I.; Rao, J.S. Disturbed choline plasmalogen and phospholipid fatty acid concentrations in Alzheimer’s disease prefrontal cortex. J. Alzheimer’s Dis. 2011, 24, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Uruno, A.; Matsumaru, D.; Ryoke, R.; Saito, R.; Kadoguchi, S.; Saigusa, D.; Saito, T.; Saido, T.C.; Kawashima, R.; Yamamoto, M. Nrf2 suppresses oxidative stress and inflammation in app knock-in Alzheimer’s disease model mice. Mol. Cell. Biol. 2020, 40, e00467-19. [Google Scholar] [CrossRef]
- Azad, A.K.; Sheikh, A.M.; Haque, M.A.; Osago, H.; Sakai, H.; Shibly, A.Z.; Yano, S.; Michikawa, M.; Hossain, S.; Tabassum, S.; et al. Time-dependent analysis of plasmalogens in the hippocampus of an Alzheimer’s disease mouse model: A role of ethanolamine plasmalogen. Brain Sci. 2021, 11, 1603. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Abe, Y.; Ali, F.; Youssef, M.; Honsho, M.; Fujiki, Y.; Katafuchi, T. Reduction of ether-type glycerophospholipids, plasmalogens, by nf-kappab signal leading to microglial activation. J. Neurosci. 2017, 37, 4074–4092. [Google Scholar] [CrossRef] [PubMed]
- Brito, A.; Grapov, D.; Fahrmann, J.; Harvey, D.; Green, R.; Miller, J.W.; Fedosov, S.N.; Shahab-Ferdows, S.; Hampel, D.; Pedersen, T.L.; et al. The human serum metabolome of vitamin b-12 deficiency and repletion, and associations with neurological function in elderly adults. J. Nutr. 2017, 147, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Gilfix, B.M. Vitamin b12 and homocysteine. CMAJ 2005, 173, 1360. [Google Scholar] [CrossRef]
- Selhub, J. Homocysteine metabolism. Annu. Rev. Nutr. 1999, 19, 217–246. [Google Scholar] [CrossRef]
- Wurtman, R.J. A nutrient combination that can affect synapse formation. Nutrients 2014, 6, 1701–1710. [Google Scholar] [CrossRef]
- Soininen, H.; Solomon, A.; Visser, P.J.; Hendrix, S.B.; Blennow, K.; Kivipelto, M.; Hartmann, T.; LipiDiDiet clinical study, g. 24-month intervention with a specific multinutrient in people with prodromal Alzheimer’s disease (lipididiet): A randomised, double-blind, controlled trial. Lancet Neurol. 2017, 16, 965–975. [Google Scholar] [CrossRef]
- Pettegrew, J.W.; Panchalingam, K.; Hamilton, R.L.; McClure, R.J. Brain membrane phospholipid alterations in Alzheimer’s disease. Neurochem. Res. 2001, 26, 771–782. [Google Scholar] [CrossRef]
- Stokes, C.E.; Hawthorne, J.N. Reduced phosphoinositide concentrations in anterior temporal cortex of Alzheimer-diseased brains. J. Neurochem. 1987, 48, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Kosicek, M.; Zetterberg, H.; Andreasen, N.; Peter-Katalinic, J.; Hecimovic, S. Elevated cerebrospinal fluid sphingomyelin levels in prodromal Alzheimer’s disease. Neurosci. Lett. 2012, 516, 302–305. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Huang, Y.; Li, B.; Gong, C.X.; Schuchman, E.H. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef]
- Bandaru, V.V.; Troncoso, J.; Wheeler, D.; Pletnikova, O.; Wang, J.; Conant, K.; Haughey, N.J. Apoe4 disrupts sterol and sphingolipid metabolism in Alzheimer’s but not normal brain. Neurobiol. Aging 2009, 30, 591–599. [Google Scholar] [CrossRef]
