1. Introduction
Huntington’s Disease (HD) is an autosomal dominant progressive neurodegenerative disorder that affects mainly the striatum (caudate and putamen) and later the cortex [
1]. HD is characterized by psychiatric and behavioral disturbances, such as motor impairment, and psychiatric symptoms such as obsessive-compulsive disorder, depression and/or anxiety, cognitive decline and weight loss [
2,
3]. HD is caused by an abnormal expansion of cytosine-adenine-guanine (CAG) repeat at the
HTT gene, encoding for mutant huntingtin (mHTT) retaining a polyglutamine (polyQ) extension at the N-terminus [
4]. mHTT has been associated with protein conformational changes, aggregation, and abnormal protein–protein interactions [
2], which cause cytotoxicity, evidenced through changes in gene transcription, synaptic dysfunction,
N-methyl-D-aspartate receptors (NMDARs) overactivation, decreased mitochondrial calcium (Ca
2+) handling and organelle dysfunction, and increased oxidative events, leading to neuronal death [
4]. NMDARs involvement in HD pathogenesis is not completely clear. HD-affected medium spiny neurons (MSNs) show altered NMDAR currents as well as augmented ratio of surface to internal GluN2B-containing NMDARs, with augmented accumulation at extrasynaptic sites [
5]. Nonetheless, the mechanism underlying this altered NMDARs localization is largely unknown.
NMDARs are important postsynaptic receptors that contribute to normal synaptic function and cell survival. Activation of synaptic NMDARs is associated with augmented cAMP response element-binding protein (CREB)-dependent gene expression. CREB is a signal-regulated transcription factor, important for neuronal survival with important roles in several processes, namely synaptic plasticity, neurogenesis, learning, and memory. Conversely, extrasynaptic NMDARs activation promotes cell death pathways linked to dendritic blebbing, loss of mitochondrial membrane potential, and CREB shut-off pathway, with blockade expression of brain-derived neurotrophic factor (BDNF), a neurotrophin relevant for survival of striatal neurons (e.g., 15). Altered NMDAR function has been related to HD, as documented by previous studies. Milnerwood and colleagues showed that YAC128 HD mouse striatum presented increased extrasynaptic NMDAR expression and currents and reduced nuclear CREB activation, which was reversed after NMDAR inhibition with memantine [
5].
Interestingly, intracellular mechanisms involved in HD pathogenesis, as altered striatal glutamatergic synapses and mitochondrial dysfunction linked to redox changes, are also relevant neuronal pathways modulated by c-Src and Fyn, two ubiquitous proteins belonging to the Src kinase family (SKF), predominantly located in synaptic membranes [
6]. c-Src and Fyn are broadly expressed in the Central Nervous System (CNS), being enriched in striatal neurons [
7], and have been implicated in brain neuronal development, transmission, synaptic activity, and plasticity in mammalian CNS, being both kinases activated by hydrogen peroxide (H
2O
2) [
8]. Importantly, Fyn can be found in the postsynaptic density (PSD) concomitantly with components of the NMDAR complex [
9]. Fyn interacts and phosphorylates NMDARs and postsynaptic scaffold proteins, as PSD95, to regulate synaptic transmission and plasticity [
9]. Moreover, Fyn phosphorylates NMDAR GluN2B subunit at Tyr1472, increasing receptor retention at the synapse, thus controlling synaptic plasticity [
10,
11]. Several studies evidenced depressed synaptic transmission in HD. Importantly, there is a clear evidence for altered dendrite morphology in MSNs of HD patients, showing recurved endings and appendages, altered spine density and abnormalities in dendritic spine size and shape [
12]. Moreover, Murmu and coworkers showed that mHTT in R6/2 mice causes a progressive loss of persistent-type spines, important for neuronal circuitry and long-term memory in the brain [
13,
14]. Recently, we showed that c-Src/Fyn activation and total protein levels are reduced in several human and mouse HD models mainly due to autophagy degradation [
15]. Moreover, restoration of active SKF levels improves mitochondrial morphology and function, namely through improved mitochondrial transmembrane potential, mitochondrial basal respiration, and ATP production, diminishing ROS levels [
15]. Additionally, YAC128 mouse striatum revealed increased synaptic activity of striatal-enriched protein tyrosine phosphatase (STEP), correlating with decreased GluN2B phosphorylation at Tyr1472, reducing synaptic NMDARs by facilitating their movement to extrasynaptic sites [
16].
Based on these findings, in this study we aimed to explore the impact of HD-mediated altered Fyn levels on synaptic versus non-synaptic NMDARs function and localization, as well as intracellular neuroprotective pathways. Our data reveal that SKF activation is important for normal synaptic function and neuronal survival in neurons expressing mHTT by contributing for synaptic NMDARs presence and function. Thus, synaptic modulation of Src/Fyn activation/levels may constitute a therapeutic potential target in HD.
2. Materials and Methods
2.1. YAC128 Mice
YAC128 mice, previously described by Slow and co-authors (2003) [
17], express full- length mutant HTT with 128 CAG repeats from a yeast artificial chromosome (YAC) transgene (RRID: MGI_MGI:3613515). YAC128 (line HD53) and wild-type mice were housed in the animal facility of the Center for Neuroscience and Cell Biology and Faculty of Medicine at the University of Coimbra (Coimbra, Portugal), under controlled temperature (22–23 °C) and a 12 h light/12 h dark cycle with lights on at 07:00 h. Food and water were available ad libitum throughout the experiment. Animal experiments in this study were performed in accordance with the European Community directive (2010/63/EU) and protocols approved by the Faculty of Medicine, University of Coimbra (ORBEA_189_2018/11042018). All efforts were made to minimize animal suffering and to reduce the number of animals used. Animals were used at 3, 6, and 12 months of age.
2.2. Primary Striatal Neurons from YAC128 Mice
Primary striatal neurons were prepared as described previously [
18], with some minor modifications. At 16 days of gestation, pregnant female mice were sacrificed by cervical dislocation following anesthesia using (RS)-2-chloro-2-(difluoromethoxy)-1,1,1-trifluoro-ethane. Striata were dissected out from fetal mice and cells were separated by mechanical digestion using a pipette in Ca
2+- and Mg
2+-free Hank’s balanced salt solution containing 137 mM NaCl, 5.36 mM KCl, 0.44 mM KH
2PO
4, 0.34 mM Na
2HPO
4.2H
2O, 5 mM glucose, 1 mM sodium pyruvate, and 10 mM HEPES, at pH 7.2. Cells were plated at a density of 8.4 × 10
4 cells/cm
2 in poly-D-lysine-coated 6-well or 96-well plates, and at a density of 4.2 × 10
4 cells/cm
2 in poly-D-lysine coated glass coverslips for immunocytochemistry. Cells were cultured for 12 days in Neurobasal medium supplemented with 2% B27, 0.5 mM glutamine and 0.12 mg/mL gentamicin, at 95% air and 5% CO
2. To reduce glia growth, 10 μM of the mitotic inhibitor 5-fluorodeoxyuridine (5-FDU, Sigma, #F0503, St. Louis, MI, USA) was added to the culture after 72 h in culture. One half of the medium was changed with fresh medium without 5-FDU at day 7.
2.3. Constructs and Neuron Transfection
Cells were transfected with pLNCX chick SKFY527F (Addgene, plasmid #13660), empty pLNCX vector, and GFP (Origene, Rockville, MD, USA, GFP; #PS100010). Empty vector pLNCX was obtained from the SKFY527F plasmid using the restriction enzyme digestion ClaI (BioLabs, #Ro197L) according to manufacturer’s protocol. The result of digestion was visualized in 1% agarose gel; the band corresponding to the empty vector was cropped and DNA was extracted using the NucleoSpin® Gel and PCR Clean-up (Macherey-Nagel, #740609) according to the manufacturer’s protocol. Then, the blunt end and cohesive end termini of the resulting empty vector were joined using T4 DNA Ligase enzyme (BioLabs, #M0202S). Transfection was performed at 8 DIV using the Ca2+ phosphate precipitation method. Briefly, plasmid was diluted in TE (1 mM Tris-HCl pH 7.3, 1 mM EDTA), followed by the addition of CaCl2 (2.5 M CaCl2 in 10 mM HEPES, pH 7.2). The DNA solution was carefully added to 2× HEBS (12 mM dextrose, 50 mM HEPES, 10 mM KCl, 280 mM NaCl, and 1.5 mM Na2HPO4.2H2O, pH 7.2) while bubbling air through the solution with a micropipette. The mixture was then incubated for 25 min at room temperature. The precipitates were added dropwise to the coverslips in Neurobasal medium and incubated for 80 min, at 37 °C. The DNA–Ca2+-phosphate precipitates were dissolved in freshly made dissolution medium (Neurobasal medium with 20 mM HEPES, pH 6.8) and incubated for 7 min at room temperature. The transfected neurons were then washed with Neurobasal medium and transferred back to their original dishes containing conditioned culture medium.
