
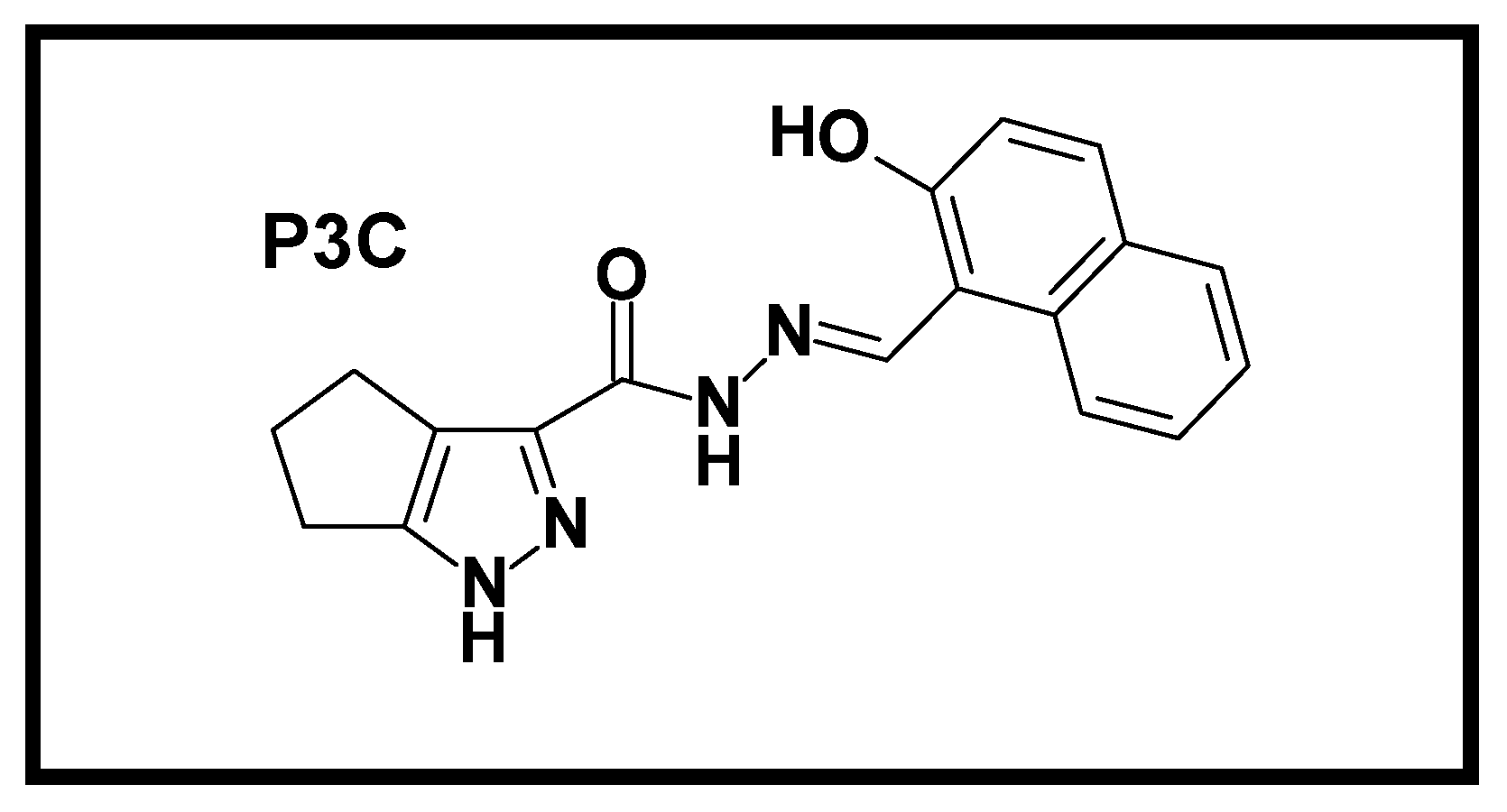
Identification of a Potent Cytotoxic Pyrazole with Anti-Breast Cancer Activity That Alters Multiple Pathways

 , , , , , ,
, , , , , ,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell lines and Culture Conditions
2.2. Differential Nuclear Staining (DNS) Assay
2.3. Cell Viability Analyses
2.4. Cytotoxic Concentration 50% (CC50) and Selective Cytotoxicity Values
2.5. Phosphatidylserine (PS) Distribution in Cell Membranes
2.6. Caspase-3/7 Activation Detection Assay
2.7. Intracellular Caspase-8 Activation Assay
2.8. Detection of Reactive Oxygen Species (ROS)
2.9. Mitochondrial Depolarization Assay
2.10. PARP-1 Cleavage Detection Assay
2.11. Analysis of the Cell Cycle Progression
2.12. Scratch-Wound Assay to Determine Cell Metastatic/Invasion Activity Using Live-Cell Microscopy
2.13. Cytoskeleton Analysis via Confocal Immunofluorescent Microscopy
2.14. Kinase Multiplex Luminex Assays
2.15. Whole Transcriptome Analysis via Next-Generation Sequencing
2.16. RNA-seq Data Analysis
2.17. Gene Ontology (GO), and Ingenuity Pathway Analyses (IPA)
2.18. Statistical Analyses
3. Results
3.1. Drug Screening Identified a Compound with Potent Cytotoxic Activity on a Panel of 27 Human Cancer Cell Lines Derived from Different Tissues
3.2. P3C Induces Cell Death via Apoptosis as Determined by PS Externalization and Caspase Activation
3.3. Induction of Reactive Oxygen Species (ROS) and Mitochondrial Depolarization by P3C
3.4. P3C Induces PARP Cleavage in MDA-MB-231 Cells
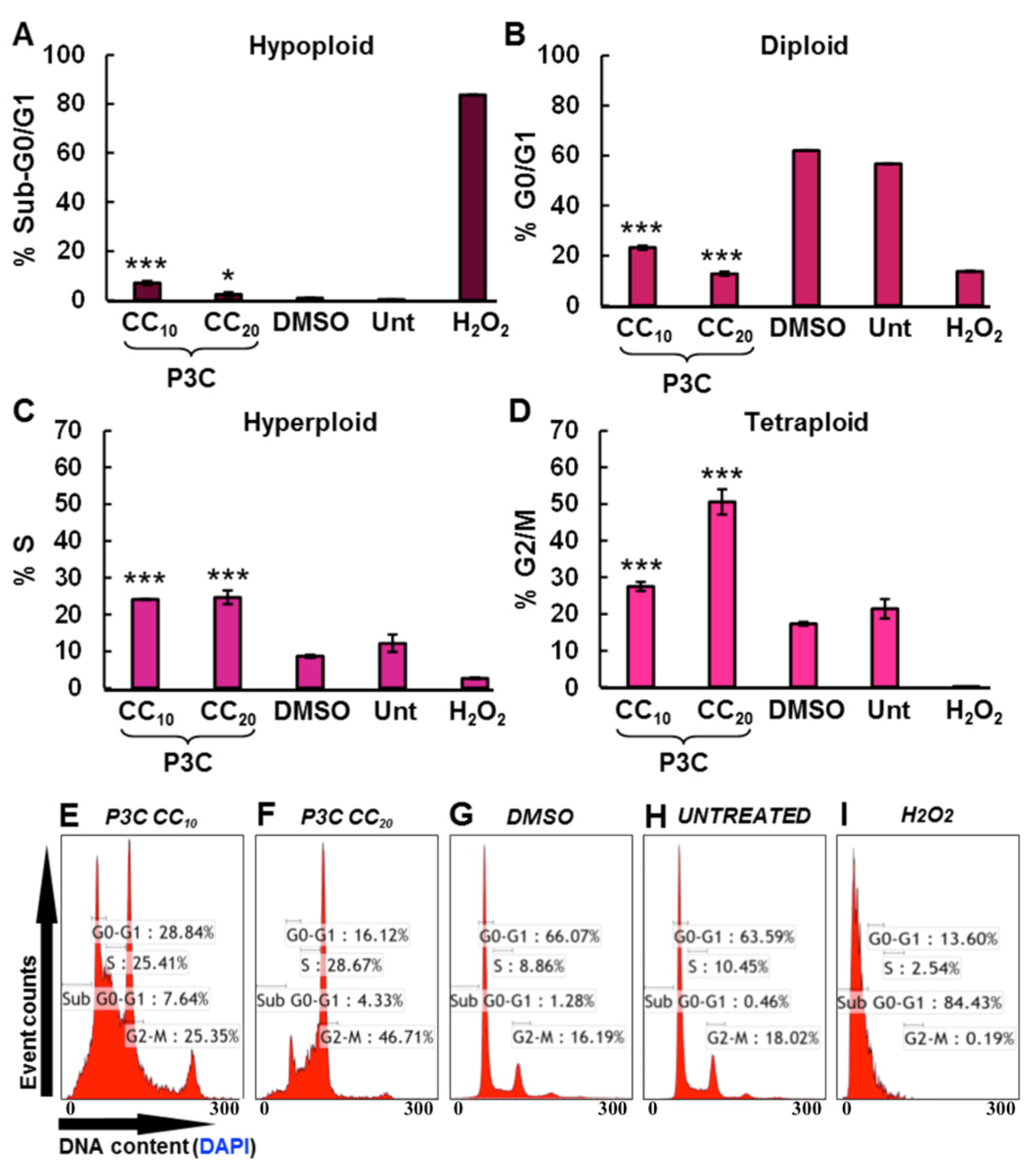
3.5. P3C Disrupts Cell Cycle Progression by Arresting Cells in S and G2-M Phases
3.6. P3C Reduced Cell Proliferation by Inhibiting Cell Migration/Invasion Activity