- Mielke, M.M.; Haughey, N.J.; Bandaru, V.V.; Weinberg, D.D.; Darby, E.; Zaidi, N.; Pavlik, V.; Doody, R.S.; Lyketsos, C.G. Plasma sphingomyelins are associated with cognitive progression in Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 27, 259–269. [Google Scholar] [CrossRef]
- Haughey, N.J.; Bandaru, V.V.; Bae, M.; Mattson, M.P. Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. Biochim. Biophys. Acta 2010, 1801, 878–886. [Google Scholar] [CrossRef]
- Malaplate-Armand, C.; Florent-Bechard, S.; Youssef, I.; Koziel, V.; Sponne, I.; Kriem, B.; Leininger-Muller, B.; Olivier, J.L.; Oster, T.; Pillot, T. Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cpla2-dependent sphingomyelinase-ceramide pathway. Neurobiol. Dis. 2006, 23, 178–189. [Google Scholar] [CrossRef]
- Grimm, M.O.; Grimm, H.S.; Patzold, A.J.; Zinser, E.G.; Halonen, R.; Duering, M.; Tschape, J.A.; De Strooper, B.; Muller, U.; Shen, J.; et al. Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat. Cell Biol. 2005, 7, 1118–1123. [Google Scholar] [CrossRef]
- Han, X.; David, M.H.; McKeel, D.W., Jr.; Kelley, J.; Morris, J.C. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: Potential role in disease pathogenesis. J. Neurochem. 2002, 82, 809–818. [Google Scholar] [CrossRef]
- Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H.V.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J. Alzheimer’s Dis. 2012, 29, 537–547. [Google Scholar] [CrossRef]
- Satoi, H.; Tomimoto, H.; Ohtani, R.; Kitano, T.; Kondo, T.; Watanabe, M.; Oka, N.; Akiguchi, I.; Furuya, S.; Hirabayashi, Y.; et al. Astroglial expression of ceramide in Alzheimer’s disease brains: A role during neuronal apoptosis. Neuroscience 2005, 130, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Katsel, P.; Li, C.; Haroutunian, V. Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: A shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochem. Res. 2007, 32, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Bandaru, V.V.; Haughey, N.J.; Xia, J.; Fried, L.P.; Yasar, S.; Albert, M.; Varma, V.; Harris, G.; Schneider, E.B.; et al. Serum ceramides increase the risk of Alzheimer disease: The women’s health and aging study ii. Neurology 2012, 79, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Papuc, E.; Rejdak, K. The role of myelin damage in Alzheimer’s disease pathology. Arch. Med. Sci. 2020, 16, 345–351. [Google Scholar] [CrossRef]
- Adamo, A.M. Nutritional factors and aging in demyelinating diseases. Genes Nutr. 2014, 9, 360. [Google Scholar] [CrossRef]
- Calderon-Ospina, C.A.; Nava-Mesa, M.O. B vitamins in the nervous system: Current knowledge of the biochemical modes of action and synergies of thiamine, pyridoxine, and cobalamin. CNS Neurosci. Ther. 2020, 26, 5–13. [Google Scholar] [CrossRef]
- Herrmann, W.; Obeid, R. Causes and early diagnosis of vitamin b12 deficiency. Dtsch. Arztebl. Int. 2008, 105, 680–685. [Google Scholar] [CrossRef]
- Briani, C.; Dalla Torre, C.; Citton, V.; Manara, R.; Pompanin, S.; Binotto, G.; Adami, F. Cobalamin deficiency: Clinical picture and radiological findings. Nutrients 2013, 5, 4521–4539. [Google Scholar] [CrossRef]
