

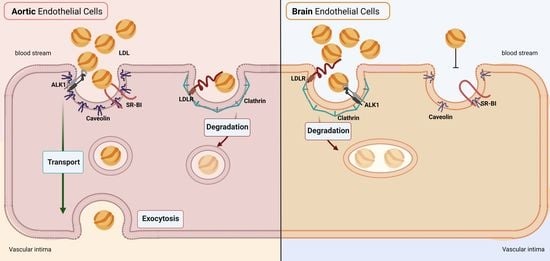
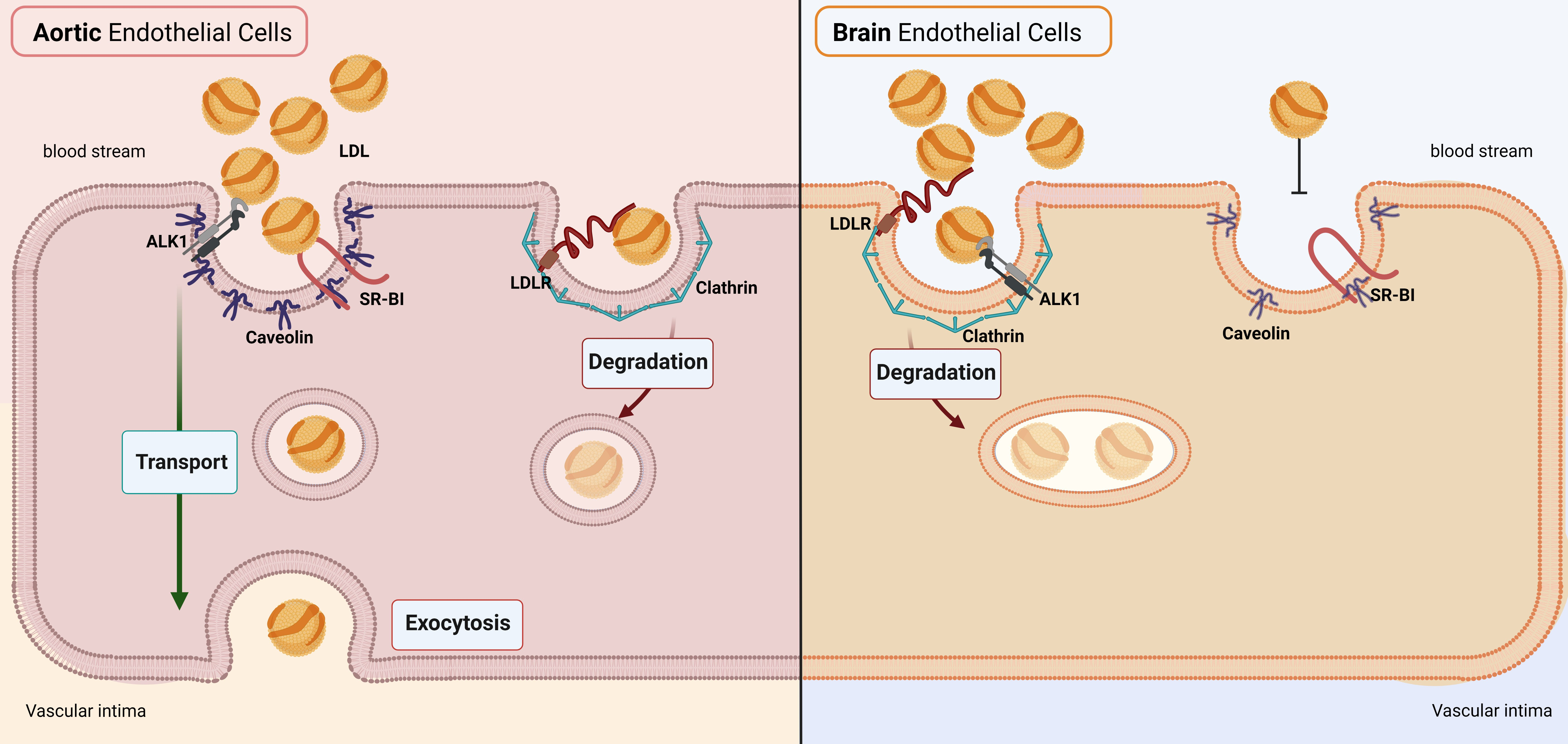
Brain Endothelial Cells in Contrary to the Aortic Do Not Transport but Degrade Low-Density Lipoproteins via Both LDLR and ALK1

 ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. LDL Isolation and Labeling
2.3. LDL Uptake
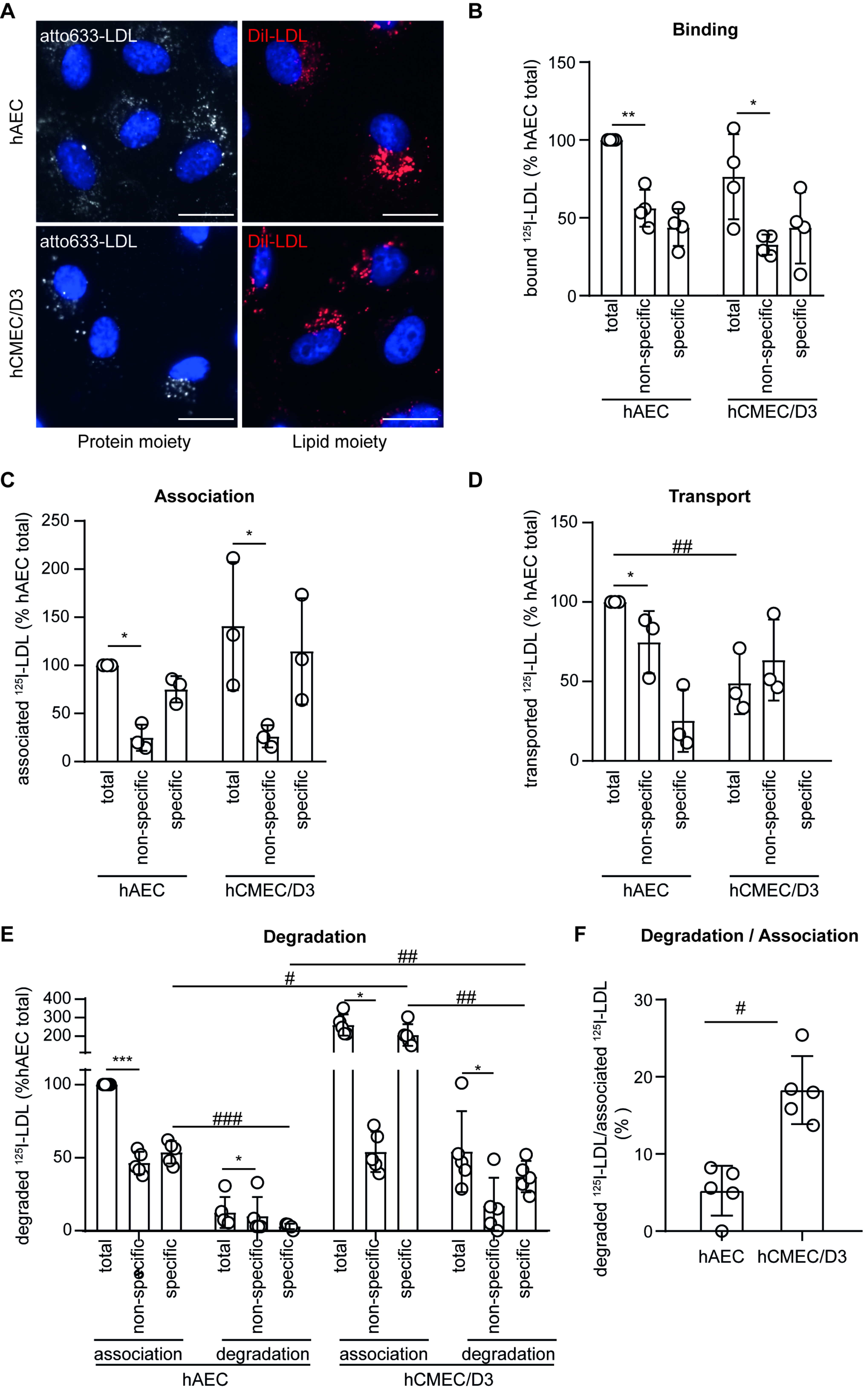
2.4. LDL Cell Binding, Association, and Transport
2.5. LDL Degradation Assays
2.6. siRNA-Mediated and Pharmacological Interferences
2.7. Generation of ALK1 Overexpressing hCMEC/D3
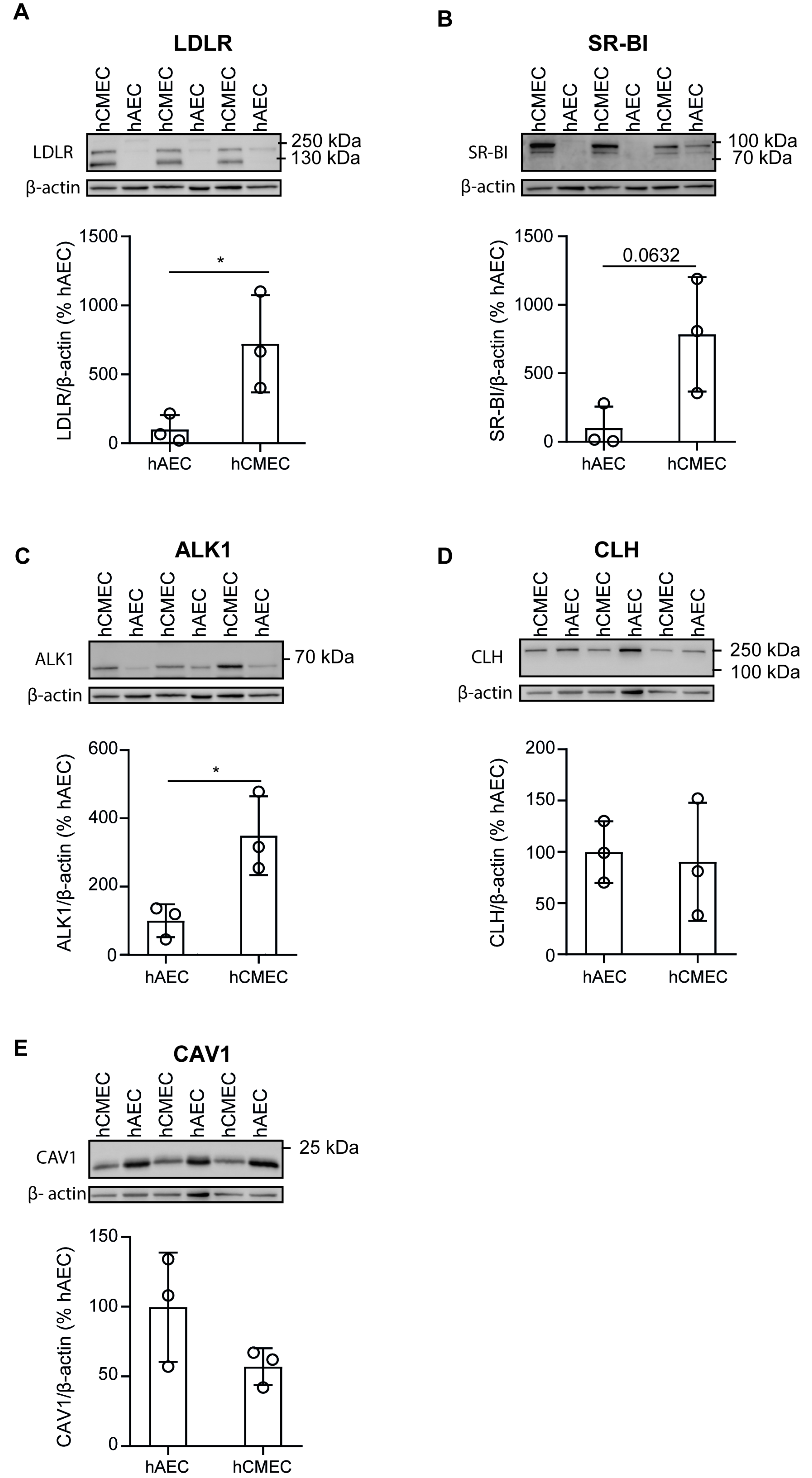
2.8. SDS-PAGE and Western Blot
2.9. Quantitative Reverse Transcription PCR
2.10. Statistical Analysis
3. Results
3.1. LDL Is Not Transported through but Rather Degraded by Brain Endothelial Cells
3.2. hCMEC/D3 Express the LDL Binding Proteins LDLR, SR-BI, and ALK1
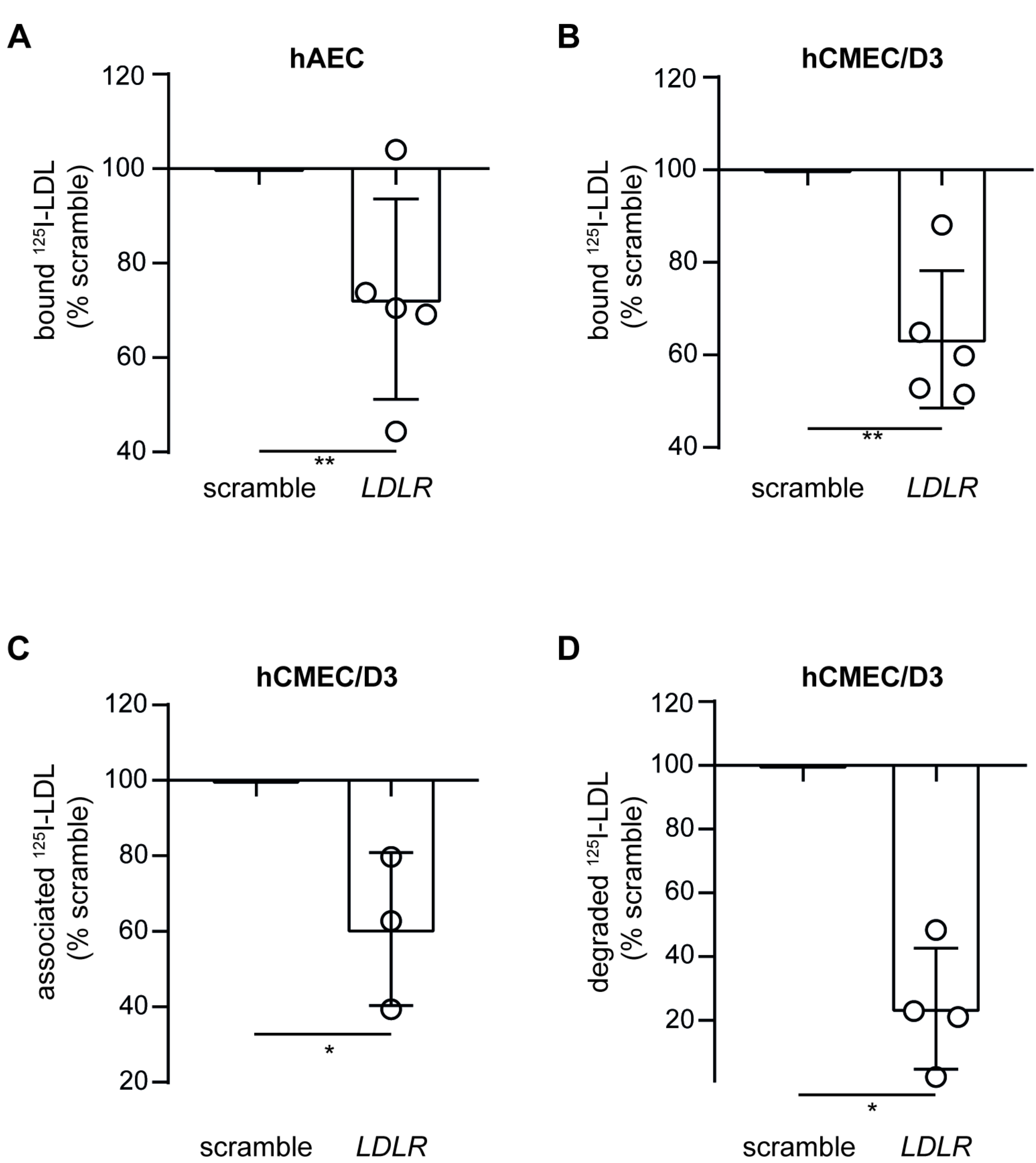
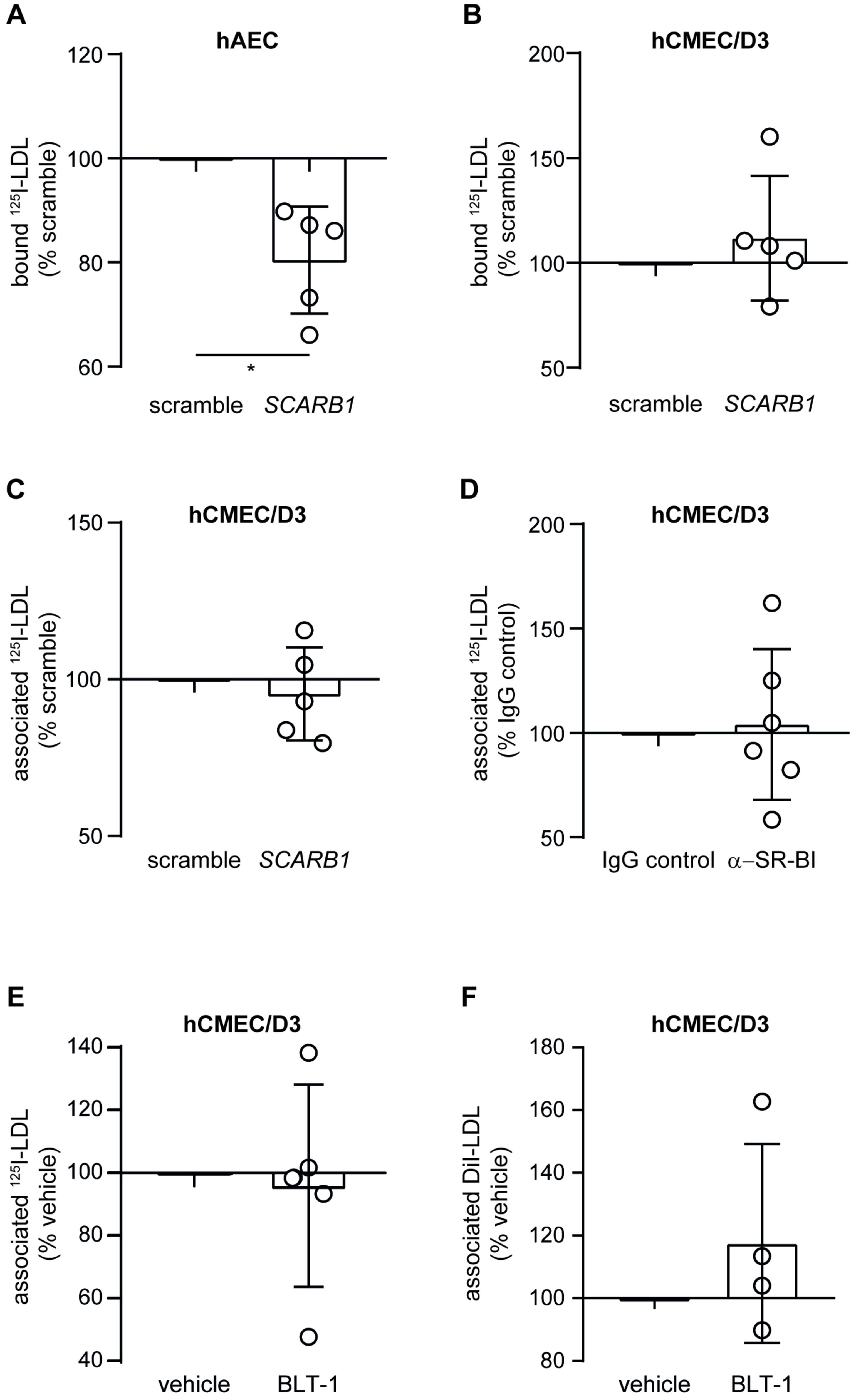
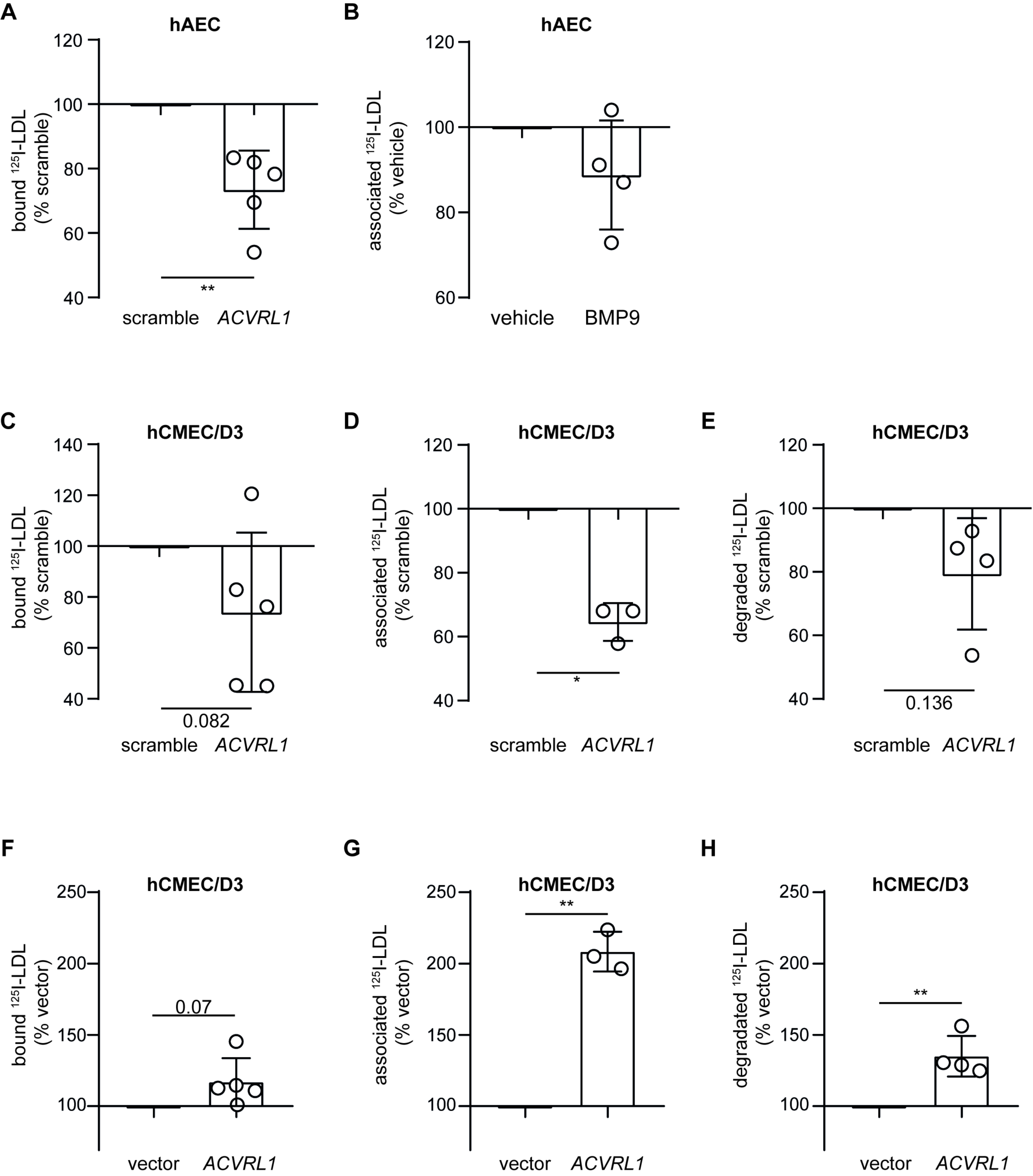
3.3. Loss of LDLR and ALK1 but Not SR-BI Reduces LDL Binding and Degradation in hCMEC/D3
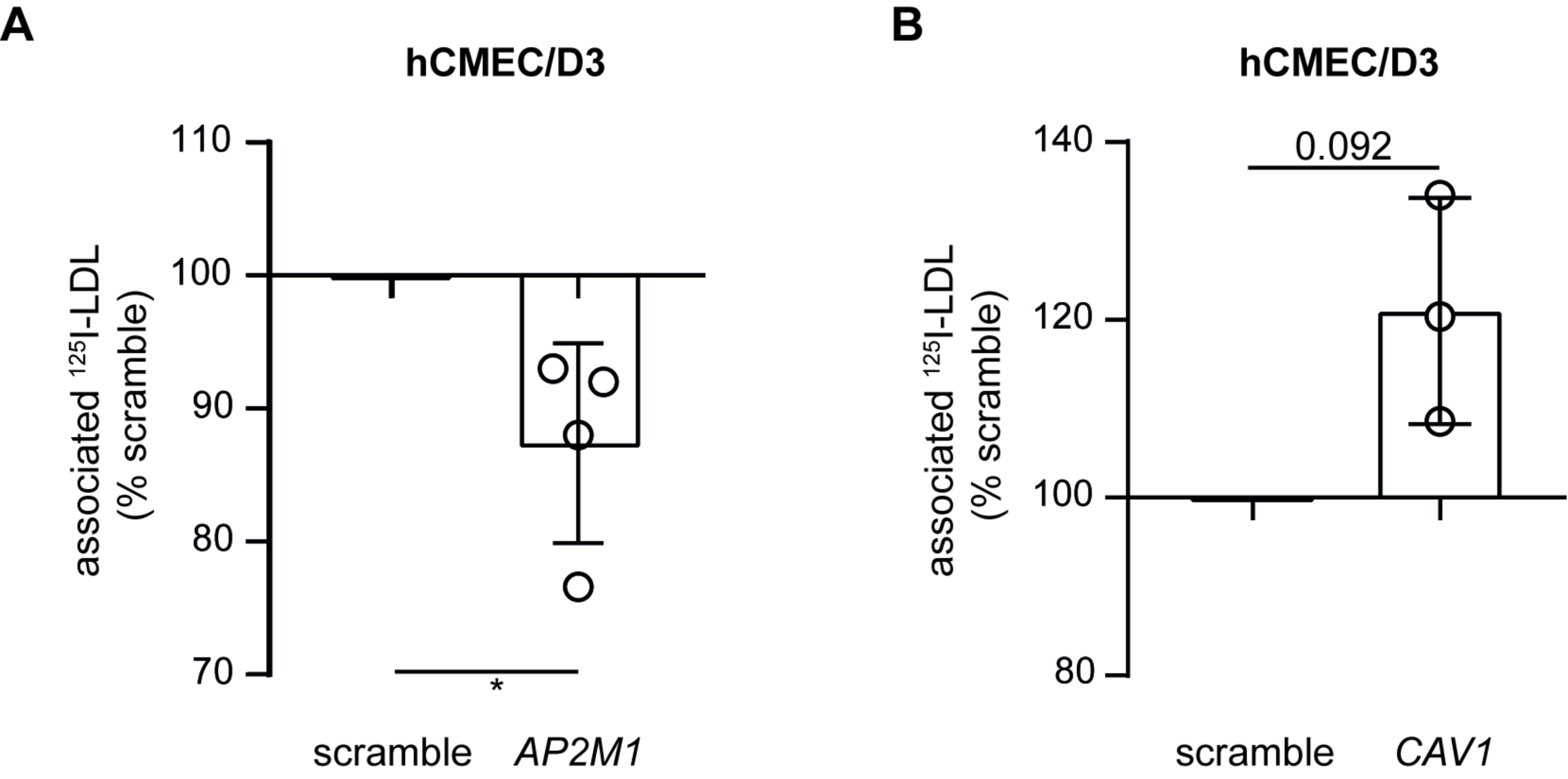
3.4. LDL Is Internalized via Clathrin- but Not Caveolin-Coated Vesicles in Brain Endothelial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALK1 and ACVRL1 | Activin-like kinase |
| AP2 and AP2M1 | Adaptor-related protein complex 2 alpha 1 |
| ApoB | Apolipoprotein B |
| ApoE | Apolipoprotein E |
| ASCVD | Atherosclerotic cardiovascular diseases |
| Aβ | Beta-amyloid |
| BBB | Blood–brain barrier |
| bBMEC | Bovine brain microvascular endothelial cells |
| bEnd.3 | Mouse brain endothelial cells clone 3 |
| BMP-9 | Bone morphogenetic protein-9 |
| CAV1 | Caveolin 1 |
| CLH | Clathrin heavy chain |
| CSF | Cerebrospinal fluid |
| Dil | 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate |
| DMEM | Dulbecco’s modified Eagle’s medium |
| eNOS | Endothelial nitric oxide synthase |
| EOAD | Early-onset Alzheimer’s disease |
| FBS | Fetal bovine serum |
| hAEC | Human aortic endothelial cells |
| hBMEC | Human brain microvascular endothelial cells |
| hCMEC/D3 | Human cortical microvascular endothelial cells/D3 |
| HDL | High-density lipoprotein |
| HMGCR | 3-hydroxy-3-methylglutaryl-CoA reductase |
| hUVEC | Human umbilical vein endothelial cells |
| LDL | Low-density lipoprotein |
| LDL-C | LDL cholesterol |
| LDLR | Low-density lipoprotein receptor |
| LOAD | Late-onset Alzheimer’s disease |
| MMSE | Mini Mental State Examination |
| NO | Nitric oxide |
| SR-BI and SCARB1 | Scavenger receptor B-I |
References
- Jang, E.; Robert, J.; Rohrer, L.; von Eckardstein, A.; Lee, W.L. Transendothelial Transport of Lipoproteins. Atherosclerosis 2020, 315, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.M.; Sugiyama, M.G.; Fung, K.Y.Y.; Gao, Y.; Wang, C.; Levy, A.S.; Azizi, P.; Roufaiel, M.; Zhu, S.N.; Neculai, D.; et al. A Novel Assay Uncovers an Unexpected Role for SR-BI in LDL Transcytosis. Cardiovasc. Res. 2015, 108, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Kraehling, J.R.; Chidlow, J.H.; Rajagopal, C.; Sugiyama, M.G.; Fowler, J.W.; Lee, M.Y.; Zhang, X.; Ramírez, C.M.; Park, E.J.; Tao, B.; et al. Genome-Wide RNAi Screen Reveals ALK1 Mediates LDL Uptake and Transcytosis in Endothelial Cells. Nat. Commun. 2016, 7, 13516. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.W.; Zu, X.Y.; Tuo, Q.H.; Chen, L.X.; Lei, X.Y.; Li, K.; Tang, C.K.; Liao, D.F. Caveolae and Caveolin-1 Mediate Endocytosis and Transcytosis of Oxidized Low Density Lipoprotein in Endothelial Cells. Acta Pharmacol. Sin. 2010, 31, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. The LDL Receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, M.; Khan, S. Plasma Lipids as Biomarkers for Alzheimer’s Disease: A Systematic Review. Cureus 2020, 12, e12008. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.M.; Emmerling, M.R.; Bisgaier, C.L.; Essenburg, A.D.; Lampert, H.C.; Drumm, D.; Roher, A.E. Elevated Low-Density Lipoprotein in Alzheimer’s Disease Correlates with Brain Abeta 1-42 Levels. Biochem. Biophys. Res. Commun. 1998, 252, 711–715. [Google Scholar] [CrossRef]
- Qian, C.; Tan, F. Ratio of ApoB/LDL: A Potential Clinical Index for Vascular Cognitive Impairment. BMC Neurol. 2016, 16, 243. [Google Scholar] [CrossRef]
- Wingo, T.S.; Cutler, D.J.; Wingo, A.P.; Le, N.A.; Rabinovici, G.D.; Miller, B.L.; Lah, J.J.; Levey, A.I. Association of Early-Onset Alzheimer Disease with Elevated Low-Density Lipoprotein Cholesterol Levels and Rare Genetic Coding Variants of APOB. JAMA Neurol. 2019, 76, 809–817. [Google Scholar] [CrossRef]
- Lénárt, N.; Szegedi, V.; Juhász, G.; Kasztner, A.; Horváth, J.; Bereczki, E.; Tóth, M.E.; Penke, B.; Sántha, M. Increased Tau Phosphorylation and Impaired Presynaptic Function in Hypertriglyceridemic ApoB-100 Transgenic Mice. PLoS ONE 2012, 7, e0046007. [Google Scholar] [CrossRef] [Green Version]
- Bereczki, E.; Bernat, G.; Csont, T.; Ferdinandy, P.; Scheich, H.; Sántha, M. Overexpression of Human Apolipoprotein B-100 Induces Severe Neurodegeneration in Transgenic Mice. J. Proteome Res. 2008, 7, 2246–2252. [Google Scholar] [CrossRef]
- Hoyk, Z.; Tóth, M.E.; Lénárt, N.; Nagy, D.; Dukay, B.; Csefová, A.; Zvara, Á.; Seprényi, G.; Kincses, A.; Walter, F.R.; et al. Cerebrovascular Pathology in Hypertriglyceridemic APOB-100 Transgenic Mice. Front. Cell. Neurosci. 2018, 12, 380. [Google Scholar] [CrossRef]
- Löffler, T.; Flunkert, S.; Havas, D.; Sántha, M.; Hutter-Paier, B.; Steyrer, E.; Windisch, M. Impact of ApoB-100 Expression on Cognition and Brain Pathology in Wild-Type and HAPPsl Mice. Neurobiol. Aging 2013, 34, 2379–2388. [Google Scholar] [CrossRef]
- Roheim, P.S.; Carey, M.; Forte, T.; Vega, G.L. Apolipoproteins in Human Cerebrospinal Fluid. Proc. Natl. Acad. Sci. USA 1979, 76, 4646–4649. [Google Scholar] [CrossRef]
- Picard, C.; Nilsson, N.; Labonté, A.; Auld, D.; Rosa-Neto, P.; Ashton, N.J.; Zetterberg, H.; Blennow, K.; Breitner, J.C.B.; Villeneuve, S.; et al. Apolipoprotein B Is a Novel Marker for Early Tau Pathology in Alzheimer’s Disease. Alzheimer’s Dement. 2022, 18, 875–887. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064. [Google Scholar] [CrossRef]
- Siegenthaler, J.A.; Sohet, F.; Daneman, R. “Sealing off the CNS”: Cellular and Molecular Regulation of Blood-Brain Barriergenesis. Curr. Opin. Neurobiol. 2013, 23, 1057–1064. [Google Scholar] [CrossRef]
- McFarlane, A. Efficient Trace-Labelling of Proteins with Iodine. Nature 1958, 182, 4627. [Google Scholar] [CrossRef]
- Rohrer, L.; Ohnsorg, P.M.; Lehner, M.; Landolt, F.; Rinninger, F.; von Eckardstein, A. High-Density Lipoprotein Transport through Aortic Endothelial Cells Involves Scavenger Receptor BI and ATP-Binding Cassette Transporter G1. Circ. Res. 2009, 104, 1142–1150. [Google Scholar] [CrossRef]
- Reynolds, G.D.; St Clair, R.W. A Comparative Microscopic and Biochemical Study of the Uptake of Fluorescent and 125I-Labeled Lipoproteins by Skin Fibroblasts, Smooth Muscle Cells, and Peritoneal Macrophages in Culture. Am. J. Pathol. 1985, 121, 200–211. [Google Scholar]
- Radhakrishnan, A.; Goldstein, J.L.; McDonald, J.G.; Brown, M.S. Switch-like Control of SREBP-2 Transport Triggered by Small Changes in ER Cholesterol: A Delicate Balance. Cell Metab. 2008, 8, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, P.G. Endothelial Caveolae and Caveolin-1 as Key Regulators of Atherosclerosis. Am. J. Pathol. 2010, 177, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Andreone, B.J.; Chow, B.W.; Tata, A.; Lacoste, B.; Ben-Zvi, A.; Bullock, K.; Deik, A.A.; Ginty, D.D.; Clish, C.B.; Gu, C. Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron 2017, 94, 581–594.e5. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Gupta, S.; Yi, M.; Michaely, P.; Hobbs, H.H.; Cohen, J.C. ARH Is a Modular Adaptor Protein that Interacts with the LDL Receptor, Clathrin, and AP-2. J. Biol. Chem. 2002, 277, 44044–44049. [Google Scholar] [CrossRef]
- Zhang, X.; Sessa, W.C.; Fernández-Hernando, C. Endothelial Transcytosis of Lipoproteins in Atherosclerosis. Front. Cardiovasc. Med. 2018, 5, 130. [Google Scholar] [CrossRef]
- Tao, B.; Kraehling, J.R.; Ghaffari, S.; Ramirez, C.M.; Lee, S.; Fowler, J.W.; Lee, W.L.; Fernandez-Hernando, C.; Eichmann, A.; Sessa, W.C. BMP-9 and LDL Crosstalk Regulates ALK-1 Endocytosis and LDL Transcytosis in Endothelial Cells. J. Biol. Chem. 2020, 295, 18179–18188. [Google Scholar] [CrossRef]
- Stender, S.; Zilversmit, D.B. Transfer of Plasma Lipoprotein Components and of Plasma Proteins into Aortas of Cholesterol-Fed Rabbits. Molecular Size as a Determinant of Plasma Lipoprotein Influx. Arteriosclerosis 1981, 1, 38–49. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Hjelms, E.; Slender, S.; Kjeldsen, K. Different Efflux Pathways for High and Low Density Lipoproteins from Porcine Aortic Intima. Arterioscler. Thromb. Vasc. Biol. 1990, 10, 477–485. [Google Scholar] [CrossRef]
- Shaikh, M.; Wootton, R.; Nordestgaard, B.G.; Baskerville, P.; Lumley, J.S.; La Ville, A.E.; Quiney, J.; Lewis, B. Quantitative Studies of Transfer in Vivo of Low Density, Sf 12-60, and Sf 60-400 Lipoproteins between Plasma and Arterial Intima in Humans. Arterioscler. Thromb. 1991, 11, 569–577. [Google Scholar] [CrossRef]
- Huang, L.; Chambliss, K.L.; Gao, X.; Yuhanna, I.S.; Behling-Kelly, E.; Bergaya, S.; Ahmed, M.; Michaely, P.; Luby-Phelps, K.; Darehshouri, A.; et al. SR-B1 Drives Endothelial Cell LDL Transcytosis via DOCK4 to Promote Atherosclerosis. Nature 2019, 569, 565–569. [Google Scholar] [CrossRef]
- Frank, P.G.; Pavlides, S.; Cheung, M.W.C.; Daumer, K.; Lisanti, M.P. Role of Caveolin-1 in the Regulation of Lipoprotein Metabolism. Am. J. Physiol.—Cell Physiol. 2008, 295. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Hernando, C.; Yu, J.; Dávalos, A.; Prendergast, J.; Sessa, W.C. Endothelial-Specific Overexpression of Caveolin-1 Accelerates Atherosclerosis in Apolipoprotein E-Deficient Mice. Am. J. Pathol. 2010, 177, 998–1003. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic Heterogeneity of the Endothelium: II. Representative Vascular Beds. Circ. Res. 2007, 100, 174–190. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic Heterogeneity of the Endothelium: I. Structure, Function, and Mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. A Molecular Atlas of Cell Types and Zonation in the Brain Vasculature. Nature 2018, 554, 475–480. [Google Scholar] [CrossRef]
- De Vries, H.E.; Breedveld, B.; Kuiper, J.; De Boer, A.G.; Van Berkel, T.J.C.; Breimer, D.D. High-Density Lipoprotein and Cerebral Endothelial Cells in Vitro: Interactions and Transport. Biochem. Pharmacol. 1995, 50, 271–273. [Google Scholar] [CrossRef]
- Dehouck, B.; Fenart, L.; Dehouck, M.P.; Pierce, A.; Torpier, G.; Cecchelli, R. A New Function for the LDL Receptor: Transcytosis of LDL across the Blood–Brain Barrier. J. Cell Biol. 1997, 138, 877–889. [Google Scholar] [CrossRef]
- Candela, P.; Gosselet, F.; Miller, F.; Buee-Scherrer, V.; Torpier, G.; Cecchelli, R.; Fenart, L. Physiological Pathway for Low-Density Lipoproteins across the Blood-Brain Barrier: Transcytosis through Brain Capillary Endothelial Cells In Vitro. Endothelium 2008, 15, 254–264. [Google Scholar] [CrossRef]
- Molino, Y.; David, M.; Varini, K.; Jabès, F.; Gaudin, N.; Fortoul, A.; Bakloul, K.; Masse, M.; Bernard, A.; Drobecq, L.; et al. Use of LDL Receptor-Targeting Peptide Vectors for in Vitro and in Vivo Cargo Transport across the Blood-Brain Barrier. FASEB J. 2017, 31, 1807–1827. [Google Scholar] [CrossRef]
- Spencer, B.J.; Verma, I.M. Targeted Delivery of Proteins across the Blood-Brain Barrier. Proc. Natl. Acad. Sci. USA 2007, 104, 7594–7599. [Google Scholar] [CrossRef]
- Ramírez, C.M.; Zhang, X.; Bandyopadhyay, C.; Rotllan, N.; Sugiyama, M.G.; Aryal, B.; Liu, X.; He, S.; Kraehling, J.R.; Ulrich, V.; et al. Caveolin-1 Regulates Atherogenesis by Attenuating Low-Density Lipoprotein Transcytosis and Vascular Inflammation Independently of Endothelial Nitric Oxide Synthase Activation. Circulation 2019, 140, 225–239. [Google Scholar] [CrossRef]
- Balazs, Z.; Panzenboeck, U.; Hammer, A.; Sovic, A.; Quehenberger, O.; Malle, E.; Sattler, W. Uptake and Transport of High-Density Lipoprotein (HDL) and HDL-Associated α-Tocopherol by an in Vitro Blood-Brain Barrier Model. J. Neurochem. 2004, 89, 939–950. [Google Scholar] [CrossRef]
- Fung, K.Y.; Wang, C.; Nyegaard, S.; Heit, B.; Fairn, G.D.; Lee, W.L. SR-BI Mediated Transcytosis of HDL in Brain Microvascular Endothelial Cells Is Independent of Caveolin, Clathrin, and PDZK1. Front. Physiol. 2017, 8, 841. [Google Scholar] [CrossRef]
- Rhainds, D.; Brodeur, M.; Lapointe, J.; Charpentier, D.; Falstrault, L.; Brissette, L. The Role of Human and Mouse Hepatic Scavenger Receptor Class B Type I (SR-BI) in the Selective Uptake of Low-Density Lipoprotein-Cholesteryl Esters. Biochemistry 2003, 42, 7527–7538. [Google Scholar] [CrossRef] [PubMed]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of Scavenger Receptor SR-BI as a High Density Lipoprotein Receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Robert, J.; Button, E.B.; Stukas, S.; Boyce, G.K.; Gibbs, E.; Cowan, C.M.; Gilmour, M.; Cheng, W.H.; Soo, S.K.; Yuen, B.; et al. High-Density Lipoproteins Suppress Aβ-Induced PBMC Adhesion to Human Endothelial Cells in Bioengineered Vessels and in Monoculture. Mol. Neurodegener. 2017, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Tran-Dinh, A.; Levoye, A.; Couret, D.; Galle-Treger, L.; Moreau, M.; Delbosc, S.; Hoteit, C.; Montravers, P.; Amarenco, P.; Huby, T.; et al. Article High-Density Lipoprotein Therapy in Stroke: Evaluation of Endothelial Sr-Bi-Dependent Neuroprotective Effects. Int. J. Mol. Sci. 2021, 22, 106. [Google Scholar] [CrossRef]
- Weksler, B.; Romero, I.A.; Couraud, P.O. The HCMEC/D3 Cell Line as a Model of the Human Blood Brain Barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef]
- Dehouck, B.; Dehouck, M.P.; Fruchart, J.C.; Cecchelli, R. Upregulation of the Low Density Lipoprotein Receptor at the Blood-Brain Barrier: Intercommunications between Brain Capillary Endothelial Cells and Astrocytes. J. Cell Biol. 1994, 126, 465–473. [Google Scholar] [CrossRef]
- Napoli, C.; Witztum, J.L.; De Nigris, F.; Palumbo, G.; D’armiento, F.P.; Palinski, W. Intracranial Arteries of Human Fetuses Are More Resistant to Hypercholesterolemia-Induced Fatty Streak Formation Than Extracranial Arteries. Circulation 1999, 99, 2003–2010. [Google Scholar] [CrossRef]
- Labonte, A.; Picard, C.; Nilsson, N.I.V.; Rosa-Neto, P.; Breitner, J.; Villeneuve, S.; Poirier, J. CSF Apolipoprotein B Is Associated with Early Tau Pathology and Selective Activation of a Cytokine Cascade in Cognitively Unaffected Subjects with a Parental History of Alzheimer’s Disease. Alzheimer’s Dement. 2021, 17, 51354. [Google Scholar] [CrossRef]
- Poirier, J.; Nilsson, N.I.V.; Labonté, A.; Rosa-Neto, P.; Ashton, N.J.; Zetterberg, H.; Blennow, K.; Breitner, J.; Villeneuve, S.; Picard, C.; et al. CSF Apolipoprotein B Levels Predict Future Visuospatial Cognitive Decline and Synaptic Pathology in Cognitively Unimpaired Healthy Elderly with a Parental History of Alzheimer’s Disease. Alzheimer’s Dement. 2021, 17, 50342. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kakava, S.; Schlumpf, E.; Panteloglou, G.; Tellenbach, F.; von Eckardstein, A.; Robert, J. Brain Endothelial Cells in Contrary to the Aortic Do Not Transport but Degrade Low-Density Lipoproteins via Both LDLR and ALK1. Cells 2022, 11, 3044. https://doi.org/10.3390/cells11193044
Kakava S, Schlumpf E, Panteloglou G, Tellenbach F, von Eckardstein A, Robert J. Brain Endothelial Cells in Contrary to the Aortic Do Not Transport but Degrade Low-Density Lipoproteins via Both LDLR and ALK1. Cells. 2022; 11(19):3044. https://doi.org/10.3390/cells11193044
Chicago/Turabian StyleKakava, Sofia, Eveline Schlumpf, Grigorios Panteloglou, Flavia Tellenbach, Arnold von Eckardstein, and Jerome Robert. 2022. "Brain Endothelial Cells in Contrary to the Aortic Do Not Transport but Degrade Low-Density Lipoproteins via Both LDLR and ALK1" Cells 11, no. 19: 3044. https://doi.org/10.3390/cells11193044
APA StyleKakava, S., Schlumpf, E., Panteloglou, G., Tellenbach, F., von Eckardstein, A., & Robert, J. (2022). Brain Endothelial Cells in Contrary to the Aortic Do Not Transport but Degrade Low-Density Lipoproteins via Both LDLR and ALK1. Cells, 11(19), 3044. https://doi.org/10.3390/cells11193044