3.7. P3C Treatment Leads to Microtubule Structure Disruption
3.8. P3C Perturbs and Inhibits Microtubule Mitotic Spindle Formation in HeLa Cells
3.9. P3C Decreased CREB, p38, ERK, Akt, P70S6K, and STAT3 and Increased JNK and NF-kB Phosphorylation in MDA-MB-231 Cells
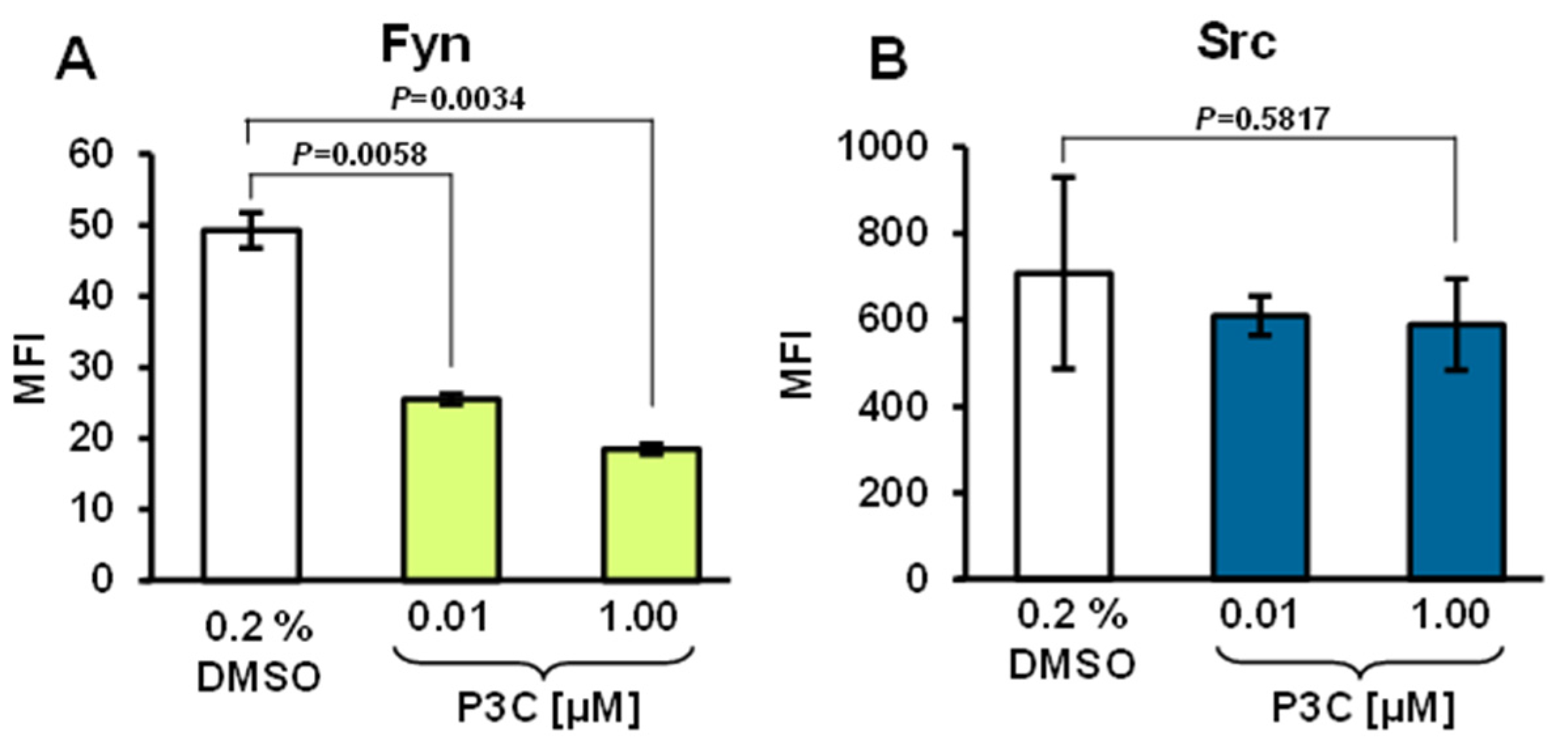
3.10. P3C Treatment Resulted in Fyn Tyrosine Kinase Dephosphorylation
3.11. Transcriptome Analysis of P3C-Treated MDA-MB-231 and MDA-MB-468 Cells Revealed 28 Differentially Expressed Genes in Common
3.12. In P3C-Treated MDA-MB-231 and MDA-MB-468 Cells, the Common DEGs Are Implicated in Apoptosis, Stress Response, MAPK Kinases Inactivation, and Microtubule Structure and Stability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Cancer. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 5 June 2020).
- American Cancer Society. Global Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2018. [Google Scholar]
- Khan, M.; Alam, M.; Verma, G.; Akhtar, W.; Akhter, M.; Shaquiquzzaman, M. The therapeutic voyage of pyrazole and its analogs: A review. Eur. J. Med. Chem. 2016, 120, 170–201. [Google Scholar] [CrossRef]
- Li, Y.; Li, C.; Liu, J.; Guo, M.; Zhang, T.; Sun, L.; Zheng, C.; Piao, H.-R. Synthesis and biological evaluation of 1,3-diaryl pyrazole derivatives as potential antibacterial and anti-inflammatory agents. Bioorg. Med. Chem. 2015, 25, 5052–5057. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, Z.; Bai, F.; Wang, X.; Zhang, X.; Xing, Y. Molecular design and the optimum synthetic route of the compounds with multipyrazole and its derivatives and the potential application in antibacterial agents. Polyhedron 2015, 2015, 59–70. [Google Scholar] [CrossRef]