- Su, X.Q.; Wang, J.; Sinclair, A.J. Plasmalogens and Alzheimer’s disease: A review. Lipids Health Dis. 2019, 18, 100. [Google Scholar] [CrossRef]
- Ginsberg, L.; Rafique, S.; Xuereb, J.H.; Rapoport, S.I.; Gershfeld, N.L. Disease and anatomic specificity of ethanolamine plasmalogen deficiency in Alzheimer’s disease brain. Brain Res. 1995, 698, 223–226. [Google Scholar] [CrossRef]
- Ellison, D.W.; Beal, M.F.; Martin, J.B. Phosphoethanolamine and ethanolamine are decreased in Alzheimer’s disease and Huntington’s disease. Brain Res. 1987, 417, 389–392. [Google Scholar] [CrossRef]
- Wood, P.L.; Mankidy, R.; Ritchie, S.; Heath, D.; Wood, J.A.; Flax, J.; Goodenowe, D.B. Circulating plasmalogen levels and Alzheimer disease assessment scale-cognitive scores in Alzheimer patients. J. Psychiatry Neurosci. 2010, 35, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Kiko, T.; Fujiwara, H.; Hashimoto, M.; Nakagawa, K.; Kinoshita, M.; Furukawa, K.; Arai, H.; Miyazawa, T. Alterations in the levels of amyloid-beta, phospholipid hydroperoxide, and plasmalogen in the blood of patients with Alzheimer’s disease: Possible interactions between amyloid-beta and these lipids. J. Alzheimer’s Dis. 2016, 50, 527–537. [Google Scholar] [CrossRef]
- Goodenowe, D.B.; Cook, L.L.; Liu, J.; Lu, Y.; Jayasinghe, D.A.; Ahiahonu, P.W.; Heath, D.; Yamazaki, Y.; Flax, J.; Krenitsky, K.F.; et al. Peripheral ethanolamine plasmalogen deficiency: A logical causative factor in Alzheimer’s disease and dementia. J. Lipid Res. 2007, 48, 2485–2498. [Google Scholar] [CrossRef]
- Braverman, N.E.; Moser, A.B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 2012, 1822, 1442–1452. [Google Scholar] [CrossRef]
- Reiss, D.; Beyer, K.; Engelmann, B. Delayed oxidative degradation of polyunsaturated diacyl phospholipids in the presence of plasmalogen phospholipids in vitro. Biochem. J. 1997, 323 Pt 3, 807–814. [Google Scholar] [CrossRef]
- Broniec, A.; Klosinski, R.; Pawlak, A.; Wrona-Krol, M.; Thompson, D.; Sarna, T. Interactions of plasmalogens and their diacyl analogs with singlet oxygen in selected model systems. Free Radic. Biol. Med. 2011, 50, 892–898. [Google Scholar] [CrossRef]
- Katafuchi, T.; Ifuku, M.; Mawatari, S.; Noda, M.; Miake, K.; Sugiyama, M.; Fujino, T. Effects of plasmalogens on systemic lipopolysaccharide-induced glial activation and beta-amyloid accumulation in adult mice. Ann. N. Y. Acad. Sci. 2012, 1262, 85–92. [Google Scholar] [CrossRef]
- Grimm, M.O.; Kuchenbecker, J.; Rothhaar, T.L.; Grosgen, S.; Hundsdorfer, B.; Burg, V.K.; Friess, P.; Muller, U.; Grimm, H.S.; Riemenschneider, M.; et al. Plasmalogen synthesis is regulated via alkyl-dihydroxyacetonephosphate-synthase by amyloid precursor protein processing and is affected in Alzheimer’s disease. J. Neurochem. 2011, 116, 916–925. [Google Scholar] [CrossRef]
- De Wilde, M.C.; Overk, C.R.; Sijben, J.W.; Masliah, E. Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimer’s Dement. 2016, 12, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.S. Homocysteine and Alzheimer’s disease. Lancet Neurol. 2003, 2, 425–428. [Google Scholar] [CrossRef]