2.4. Sample Preparation and Western Blotting
Total extracts were obtained from primary striatal neurons or striatal or cortical brain areas from YAC128 mice at 3, 6, or 12 months of age. The cells were scraped and brain areas suspended in Ripa buffer (containing 150 mM NaCl, 50 mM Tris HCl, 5 mM EGTA, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate, pH 7.5) supplemented with 100 nM okadaic acid, 1 mM PMSF, 25 mM NaF, 1 mM Na3VO4, 1 mM DTT, and 1 μg/mL protease inhibitor cocktail (chymostatin, pepstatin A, leupeptin and antipain). Total homogenates were lysed in an ultrasonic bath (UCS 300-THD; at heater power 200 W and frequency 45 kHz) during 10 s and centrifuged at 4 °C for 10 min at 20,800× g to remove cell debris. The supernatant was collected, and protein content was determined using the Bio-Rad protein assay reagent based on the Bradford dye-binding procedure (Bio-Rad, Hercules, CA, USA). Then, protein extracts were denatured with 6× concentrated loading buffer (containing 300 mM Tris-HCl pH 6.8, 12% SDS, 30% glycerol, 600 mM DTT, 0.06% bromophenol blue) at 95 °C, for 5 min. Equivalent amounts of protein samples (15 μg–30 μg) were separated by 8–12% SDS-PAGE and electroblotted onto polyvinylidene difluoride (PVDF) membrane (Millipore, Burlington, MA, USA). Membranes were further blocked with 5% (w/v) BSA (Santa Cruz Biotechnology, Santa Cruz, CA, USA) in Tris Buffered Saline ((TBS) containing 250 mM, 150 mM NaCl, pH7.6) plus 0.1% Tween 20 before incubation with the specific antibody against: GluN2A (1:1000, Millipore, Burlington, MA, USA, #07-632), p(Tyr1472)GluN2B (1:1000, Cell Signaling, Danvers, MA, USA, #4208S), GluN2B C-terminal (1:1000, Millipore, Burlington, MA, USA, MAB #5778), βActin (1:5000, Sigma, St. Louis, MI, USA, #A5316) overnight, at 4 °C. βActin was used as a control of protein loading of total extracts. An anti-rabbit (1:20,000; Thermo Fisher Scientific, Waltham, MA, USA, #31340) or anti-mouse (1:20,000; Thermo Fisher Scientific, Waltham, MA, USA #31320) IgG secondary antibodies conjugated to the alkaline phosphatase, prepared in 1% (w/v) BSA in TBS-T, were used for 1 h at room temperature. Immunoreactive bands were visualized by alkaline phosphatase activity after incubation for 5 min with ECF reagent (GE Healthcare Bio-Sciences, Piscataway, NJ, USA) on Bio-Rad ChemiDoc Touch Imaging System (Bio-Rad, Hercules, CA, USA) and quantified using Image Lab analysis software (Bio-Rad, Hercules, CA, USA).
2.5. Immunocytochemistry
To assess Fyn, P(Tyr416)SKF, GluN2B, or P(Tyr1472)GluN2B levels, primary neurons from WT and YAC128 mice were cultured on glass coverslips. When applicable, 24 h after transfection, cells were fixed with 4% paraformaldehyde (pre-warmed at 37 °C) for 20 min and permeabilized in 0.1% Triton X-100 in PBS for 2 min. Then, cells were blocked for 1 h at room temperature with 3% (w/v) BSA in PBS and further incubated with the primary antibody prepared in blocking solution, overnight, at 4 °C (antibodies referred above, and against CREB (Abcam, Cambridge, UK, #ab32515), P(Ser133)CREB (Cell Signaling, Danvers, MA, USA, #9196), PSD-95 (Thermo Scientific, Waltham, MA, USA #7E3-1B8), and caspase3-cleaved (Cell Signaling, Danvers, MA, USA #9664), at 1:200). Cells were then washed with PBS and incubated with the adequate secondary antibody (Alexa Fluor-594 goat anti-rabbit (Invitrogen, Carlsbad, CA, USA #R37117), Alexa Fluor-488 donkey anti-rabbit (Invitrogen, Carlsbad, CA, USA #R37118) and Alexa Fluor-488 donkey anti-mouse (Invitrogen, Carlsbad, CA, USA #R37114)) at 1:300 in blocking solution for 1 h at room temperature. Nuclei were stained with 1 μg/mL Hoechst 33342 in PBS (Invitrogen, Carlsbad, CA, USA) for 10 min and coverslips were mounted using Mowiol 40-88 (Sigma Chemical, St. Louis, MI, USA). Confocal images were obtained using a Plan-Apochromat/1.4NA 63x lens on an Axio Observer.Z1 confocal microscope (Zeiss Microscopy, Oberkochen, Germany) with Zeiss LSM 710 software.
2.6. Electrophysiological Recordings
NMDA-induced currents were recorded in transfected primary striatal neurons (DIV 11) at −60 mV by whole-cell patch clamping using an AxonPatch 200B amplifier (Molecular Devices, San Jose, CA, USA). The borosilicate glass micropipettes used had a resistance of 4–6 MΩ and were filled with the following internal solution (in mM): CsMeSO4 130, CsCl 10, CaCl2 0.5, EGTA 5, HEPES 10, and NaCl 10 (pH 7.3 adjusted with CsOH). Cells were perfused with extracellular solution containing 140 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, 10 mM HEPES, and 15 mM glucose supplemented with 10 μM glycine (pH 7.4 adjusted with NaOH). NMDA (100 μM) was diluted in the extracellular solution and rapidly perfused with a six-channel perfusion valve control system VC-77SP/perfusion fast-step SF-77B (Warner Instruments, Hamden, CT, USA). All experiments were performed at RT (22–25 °C). The currents were filtered at 1 kHz (4-pole low-pass Bessel filter) and digitized at a sampling rate of 10 kHz to a personal computer and analyzed with pClamp 10.7 software (Molecular Devices, San Jose, CA, USA).
2.7. Measurement of Intracellular Calcium Levels
Twenty-four hours after transfection with empty or empty+ SKFY527F, primary striatal neurons were incubated in experimental media (in mM: 132 NaCl, 4 KCl, 1 CaCl2, 1.2 NaH2PO4.H2O, 1.4 MgCl2, 6 Glucose, 10 HEPES, pH 7.4) plus 2 μM Fluo4-AM (Thermo Fisher Sci., #F14201) for 45 min, at 37 °C. Cells were then washed, and the experiment was recorded in experimental media without Mg2+ and supplemented with glycine (20 μM) and serine (30 μM). Fluo4 fluorescence was monitored before and after exposure to 100 μM NMDA in primary striatal neurons from WT and YAC128 mice, using an Axio Observer Z1 system, a fully motorized inverted widefield microscope (Zeiss, Jena, Germany) equipped with a large stage incubator for temperature and humidity control and EC plan-neofluar/1.3NA 63x lens. Fluo4 fluorescence was imaged along time at 494 nm excitation and 506 nm emission, respectively. Fluorescence intensities were calculated using Fiji software (Zurich, Switzerland).
2.8. Statistical Analyses
Data were analyzed by using Excel (Microsoft, Seattle, WA, USA) and GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA) software, and are expressed as the mean ± S.E.M. of the number of independent experiments or cells indicated in figure legends. Comparisons among multiple groups were performed by one-way ANOVA followed by the Bonferroni or Dunnett’s nonparametric Multiple Comparison post-hoc tests or by two-way ANOVA, followed by Sidak’s Multiple Comparison as post-hoc test. Unpaired non-parametric Mann–Whitney test was also performed for comparison between two Gaussian populations, when applicable, as described in figure legends. Significance was defined as p < 0.05.
4. Discussion
NMDARs are important synaptic receptors that contribute to normal synaptic function, cell survival, learning, and memory [
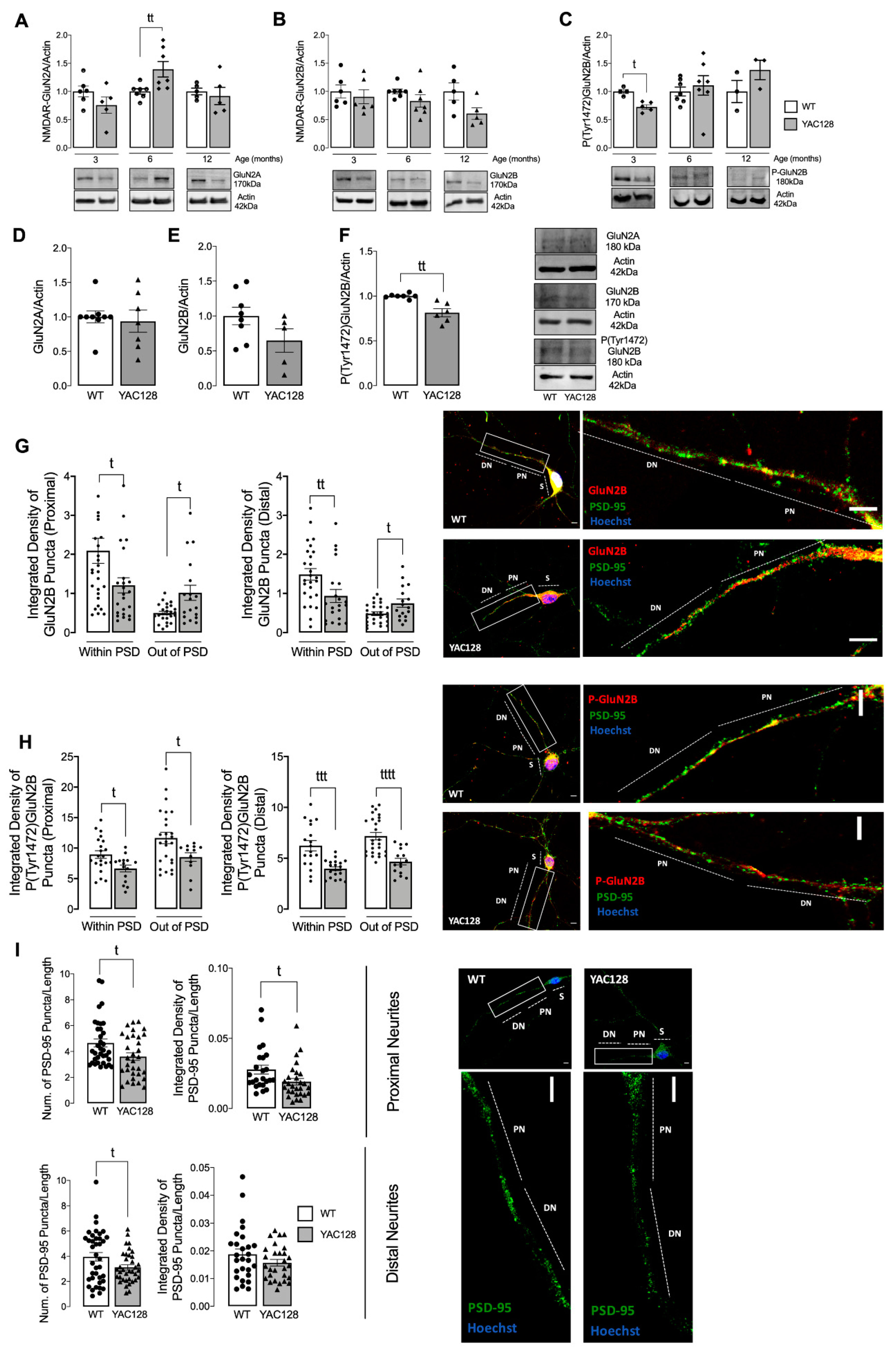
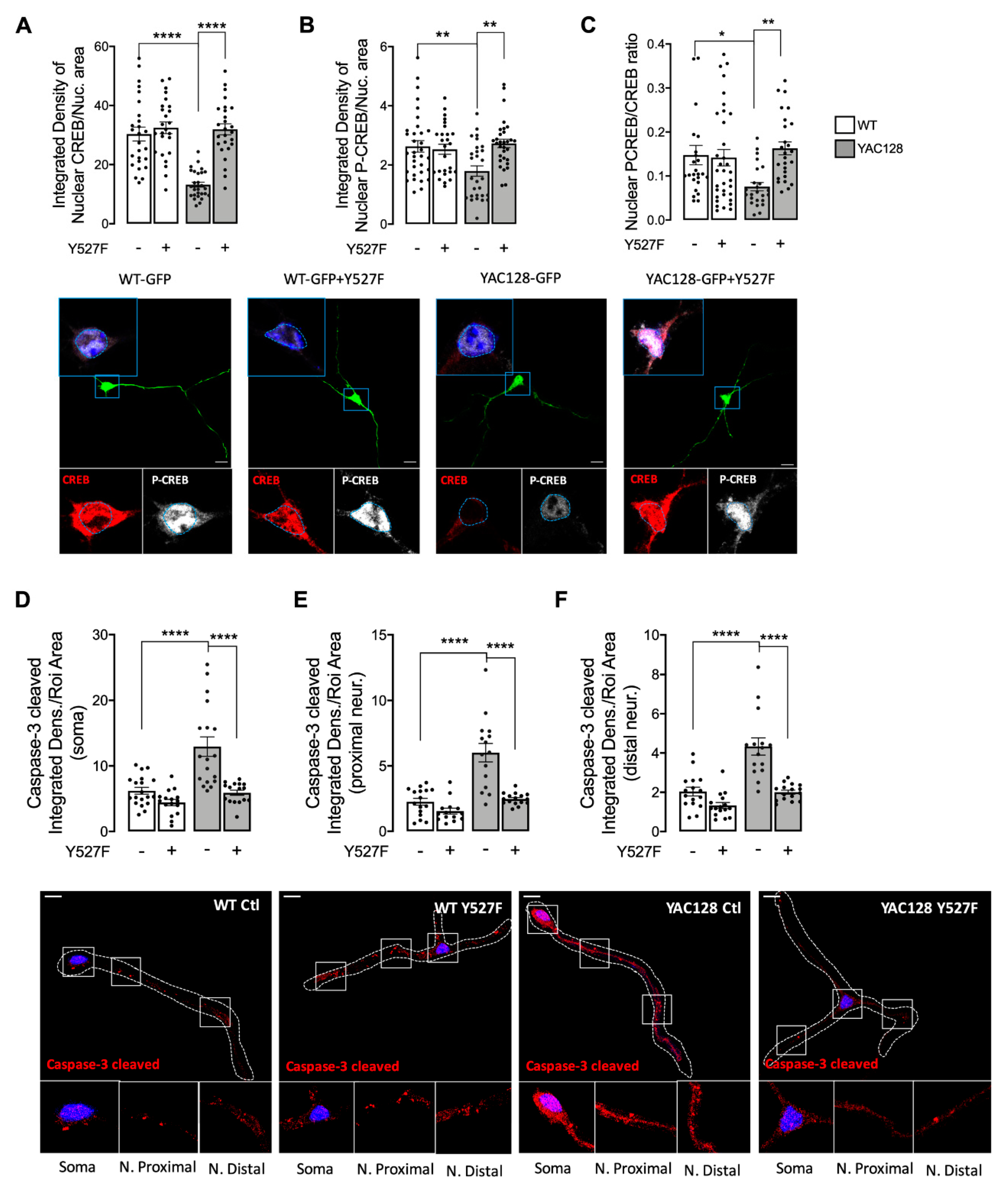
28]. Our study shows augmented extrasynaptic GluN2B-composed NMDARs in HD mouse neurons (
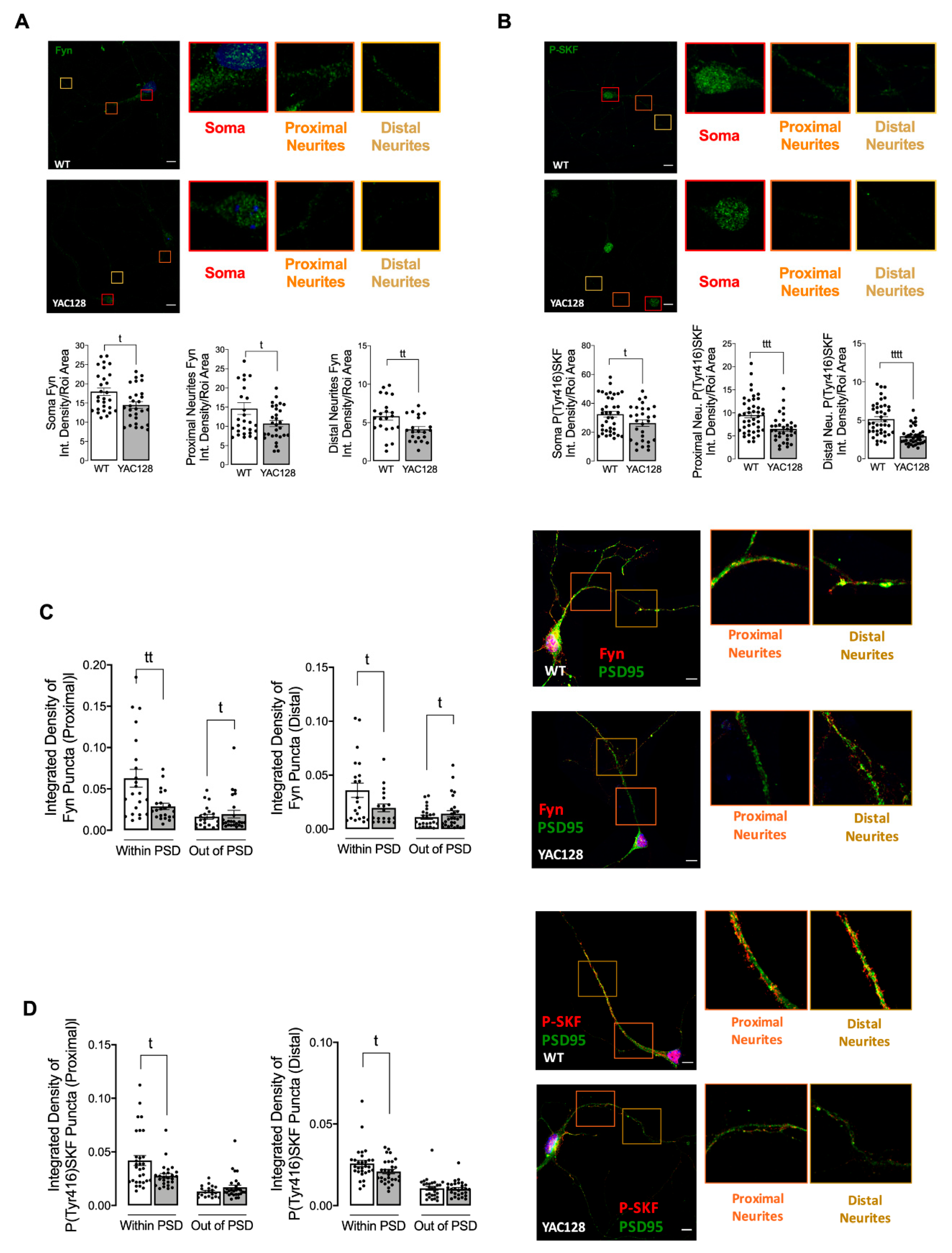
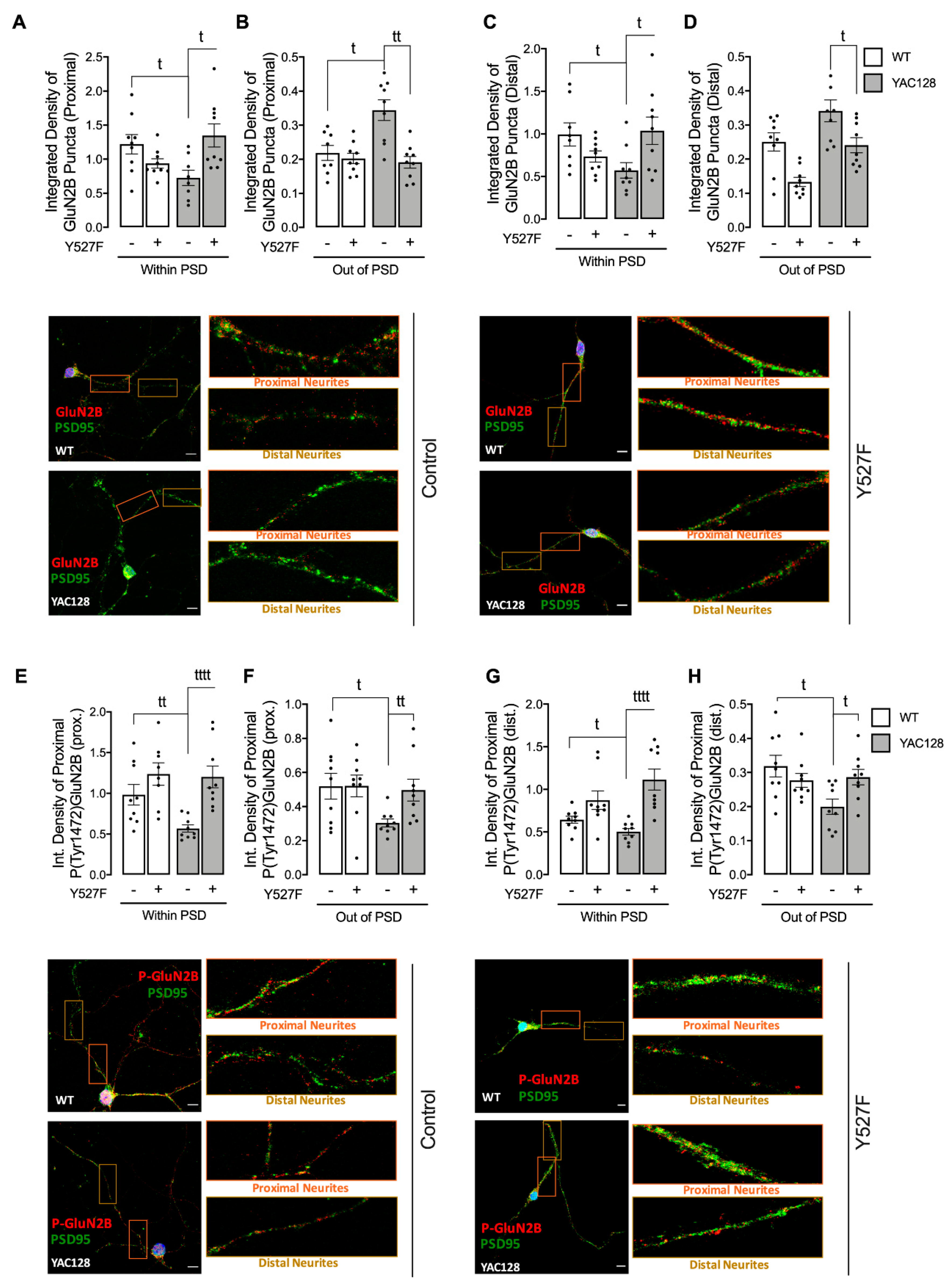
Figure 1), while expression of a constitutive active form of SKF contributed to reestablish NMDARs localization and activity at PSD (
Figure 3). Indeed, reduced Fyn activity in YAC128 PSDs (
Figure 2) contributes to reduced GluN2B phosphorylation at Tyr1472, which decreases synaptic NMDAR retention and may facilitate movement to extrasynaptic sites (
Figure 4). Apart from restoring NMDARs phosphorylated levels at PSD, enhanced SKF levels and activity promote CREB activation and reduce apoptosis in YAC128 primary striatal neurons (
Figure 5).
Previous studies showed that GluN2B-composed NMDARs function and trafficking are altered in HD [
5,
29,
30,
31]. Importantly, synaptic or extrasynaptic NMDARs activation are related to survival or apoptosis activation, respectively [
32]. Striatal neurons showed faster NMDAR trafficking to the surface membrane induced by mHTT [
29], whereas this receptor accumulated at extrasynaptic sites of YAC128 mice at early age [
5]. Concordantly, our results evidence a reduction in GluN2B-composed NMDARs levels in PSD, whereas these levels were augmented at extrasynaptic sites is YAC128 mouse striatal neurons. Moreover, in accordance with our data, other studies showed augmented NMDARs currents in HD mouse models [
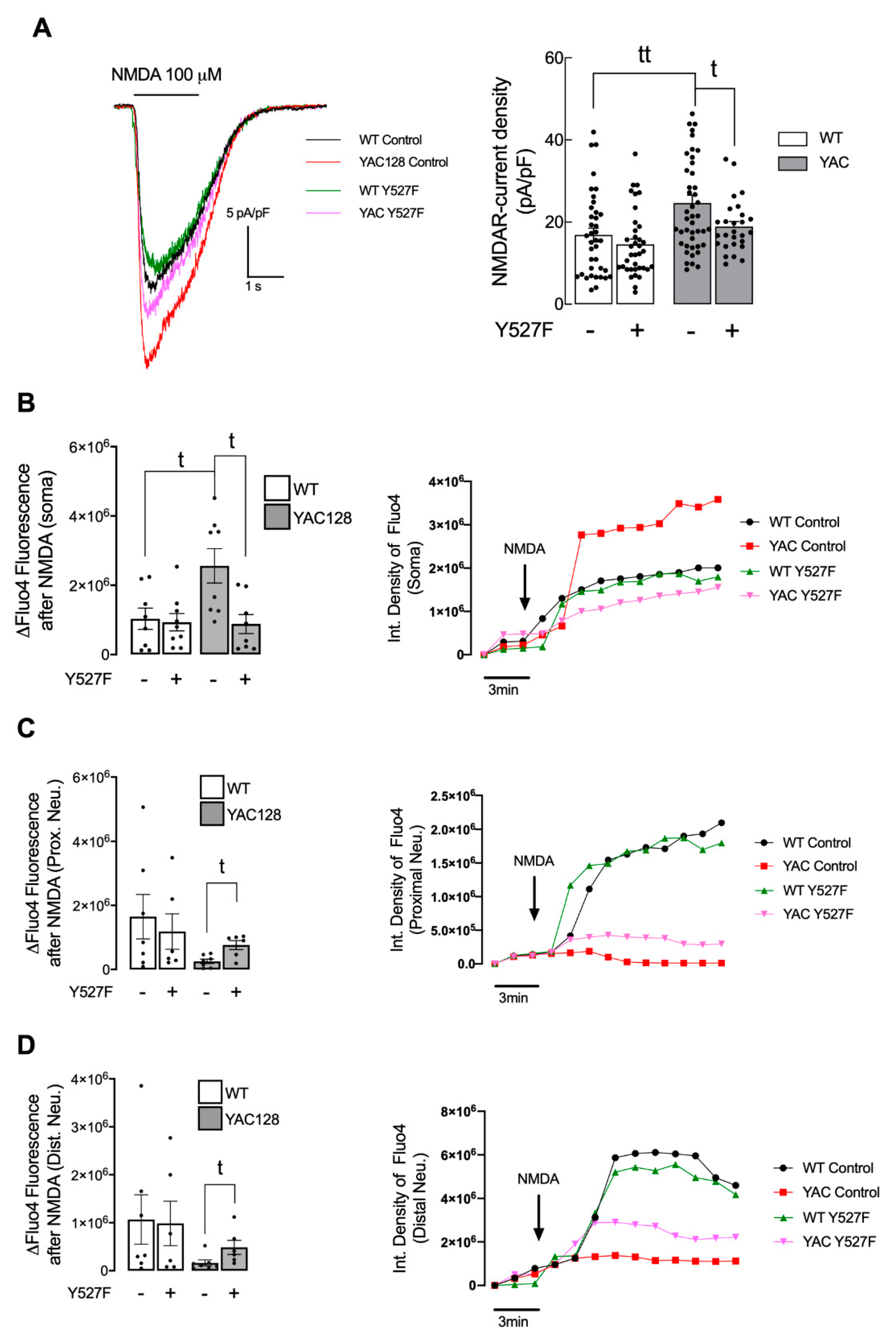
5]. However, this alteration is only measured in HD neuronal soma. In contrast, in proximal and distal neurites, altered PSD number, as well as reduced SKF levels and activity may result in abnormal synaptic function in HD, which may explain the reduction in NMDARs activity. Importantly, altered NMDARs function in proximal and distal neurites can be partially restored by augmenting active SKF levels.
Additionally, GluN2B Tyr1472 phosphorylation levels were decreased both in synaptic and extrasynaptic sites, which is in accordance with previous studies. Gladding and coworkers showed increased synaptic STEP activity in YAC128 striatum, correlated with decreased Tyr1472-GluN2B phosphorylation in YAC128 non-PSD and PSD fractions similar to what we observed in the present study, which facilitates NMDAR movement to extrasynaptic sites, reducing synaptic NMDAR retention [
16]. Indeed, GluN2B-composed NMDAR Tyr1472 phosphorylation by SKF is associated with enrichment of synaptic NMDARs [
6], indicating that NMDAR lateral diffusion between synaptic and extrasynaptic sites is modulated by Tyr1472 phosphorylation. We previously showed that SKF members, specifically c-Src and Fyn proteins, are reduced in several HD models due to augmented degradation by autophagy [
15]. Concordantly with these data, here we show that Fyn total and phosphorylated levels are reduced at both PSD and non-PSD compartments, which may contribute to decrease Tyr1472-GluN2B phosphorylation at PSD and thus reduce synaptic NMDAR retention.
Fyn influence and its fundamental role in PSD has long been recognized. Several studies have previously shown c-Src and Fyn roles in synapse development, plasticity, learning, and memory [
22,
23]. Takasu and colleagues showed that Ca
2+ influx mediated by NMDARs activation upon its Tyr1472 phosphorylation by SKF resulted in gene transcription required for the remodeling of synaptic connections in excitatory synapses of primary cortical neurons [
33]. In the present study we show, for the first time, that expression of a constitutive active form of SKF reestablishes NMDARs localization and function at PSD in YAC128 mouse striatal neurons, restoring neuronal function.
Decreased synaptic NMDARs and augmented extrasynaptic currents are associated with reduced nuclear CREB activation, reduced neuronal expression of the survival factor BDNF, and caused dysfunctional mitochondria, low energy levels, and cell death in HD mouse striatum, before and after phenotype onset [
5]. Extrasynaptic NMDARs activity is linked to cell death signaling cascades due to reduced phosphorylation/activation of CREB [
26]. Concordantly with previous studies showing decreased CREB activation in striatum of 1 and 4 months of age YAC128 mice [
5], our results show reduced nuclear CREB activation in YAC128 primary striatal neurons (
Figure 5A,B). Interestingly, decreased CREB activity and augmented active caspase-3 found in YAC128 striatal neurons were reversed following expression of the constitutive active form of SKF, SKF
Y527F (
Figure 5), previously shown by our group to be neuroprotective against mHTT-induced mitochondrial dysfunction and enhanced ROS levels [
15].
Our study evidence, for the first time, that Fyn protein has an important role in synaptic GluN2B-composed NMDARs phosphorylation and activity, restoring CREB activation and decreasing caspase-3 levels, indicative of a decrease in cell death by apoptosis in HD. Decreased Fyn PSD co-localization correlates with HD-related reduced Tyr1472 GluN2B phosphorylation and augmented extrasynaptic NMDARs currents, as well as decreased CREB activation and cell death. Interestingly, reestablished active SKF levels partially restored NMDARs currents, NMDAR-dependent Ca2+ levels, CREB activation, and reduced caspase-3 cleavage. Hence, we describe a potential mechanism involving Fyn restored levels and activity that may constitute a potential HD therapeutic target to promote striatal glutamatergic synaptic function and survival.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