- Manvar, D.; Pelliccia, S.; La Regina, G.; Famiglini, V.; Coluccia, A.; Ruggieri, A.; Anticoli, S.; Lee, J.; Basu, A.; Cevik, O.; et al. New 1-phenyl-5-(1H-pyrrol-1-yl)-1H-pyrazole-3-carboxamides inhibit hepatitis C virus A. Shafiee, New tetracyclic tacrine analogs containing pyrano[2,3-c]pyrazole: Efficient synthesis, biological assessment and docking simulation study. Eur. J. Med. Chem. 2015, 89, 269–303. [Google Scholar]
- Bekhit, A.; Hassan, A.; Abd El Razik, H.A.; El-Miligy, M.; El-Agroudy, E.J.; Bekhit, A.E.D.A. New heterocyclic hybrids of pyrazole and its bioisosteres: Design, synthesis and biological evaluation as dual acting antimalarial-antileishmanial agents. Eur. J. Med. Chem. 2015, 94, 30–44. [Google Scholar] [CrossRef]
- Barcelo, M.; Ravina, E.; Masaguer, C.; Dominguez, E.; Areias, F.; Brea, J.; Loza, M. Synthesis and binding affinity of new pyrazole and isoxazole derivatives as potential atypical antipsychotics. Bioorg. Med. Chem. 2007, 17, 4873–4877. [Google Scholar] [CrossRef] [PubMed]
- Khloya, P.; Ceruso, M.; Ram, S.; Supuran, C.; Sharma, P.K. Sulfonamide bearing pyrazolylpyrazolines as potent inhibitors of carbonic anhydrase isoforms I, II, IX and XII. Bioorg. Med. Chem. 2015, 25, 3208–3212. [Google Scholar] [CrossRef]
- Hasui, T.; Ohyabu, N.; Ohra, T.; Fuji, K.; Sugimoto, T.; Fujimoto, J.; Asano, K.; Oosawa, M.; Shiotani, S.; Nishigaki, N.; et al. Discovery of 6-[5-(4-fluorophenyl)-3-methyl-pyrazol-4-yl]-benzoxazin-3-one derivatives as novel selective nonsteroidal mineralocorticoid receptor antagonists. Bioorg. Med. Chem. 2014, 22, 5428–5445. [Google Scholar] [CrossRef]
- Han, Y.; Kim, M.; Choi, G.; An, H.; Son, D.; Kim, H.; Ha, H.; Son, J.; Chung, S.; Park, H.; et al. Pyrazole-5-carboxamides, novel inhibitors of receptor for advanced glycation end products (RAGE). Eur. J. Med. Chem. 2014, 79, 128–142. [Google Scholar] [CrossRef]
- Chan, B.; Estrada, A.; Chen, H.; Atherall, J.; Baker-Glenn, C.; Beresford, A.; Burdick, D.; Chambers, M.; Dominguez, S.; Drummond, J.; et al. Discovery of a highly selective, brain-penetrant aminopyrazole LRRK2 inhibitor. Med. Chem. 2013, 4, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Saini, D.; Jain, S.; Jain, N. Pyrazole scaffold: A remarkable tool in the development of anticancer agents. Eur. J. Med. Chem. 2013, 70, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Bao, Y.; Niu, Q.; Yang, J.; Fan, Y.; Li, J.; Jing, Y.; Zhao, L.; Liu, D. Identification of N-(6-mercaptohexyl)-3-(4-pyridyl)-1H-pyrazole-5-carboxamide and its disulfide prodrug as potent histone deacetylase inhibitors with in vitro and in vivo anti-tumor efficacy. Eur. J. Med. Chem. 2016, 109, 350–359. [Google Scholar] [CrossRef]
- Balbi, A.; Anzaldi, M.; Macciò, C.; Aiello, C.; Mazzei, M.; Gangemi, R.; Castagnola, P.; Miele, M.; Rosano, C.; Viale, M. Synthesis and biological evaluation of novel pyrazole derivatives with anticancer activity. Eur. J. Med. Chem. 2011, 46, 5293–5309. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.; Draghici, C.; Missir, A. Synthesis of new pyrazole derivatives and their anticancer evaluation. Eur. J. Med. Chem. 2010, 45, 4914–4919. [Google Scholar] [CrossRef]
- Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman. Review: Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Lema, C.; Varela-Ramirez, A.; Aguilera, R.J. Differential nuclear staining assay for high-throughput screening to identify cytotoxic compounds. Curr. Cell Biochem. 2011, 1, 1–14. [Google Scholar] [PubMed]
- Ruiz-Medina, B.; Lerma, D.; Hwang, M.; Ross, J.; Skouta, R.; Aguilera, R.J.; Kirken, R.; Varela-Ramirez, A.; Robles-Escajeda, E. Green barley mitigates cytotoxicity in human lymphocytes undergoing aggressive oxidative stress, via activation of both the Lyn/PI3K/Akt and MAPK/ERK pathways. Sci. Rep. 2019, 9, 6005. [Google Scholar] [CrossRef]
- Gutierrez, D.A.; DeJesus, R.; Contreras, L.; Rodriguez-Palomares, I.; Villanueva, P.; Balderrama, K.; Monterroza, L.; Larragoity, M.; Varela-Ramirez, A.; Aguilera, R.J. A new pyridazinone exhibits potent cytotoxicity on human cancer cells via apoptosis and poly-ubiquitinated protein accumulation. Cell Biol. Toxicol. 2019, 35, 503–519. [Google Scholar] [CrossRef]
- Varela-Ramirez, A.; Costanzo, M.; Carrasco, Y.; Pannell, K.; Aguilera, R.J. Cytotoxic effects of two organotin compounds and their mode of inflicting cell death on four mammalian cancer cells. Cell Biol. Toxicol. 2011, 27, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robles-Escajeda, E.; Das, U.; Ortega, N.; Parra, K.; Francia, G.; Dimmock, J.; Varela-Ramirez, A.; Aguilera, R.J. A novel curcumin-like dienone induces apoptosis in triple-negative breast cancer cells. Cell Oncol. 2016, 39, 265–277. [Google Scholar] [CrossRef]
- Robles-Escajeda, E.; Lerma, D.; Nyakeriga, A.M.; Ross, J.A.; Kirken, R.A.; Aguilera, R.J.; Varela-Ramirez, A. Searching in mother nature for anti-cancer activity: Anti-proliferative and pro-apoptotic effect elicited by green barley on leukemia/lymphoma cells. PLoS ONE 2013, 8, 73508. [Google Scholar]
- Nakamura-Bencomo, S.; Gutierrez, D.A.; Robles-Escajeda, E.; Iglesias-Figueroa, B.; Siqueiros-Cendón, T.S.; Espinoza-Sánchez, E.A.; Arévalo-Gallegos, S.; Aguilera, R.J.; Rascón-Cruz, Q.; Varela-Ramirez, A. Recombinant human lactoferrin carrying humanized glycosylation exhibits antileukemia selective cytotoxicity, microfilament disruption, cell cycle arrest, and apoptosis activities. Invest. New Drugs 2021, 39, 15. [Google Scholar] [CrossRef] [PubMed]
- Varela, J.; De Chatterjee, A.; Guevara, P.; Ramirez, V.; Metta-Magaña, A.; Villagrán, D.; Varela-Ramirez, A.; Das, S.; Nuñez, J. Synthesis, characterization, and evaluation of cis-diphenyl pyridineamine platinum(II) complexes as potential anti-breast cancer agents. Biol. Inorg. Chem. 2014, 19, 967–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, A.; Jänicke, R. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Van Raam, B.; Salvesen, G.S. (Eds.) Handbook of Proteolytic Enzymes, Caspase-8 and Caspase-10, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2013; Volume 1. [Google Scholar]
- Redza-Dutordoir, M.; Averill-Bates, D. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.; Jansen, E.; Sanchez, W.; Waterhouse, N. Flow cytometry based assays for the measurement of apoptosis-associated mitochondrial membrane depolarisation and cytochrome c release. Methods 2013, 61, 138–145. [Google Scholar] [CrossRef]
- Murphy, M.P. Mitochondrial Dysfunction Indirectly Elevates ROS Production by the Endoplasmic Reticulum. Cell Metab. 2013, 18, 145–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, M.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Morales, J.; Li, L.; Fattah, F.; Dong, Y.; Bey, E.; Patel, M.; