

Therapeutic Drug-Induced Metabolic Reprogramming in Glioblastoma

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Glucose Metabolism as a Central Component of GBM and Tumor Growth
3. Major Metabolism Pathways in GBM Model Systems upon Therapy
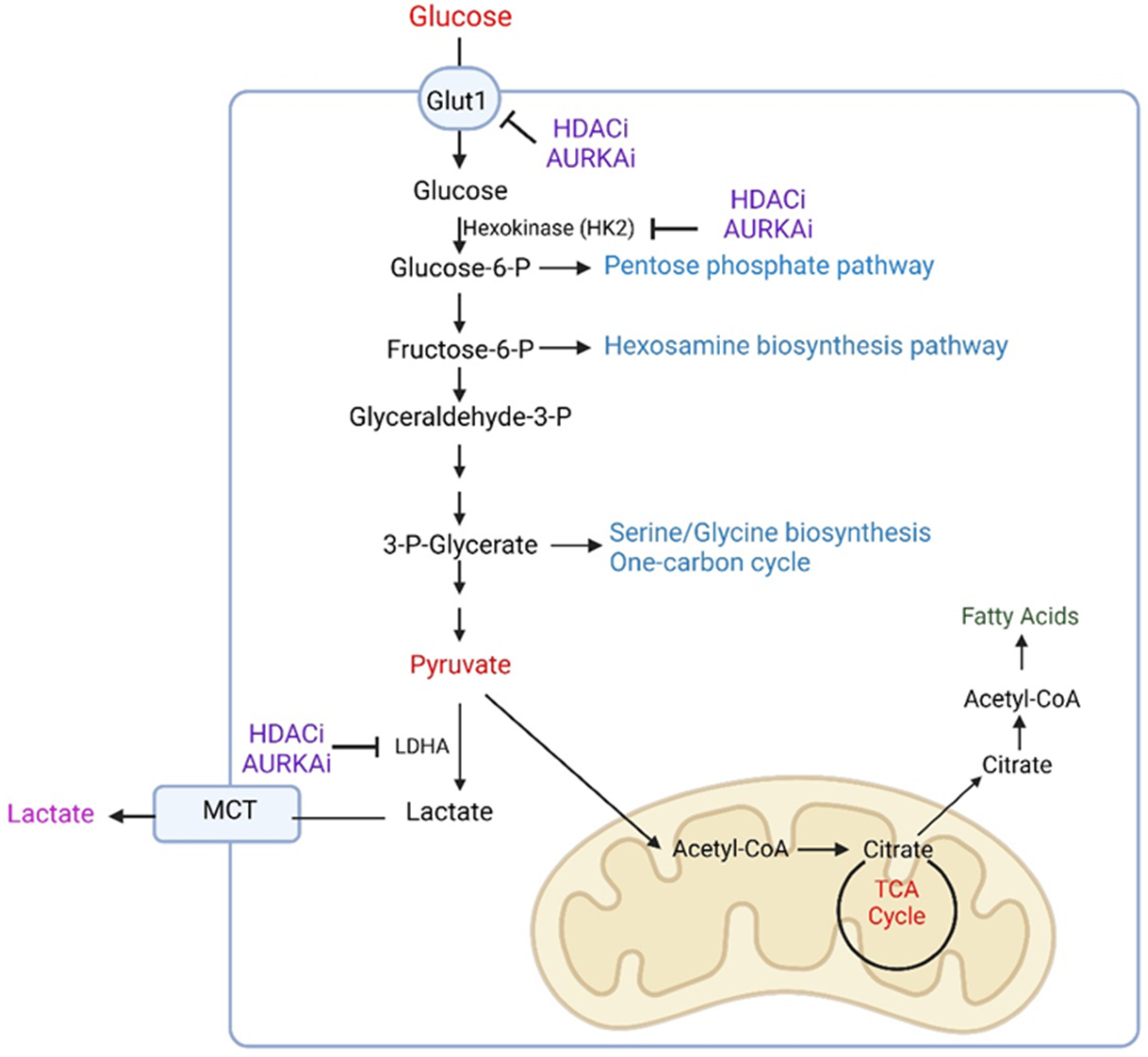
3.1. Metabolic Reprogramming Related to Glycolysis
3.1.1. Glycolysis in the Context of Different Microenvironments
3.1.2. Drug-Induced Reprogramming of Glycolysis in GBM Model Systems
3.2. Metabolic Reprogramming of the Pentose Phosphate Pathway
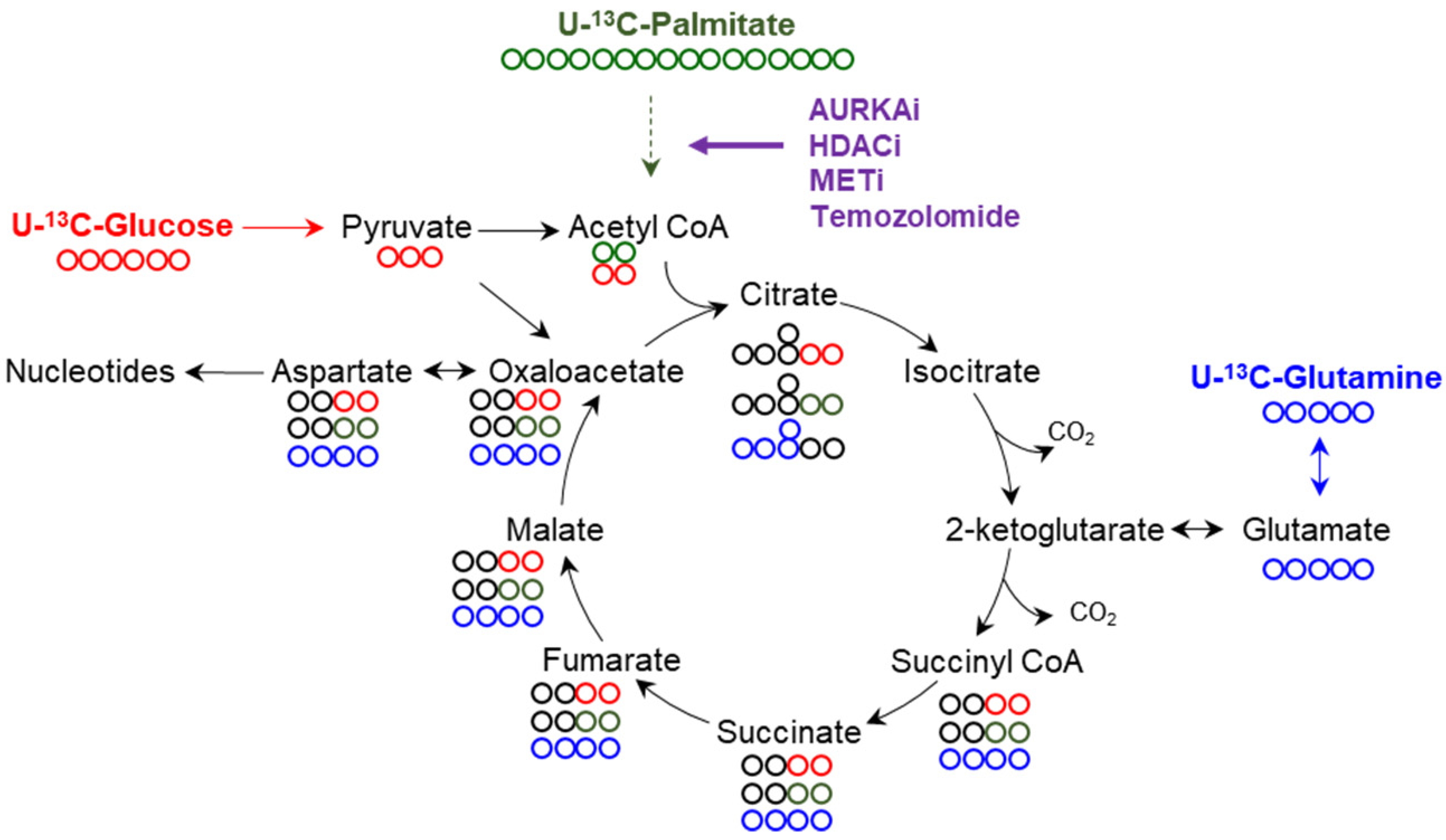
3.3. The Tricarboxylic Acid (TCA) Cycle and Oxidative Phosphorylation (OXPHOS)
3.3.1. Basic Functions of the TCA Cycle and Cellular Respiration
3.3.2. Drug Compounds That Interfere with the TCA-Cycle and OXPHOS
3.3.3. Loss of Function of HDAC1/2, AURKA and MET Drives Oxidative Energy Metabolism
4. A Novel Fuel for the TCA-Cycle in GBM
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guo, G.; Gong, K.; Beckley, N.; Zhang, Y.; Yang, X.; Chkheidze, R.; Hatanpaa, K.J.; Garzon-Muvdi, T.; Koduru, P.; Nayab, A.; et al. EGFR ligand shifts the role of EGFR from oncogene to tumour suppressor in EGFR-amplified glioblastoma by suppressing invasion through BIN3 upregulation. Nat. Cell Biol. 2022, 24, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Filippo, B.; Cusimano, M.; Federico, R.; Stefano, B.; Paola, M.V.R.; Anna, R.; Stefano, C.; Barbara, C.; Peter, A.; Francesca, S.; et al. Targeted inducible delivery of immunoactivating cytokines reprograms glioblastoma microenvironment and inhibits growth in mouse models. Sci. Transl. Med 2022, 14, eabl4106. [Google Scholar]
- Sun, X.; Turcan, S. From Laboratory Studies to Clinical Trials: Temozolomide Use in IDH-Mutant Gliomas. Cells 2021, 10, 1225. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Wang, F.; Chen, G.; Zhang, H.; Feng, L.; Wang, L.; Colman, H.; Keating, M.J.; Li, X.; Xu, R.H.; et al. Effective elimination of cancer stem cells by a novel drug combination strategy. Stem Cells 2013, 31, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Yan, Q.; Zhang, Y.; Fang, X.; Liu, B.; Guan, X. Cancer cell reprogramming: A promising therapy converting malignancy to benignity. Cancer Commun. 2019, 39, 48. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Sylow, L.; Tokarz, V.L.; Richter, E.A.; Klip, A. The many actions of insulin in skeletal muscle, the paramount tissue determining glycemia. Cell Metab. 2021, 33, 758–780. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Insulin-PI3K signalling: An evolutionarily insulated metabolic driver of cancer. Nat. Rev. Endocrinol. 2020, 16, 276–283. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Carbohydrate Metabolism. Cold Spring Harb. Perspect. Biol. 2021, 13, a040568. [Google Scholar] [CrossRef]
- Morrow, D.; Minami, J.; Nathanson, D.A. Metabolic Vulnerabilities in Brain Cancer. Neurosurg. Clin N. Am. 2021, 32, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Trefts, E.; Shaw, R.J. AMPK: Restoring metabolic homeostasis over space and time. Mol. Cell 2021, 81, 3677–3690. [Google Scholar] [CrossRef] [PubMed]
- Ciraku, L.; Bacigalupa, Z.A.; Ju, J.; Moeller, R.A.; Le Minh, G.; Lee, R.H.; Smith, M.D.; Ferrer, C.M.; Trefely, S.; Izzo, L.T.; et al. O-GlcNAc transferase regulates glioblastoma acetate metabolism via regulation of CDK5-dependent ACSS2 phosphorylation. Oncogene 2022, 41, 2122–2136. [Google Scholar] [CrossRef]
- Otto, W.; Wind, F.; Erwin, N. Metabolism of tumors in body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef]
- Yokota, H.; Guo, J.; Matoba, M.; Higashi, K.; Tonami, H.; Nagao, Y. Lactate, choline, and creatine levels measured by vitro 1H-MRS as prognostic parameters in patients with non-small-cell lung cancer. J. Magn. Reson. Imaging 2007, 25, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Brizel, D.M.; Schroeder, T.; Scher, R.L.; Walenta, S.; Clough, R.W.; Dewhirst, M.W.; Mueller-Klieser, W. Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int. J. Radiation Oncology Biol. Phys. 2001, 51, 349–353. [Google Scholar] [CrossRef]
- Certo, M.; Llibre, A.; Lee, W.; Mauro, C. Understanding lactate sensing and signalling. Trends Endocrinol. Metab. 2022, 7, 4. [Google Scholar] [CrossRef]
- Torrini, C.; Nguyen, T.T.T.; Shu, C.; Mela, A.; Humala, N.; Mahajan, A.; Seeley, E.H.; Zhang, G.; Westhoff, M.A.; Karpel-Massler, G.; et al. Lactate is an epigenetic metabolite that drives survival in model systems of glioblastoma. Mol. Cell 2022, 82, 3061–3076.e3066. [Google Scholar] [CrossRef]
- Aurora, A.B.; Khivansara, V.; Leach, A.; Gill, J.G.; Martin-Sandoval, M.; Yang, C.; Kasitinon, S.Y.; Bezwada, D.; Tasdogan, A.; Gu, W.; et al. Loss of glucose 6-phosphate dehydrogenase function increases oxidative stress and glutaminolysis in metastasizing melanoma cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2120617119. [Google Scholar] [CrossRef]
- Hosios, A.M.; Hecht, V.C.; Danai, L.V.; Johnson, M.O.; Rathmell, J.C.; Steinhauser, M.L.; Manalis, S.R.; Vander Heiden, M.G. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell 2016, 36, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Alfarouk, K.O.; Ahmed, S.B.M.; Elliott, R.L.; Benoit, A.; Alqahtani, S.S.; Ibrahim, M.E.; Bashir, A.H.H.; Alhoufie, S.T.S.; Elhassan, G.O.; Wales, C.C.; et al. The Pentose Phosphate Pathway Dynamics in Cancer and Its Dependency on Intracellular pH. Metabolites 2020, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Kruger, N.J.; von Schaewen, A. The oxidative pentose phosphate pathway: Structure and organisation. Curr. Opin. Plant Biol. 2003, 6, 236–246. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Pan, S.; Fan, M.; Liu, Z.; Li, X.; Wang, H. Serine, glycine and onecarbon metabolism in cancer. Int. J. Oncol. 2021, 58, 158–170. [Google Scholar] [CrossRef]
- Newman, A.C.; Maddocks, O.D.K. Serine and Functional Metabolites in Cancer. Trends Cell Biol 2017, 27, 645–657. [Google Scholar] [CrossRef]
- Fantus, I.G.; Goldberg, H.J.; Whiteside, C.I.; Topic, D. The Hexosamine Biosynthesis Pathway. In The Diabetic Kidney; Cortes, P., Mogensen, C.E., Eds.; Contemporary Diabetes; Humana Press: Totowa, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Taparra, K.; Tran, P.T.; Zachara, N.E. Hijacking the Hexosamine Biosynthetic Pathway to Promote EMT-Mediated Neoplastic Phenotypes. Front. Oncol. 2016, 6, 85. [Google Scholar] [CrossRef] [Green Version]
- de Queiroz, R.M.; Oliveira, I.A.; Piva, B.; Bouchuid Catao, F.; da Costa Rodrigues, B.; da Costa Pascoal, A.; Diaz, B.L.; Todeschini, A.R.; Caarls, M.B.; Dias, W.B. Hexosamine Biosynthetic Pathway and Glycosylation Regulate Cell Migration in Melanoma Cells. Front. Oncol. 2019, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Akella, N.M.; Ciraku, L.; Reginato, M.J. Fueling the fire: Emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Morfouace, M.; Lalier, L.; Bahut, M.; Bonnamain, V.; Naveilhan, P.; Guette, C.; Oliver, L.; Gueguen, N.; Reynier, P.; Vallette, F.M. Comparison of spheroids formed by rat glioma stem cells and neural stem cells reveals differences in glucose metabolism and promising therapeutic applications. J. Biol. Chem. 2012, 287, 33664–33674. [Google Scholar] [CrossRef] [PubMed]
- Duan, K.; Liu, Z.J.; Hu, S.Q.; Huo, H.Y.; Xu, Z.R.; Ruan, J.F.; Sun, Y.; Dai, L.P.; Yan, C.B.; Xiong, W.; et al. Lactic acid induces lactate transport and glycolysis/OXPHOS interconversion in glioblastoma. Biochem. Biophys. Res. Commun. 2018, 503, 888–894. [Google Scholar] [CrossRef]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation. Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Chao, Z.; Yang, C.; Michael, J.F.; Herui, W.; Ying, P.; Dominic, M.M.; Dongwang, Z.; Cody, L.N.; Pauline, D.; Petra, B.; et al. Vorinostat suppresses hypoxia signaling by modulating nuclear translocation of hypoxia inducible factor 1 alpha. Oncotarget 2017, 8, 56110–56125. [Google Scholar]
- Mair, R.; Wright, A.J.; Ros, S.; Hu, D.E.; Booth, T.; Kreis, F.; Rao, J.; Watts, C.; Brindle, K.M. Metabolic Imaging Detects Low Levels of Glycolytic Activity That Vary with Levels of c-Myc Expression in Patient-Derived Xenograft Models of Glioblastoma. Cancer Res. 2018, 78, 5408–5418. [Google Scholar] [CrossRef]
- Ishida, C.T.; Zhang, Y.; Bianchetti, E.; Shu, C.; Nguyen, T.T.T.; Kleiner, G.; Sanchez-Quintero, M.J.; Quinzii, C.M.; Westhoff, M.A.; Karpel-Massler, G.; et al. Metabolic Reprogramming by Dual AKT/ERK Inhibition through Imipridones Elicits Unique Vulnerabilities in Glioblastoma. Clin. Cancer Res. 2018, 24, 5392–5406. [Google Scholar] [CrossRef]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef]
- Tateishi, K.; Iafrate, A.J.; Ho, Q.; Curry, W.T.; Batchelor, T.T.; Flaherty, K.T.; Onozato, M.L.; Lelic, N.; Sundaram, S.; Cahill, D.P.; et al. Myc-Driven Glycolysis Is a Therapeutic Target in Glioblastoma. Clin. Cancer Res. 2016, 22, 4452–4465. [Google Scholar] [CrossRef] [PubMed]
- Said. Oxygen-dependent regulation of NDRG1 in human glioblastoma cells in vitro and in vivo. Oncol. Rep. 1994, 21, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J. Clin. Invest. 2020, 130, 3699–3716. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Shang, E.; Shu, C.; Kim, S.; Mela, A.; Humala, N.; Mahajan, A.; Yang, H.W.; Akman, H.O.; Quinzii, C.M.; et al. Aurora kinase A inhibition reverses the Warburg effect and elicits unique metabolic vulnerabilities in glioblastoma. Nat. Commun. 2021, 12, 5203. [Google Scholar] [CrossRef] [PubMed]
- Aguer, C.; Gambarotta, D.; Mailloux, R.J.; Moffat, C.; Dent, R.; McPherson, R.; Harper, M.E. Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS ONE 2011, 6, e28536. [Google Scholar] [CrossRef]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.H.Y.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 suppression upregulates beta-catenin and c-Myc to abrogate KRas-dependent tumors. Nat. Commun. 2018, 9, 5154. [Google Scholar] [CrossRef]
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J. Biol. Chem. 2003, 278, 51606–51612. [Google Scholar] [CrossRef]
- Keibler, M.A.; Wasylenko, T.M.; Kelleher, J.K.; Iliopoulos, O.; Vander Heiden, M.G.; Stephanopoulos, G. Metabolic requirements for cancer cell proliferation. Cancer Metab. 2016, 4, 16. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chen, Y.; Shi, X.; Zhou, M.; Bao, L.; Hatanpaa, K.J.; Patel, T.; DeBerardinis, R.J.; Wang, Y.; Luo, W. Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cell Mol. Life Sci. 2021, 78, 195–206. [Google Scholar] [CrossRef]
- Liu, R.; Li, W.; Tao, B.; Wang, X.; Yang, Z.; Zhang, Y.; Wang, C.; Liu, R.; Gao, H.; Liang, J.; et al. Tyrosine phosphorylation activates 6-phosphogluconate dehydrogenase and promotes tumor growth and radiation resistance. Nat. Commun. 2019, 10, 991. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Dixit, D.; Sharma, V.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Nrf2-driven TERT regulates pentose phosphate pathway in glioblastoma. Cell Death Dis. 2016, 7, e2213. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR Signaling Through an Akt-SREBP-1–Dependent, Rapamycin-Resistant Pathway Sensitizes Glioblastomas to Antilipogenic Therapy. Cancer Cell 2009, 2, ra82. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol. 2017, 19, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Duraj, T.; Garcia-Romero, N.; Carrion-Navarro, J.; Madurga, R.; Mendivil, A.O.; Prat-Acin, R.; Garcia-Canamaque, L.; Ayuso-Sacido, A. Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef]
- Anderson, R.; Miller, L.D.; Isom, S.; Chou, J.W.; Pladna, K.M.; Schramm, N.J.; Ellis, L.R.; Howard, D.S.; Bhave, R.R.; Manuel, M.; et al. Phase II trial of cytarabine and mitoxantrone with devimistat in acute myeloid leukemia. Nat. Commun. 2022, 13, 1673. [Google Scholar] [CrossRef] [PubMed]
- Pardee, T.S.; Anderson, R.G.; Pladna, K.M.; Isom, S.; Ghiraldeli, L.P.; Miller, L.D.; Chou, J.W.; Jin, G.; Zhang, W.; Ellis, L.R.; et al. A Phase I Study of CPI-613 in Combination with High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 2060–2073. [Google Scholar] [CrossRef]
- Bonner, E.R.; Waszak, S.M.; Grotzer, M.A.; Mueller, S.; Nazarian, J. Mechanisms of imipridones in targeting mitochondrial metabolism in cancer cells. Neuro Oncol. 2021, 23, 542–556. [Google Scholar] [CrossRef]
- Prabhu, V.V.; Morrow, S.; Kawakibi, A.R.; Zhou, L.; Ralff, M.; Ray, J.; Jhaveri, A.; Ferrarini, I.; Lee, Y.; Parker, C.; et al. ONC201 and imipridones: Anti-cancer compounds with clinical efficacy. NeoPlasia 2020, 22, 725–744. [Google Scholar] [CrossRef]
- Allen, J.E.; Kline, C.L.B.; Prabhu, V.V.; Wagner, J.; Ishizawa, J.; Madhukar, N.; Lev, A.; Baumeister, M.; Zhou, L.; Lulla, A.; et al. Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget 2016, 7, 74380–74392. [Google Scholar] [CrossRef]
- Pruss, M.; Dwucet, A.; Tanriover, M.; Hlavac, M.; Kast, R.E.; Debatin, K.M.; Wirtz, C.R.; Halatsch, M.E.; Siegelin, M.D.; Westhoff, M.A.; et al. Dual metabolic reprogramming by ONC201/TIC10 and 2-Deoxyglucose induces energy depletion and synergistic anti-cancer activity in glioblastoma. Br. J. Cancer 2020, 122, 1146–1157. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Shang, E.; Schiffgens, S.; Torrini, C.; Shu, C.; Akman, H.O.; Prabhu, V.V.; Allen, J.E.; Westhoff, M.A.; Karpel-Massler, G.; et al. Induction of Synthetic Lethality by Activation of Mitochondrial ClpP and Inhibition of HDAC1/2 in Glioblastoma. Clin. Cancer Res. 2022, 28, 1881–1895. [Google Scholar] [CrossRef]
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737.e729. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Lee, J.H.; Oh, Y.; Koh, I.; Shim, J.K.; Park, J.; Choi, J.; Yun, M.; Jeon, J.Y.; Huh, Y.M.; et al. Inhibition of glioblastoma tumorspheres by combined treatment with 2-deoxyglucose and metformin. Neuro Oncol. 2017, 19, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995, 9, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Wójcicka, G.; Jamroz-Wiśniewska, A.; Horoszewicz, K.; Bełtowski, J. Liver X receptors (LXRs). Part I: Structure, function, regulation of activity, and role in lipid metabolism. Postępy Hig. I Med. Doświadczalnej 2007, 61, 736–759. [Google Scholar]
- Nguyen, T.T.T.; Ishida, C.T.; Shang, E.; Shu, C.; Torrini, C.; Zhang, Y.; Bianchetti, E.; Sanchez-Quintero, M.J.; Kleiner, G.; Quinzii, C.M.; et al. Activation of LXRbeta inhibits tumor respiration and is synthetically lethal with Bcl-xL inhibition. EMBO Mol. Med. 2019, 11, e10769. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.T.; Ishida, C.T.; Shang, E.; Shu, C.; Bianchetti, E.; Karpel-Massler, G.; Siegelin, M.D. Activation of LXR Receptors and Inhibition of TRAP1 Causes Synthetic Lethality in Solid Tumors. Cancer 2019, 11, 788. [Google Scholar] [CrossRef]
- Chae, Y.C.; Caino, M.C.; Lisanti, S.; Ghosh, J.C.; Dohi, T.; Danial, N.N.; Villanueva, J.; Ferrero, S.; Vaira, V.; Santambrogio, L.; et al. Control of tumor bioenergetics and survival stress signaling by mitochondrial HSP90s. Cancer Cell 2012, 22, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., II; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Gene Dev. 1981, 29, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Xu, H.; Xia, H.; Tang, Q.; Bi, F. Simultaneously targeting SOAT1 and CPT1A ameliorates hepatocellular carcinoma by disrupting lipid homeostasis. Cell Death Discov. 2021, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Raud, B.; Roy, D.G.; Divakaruni, A.S.; Tarasenko, T.N.; Franke, R.; Ma, E.H.; Samborska, B.; Hsieh, W.Y.; Wong, A.H.; Stuve, P.; et al. Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell Metab. 2018, 28, 504–515.e507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Quinzii, C.M.; Westhoff, M.A.; Karpel-Massler, G.; Siegelin, M.D. Inhibition of HDAC1/2 Along with TRAP1 Causes Synthetic Lethality in Glioblastoma Model Systems. Cells 2020, 9, 1661. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Siegelin, M.D.; Vaira, V.; Faversani, A.; Tavecchio, M.; Chae, Y.C.; Lisanti, S.; Rampini, P.; Giroda, M.; Caino, M.C.; et al. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J. Natl. Cancer Inst. 2015, 107, dju502. [Google Scholar] [CrossRef]
- Zhang, Y.; Nguyen, T.T.T.; Shang, E.; Mela, A.; Humala, N.; Mahajan, A.; Zhao, J.; Shu, C.; Torrini, C.; Sanchez-Quintero, M.J.; et al. MET Inhibition Elicits PGC1alpha-Dependent Metabolic Reprogramming in Glioblastoma. Cancer Res. 2020, 80, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Caragher, S.; Miska, J.; Shireman, J.; Park, C.H.; Muroski, M.; Lesniak, M.S.; Ahmed, A.U. Temozolomide Treatment Increases Fatty Acid Uptake in Glioblastoma Stem Cells. Cancers 2020, 12, 3126. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef]
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020, 577, 115–120. [Google Scholar] [CrossRef]
- Miranda-Goncalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate transporters (MCTs) in gliomas: Expression and exploitation as therapeutic targets. Neuro Oncol. 2013, 15, 172–188. [Google Scholar] [CrossRef]
- Voss, D.M.; Spina, R.; Carter, D.L.; Lim, K.S.; Jeffery, C.J.; Bar, E.E. Disruption of the monocarboxylate transporter-4-basigin interaction inhibits the hypoxic response, proliferation, and tumor progression. Sci. Rep. 2017, 7, 4292. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Siegelin, M.D.; Borczuk, A.C. Epidermal growth factor receptor mutations in lung adenocarcinoma. Lab. Invest. 2014, 94, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Drugs | Inhibit/Activate Target | Metabolic Pathways |
|---|---|---|
| Panobinostat | pan-HDAC | Glycolysis |
| Romidepsin | HDAC1/2 | Glycolysis |
| Alisertib | Aurora kinase A | Glycolysis |
| Devimistat (CPI-613) | Pyruvate dehydrogenase | TCA-cycle |
| Imipridones (ONC201, ONC206, ONC212) | CLPP | OXPHOS Cellular respiration |
| LXR623 | LXRβ | OXPHOS |
| Gamitrinib (G-TPP) | Hsp90/TRAP1 | OXPHOS |
| Crizotinib | MET kinase | OXPHOS |
| Metformin | Complex I of respiratory chain | OXPHOS |
| IACS-010759 | Complex I of respiratory chain | OXPHOS |
| Oligomycin | ATP synthase–Complex V | OXPHOS |
| Etomoxir | CPT1A | Fatty acid oxidation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.T.T.; Shang, E.; Westhoff, M.-A.; Karpel-Massler, G.; Siegelin, M.D. Therapeutic Drug-Induced Metabolic Reprogramming in Glioblastoma. Cells 2022, 11, 2956. https://doi.org/10.3390/cells11192956
Nguyen TTT, Shang E, Westhoff M-A, Karpel-Massler G, Siegelin MD. Therapeutic Drug-Induced Metabolic Reprogramming in Glioblastoma. Cells. 2022; 11(19):2956. https://doi.org/10.3390/cells11192956
Chicago/Turabian StyleNguyen, Trang T. T., Enyuan Shang, Mike-Andrew Westhoff, Georg Karpel-Massler, and Markus D. Siegelin. 2022. "Therapeutic Drug-Induced Metabolic Reprogramming in Glioblastoma" Cells 11, no. 19: 2956. https://doi.org/10.3390/cells11192956
APA StyleNguyen, T. T. T., Shang, E., Westhoff, M. -A., Karpel-Massler, G., & Siegelin, M. D. (2022). Therapeutic Drug-Induced Metabolic Reprogramming in Glioblastoma. Cells, 11(19), 2956. https://doi.org/10.3390/cells11192956