- Lauer, A.A.; Grimm, H.S.; Apel, B.; Golobrodska, N.; Kruse, L.; Ratanski, E.; Schulten, N.; Schwarze, L.; Slawik, T.; Sperlich, S.; et al. Mechanistic link between vitamin b12 and Alzheimer’s disease. Biomolecules 2022, 12, 129. [Google Scholar] [CrossRef] [PubMed]
- Agholme, L.; Lindstrom, T.; Kagedal, K.; Marcusson, J.; Hallbeck, M. An in vitro model for neuroscience: Differentiation of sh-sy5y cells into cells with morphological and biochemical characteristics of mature neurons. J. Alzheimer’s Dis. 2010, 20, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Slanzi, A.; Iannoto, G.; Rossi, B.; Zenaro, E.; Constantin, G. In vitro models of neurodegenerative diseases. Front. Cell Dev. Biol. 2020, 8, 328. [Google Scholar] [CrossRef] [PubMed]
- Strother, L.; Miles, G.B.; Holiday, A.R.; Cheng, Y.; Doherty, G.H. Long-term culture of sh-sy5y neuroblastoma cells in the absence of neurotrophins: A novel model of neuronal ageing. J. Neurosci. Methods 2021, 362, 109301. [Google Scholar] [CrossRef] [PubMed]
- de Medeiros, L.M.; De Bastiani, M.A.; Rico, E.P.; Schonhofen, P.; Pfaffenseller, B.; Wollenhaupt-Aguiar, B.; Grun, L.; Barbe-Tuana, F.; Zimmer, E.R.; Castro, M.A.A.; et al. Cholinergic differentiation of human neuroblastoma sh-sy5y cell line and its potential use as an in vitro model for Alzheimer’s disease studies. Mol. Neurobiol. 2019, 56, 7355–7367. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Mawatari, S.; Ohara, S.; Taniwaki, Y.; Tsuboi, Y.; Maruyama, T.; Fujino, T. Improvement of blood plasmalogens and clinical symptoms in Parkinson’s disease by oral administration of ether phospholipids: A preliminary report. Parkinson’s Dis. 2020, 2020, 2671070. [Google Scholar] [CrossRef]
- Christine, C.W.; Auinger, P.; Joslin, A.; Yelpaala, Y.; Green, R.; Parkinson Study Group, D.I. Vitamin b12 and homocysteine levels predict different outcomes in early Parkinson’s disease. Mov. Disord. 2018, 33, 762–770. [Google Scholar] [CrossRef]
- Moore, E.; Mander, A.; Ames, D.; Carne, R.; Sanders, K.; Watters, D. Cognitive impairment and vitamin b12: A review. Int. Psychogeriatr. 2012, 24, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, J.; Liang, S.H.; Xu, Y.; Moore, A.; Ran, C. Imaging hydrogen peroxide in Alzheimer’s disease via cascade signal amplification. Sci. Rep. 2016, 6, 35613. [Google Scholar] [CrossRef] [PubMed]
- Milton, N.G. Role of hydrogen peroxide in the aetiology of Alzheimer’s disease: Implications for treatment. Drugs Aging 2004, 21, 81–100. [Google Scholar] [CrossRef]
- Kaminsky, Y.G.; Kosenko, E.A. Effects of amyloid-beta peptides on hydrogen peroxide-metabolizing enzymes in rat brain in vivo. Free Radic. Res. 2008, 42, 564–573. [Google Scholar] [CrossRef]
- Wang, M.; Xu, T. Methyl b12 protects pc12 cells against cytotoxicity induced by abeta25-35. J. Cell. Biochem. 2019, 120, 11921–11930. [Google Scholar] [CrossRef]
- Alam, P.; Siddiqi, M.K.; Chaturvedi, S.K.; Zaman, M.; Khan, R.H. Vitamin b12 offers neuronal cell protection by inhibiting abeta-42 amyloid fibrillation. Int. J. Biol. Macromol. 2017, 99, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Yan, Y.; He, X.Y.; Yang, H.; Liang, B.; Wang, J.; He, Y.; Ding, Y.; Yu, H. Effects of resveratrol on the mechanisms of antioxidants and estrogen in Alzheimer’s disease. Biomed. Res. Int. 2019, 2019, 8983752. [Google Scholar] [CrossRef]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef]
- Wojsiat, J.; Zoltowska, K.M.; Laskowska-Kaszub, K.; Wojda, U. Oxidant/antioxidant imbalance in Alzheimer’s disease: Therapeutic and diagnostic prospects. Oxid. Med. Cell. Longev. 2018, 2018, 6435861. [Google Scholar] [CrossRef]
- Marcus, D.L.; Thomas, C.; Rodriguez, C.; Simberkoff, K.; Tsai, J.S.; Strafaci, J.A.; Freedman, M.L. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp. Neurol. 1998, 150, 40–44. [Google Scholar] [CrossRef]
- Rinaldi, P.; Polidori, M.C.; Metastasio, A.; Mariani, E.; Mattioli, P.; Cherubini, A.; Catani, M.; Cecchetti, R.; Senin, U.; Mecocci, P. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol. Aging 2003, 24, 915–919. [Google Scholar] [CrossRef]
- Jeon, Y.M.; Kwon, Y.; Lee, S.; Kim, S.; Jo, M.; Lee, S.; Kim, S.R.; Kim, K.; Kim, H.J. Vitamin b12 reduces tdp-43 toxicity by alleviating oxidative stress and mitochondrial dysfunction. Antioxidants 2021, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, S.; Fuso, A.; D’Anselmi, F.; Cavallaro, R.A. Presenilin 1 gene silencing by s-adenosylmethionine: A treatment for Alzheimer disease? FEBS Lett. 2003, 541, 145–148. [Google Scholar] [CrossRef]
- Fuso, A.; Seminara, L.; Cavallaro, R.A.; D’Anselmi, F.; Scarpa, S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of ps1 and bace and beta-amyloid production. Mol. Cell. Neurosci. 2005, 28, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.E.; Wei, W.; Liu, Y.H.; Peng, J.H.; Tian, Q.; Liu, G.P.; Zhang, Y.; Wang, J.Z. Hyperhomocysteinemia increases beta-amyloid by enhancing expression of gamma-secretase and phosphorylation of amyloid precursor protein in rat brain. Am. J. Pathol. 2009, 174, 1481–1491. [Google Scholar] [CrossRef]
- Fuso, A.; Nicolia, V.; Cavallaro, R.A.; Ricceri, L.; D’Anselmi, F.; Coluccia, P.; Calamandrei, G.; Scarpa, S. B-vitamin deprivation induces hyperhomocysteinemia and brain s-adenosylhomocysteine, depletes brain s-adenosylmethionine, and enhances ps1 and bace expression and amyloid-beta deposition in mice. Mol. Cell. Neurosci. 2008, 37, 731–746. [Google Scholar] [CrossRef]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.F.; Frigon, N.; Hong, J.; et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease bace. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Grimm, M.O.; Tomic, I.; Hartmann, T. Potential external source of a beta in biological samples. Nat. Cell Biol. 2002, 4, E164–E165; author reply E165–E166. [Google Scholar] [CrossRef]
- Haass, C. Take five—Bace and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef]
- Adaikalakoteswari, A.; Finer, S.; Voyias, P.D.; McCarthy, C.M.; Vatish, M.; Moore, J.; Smart-Halajko, M.; Bawazeer, N.; Al-Daghri, N.M.; McTernan, P.G.; et al. Vitamin b12 insufficiency induces cholesterol biosynthesis by limiting s-adenosylmethionine and modulating the methylation of srebf1 and ldlr genes. Clin. Epigenet. 2015, 7, 14. [Google Scholar] [CrossRef]
- Andrade, S.; Loureiro, J.A.; Pereira, M.C. Vitamin b12 inhibits abeta fibrillation and disaggregates preformed fibrils in the presence of synthetic neuronal membranes. ACS Chem. Neurosci. 2021, 12, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.E.; Tian, Q.; Wei, W.; Peng, J.H.; Liu, G.P.; Zhou, X.W.; Wang, Q.; Wang, D.W.; Wang, J.Z. Homocysteine induces tau phosphorylation by inactivating protein phosphatase 2a in rat hippocampus. Neurobiol. Aging 2008, 29, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Liu, Y.H.; Zhang, C.E.; Wang, Q.; Wei, Z.; Mousseau, D.D.; Wang, J.Z.; Tian, Q.; Liu, G.P. Folate/vitamin-b12 prevents chronic hyperhomocysteinemia-induced tau hyperphosphorylation and memory deficits in aged rats. J. Alzheimer’s Dis. 2011, 27, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, S.; Asadollahi, K.; Riazi, G.; Ahmadian, S.; Saboury, A.A. Vitamin b12 inhibits tau fibrillization via binding to cysteine residues of tau. ACS Chem. Neurosci. 2017, 8, 2676–2682. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| AGPS | ACCAGATTCCCTGGAGTTCA | GAACCACCAGGTCCTCGATA |
| GNPAT | TACAACTGGGTTCTGAAAGCC | CAGCTGCCAAAGATCGAAGT |
| PEDS1 | ACCATCGCATCCACCACGTC | AGGCGTCGCCAGAAGCCTAT |
| CHPT1 | TCCAGTTCTTGGATTTCTAGGTGGAGT | ACACTGGTGCCTGCTATAGTGGA |
| SOD | CAGCAGGCTGTACCAGTGC | ACATTGCCCAAGTCTCCAAC |
| CAT | ATTCGATCTCACCAAGGTTTG | CTTGGGTCGAAGGCTATCTG |
| PRDX2 | CACCTGGCTTGGATCAACA | GCCGTAATCCTCAGACAAGC |
| TXNRD1 | ACACAAAGCTTCAGCATGTCA | CAATTCCGAGAGCGTTCC |
| TXNRD2 | GCATGACTGGAGGAAGATGG | AAACCGTGTGCTCGTCAAC |
| RN18S1 (HKG) | GGAGTATGGTTGCAAAGCTGA | ATCTGTCAATCCTGTCCGTGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theiss, E.L.; Griebsch, L.V.; Lauer, A.A.; Janitschke, D.; Erhardt, V.K.J.; Haas, E.C.; Kuppler, K.N.; Radermacher, J.; Walzer, O.; Portius, D.; et al. Vitamin B12 Attenuates Changes in Phospholipid Levels Related to Oxidative Stress in SH-SY5Y Cells. Cells 2022, 11, 2574. https://doi.org/10.3390/cells11162574
Theiss EL, Griebsch LV, Lauer AA, Janitschke D, Erhardt VKJ, Haas EC, Kuppler KN, Radermacher J, Walzer O, Portius D, et al. Vitamin B12 Attenuates Changes in Phospholipid Levels Related to Oxidative Stress in SH-SY5Y Cells. Cells. 2022; 11(16):2574. https://doi.org/10.3390/cells11162574
Chicago/Turabian StyleTheiss, Elena Leoni, Lea Victoria Griebsch, Anna Andrea Lauer, Daniel Janitschke, Vincent Konrad Johannes Erhardt, Elodie Christiane Haas, Konstantin Nicolas Kuppler, Juliane Radermacher, Oliver Walzer, Dorothea Portius, and et al. 2022. "Vitamin B12 Attenuates Changes in Phospholipid Levels Related to Oxidative Stress in SH-SY5Y Cells" Cells 11, no. 16: 2574. https://doi.org/10.3390/cells11162574
APA StyleTheiss, E. L., Griebsch, L. V., Lauer, A. A., Janitschke, D., Erhardt, V. K. J., Haas, E. C., Kuppler, K. N., Radermacher, J., Walzer, O., Portius, D., Grimm, H. S., Hartmann, T., & Grimm, M. O. W. (2022). Vitamin B12 Attenuates Changes in Phospholipid Levels Related to Oxidative Stress in SH-SY5Y Cells. Cells, 11(16), 2574. https://doi.org/10.3390/cells11162574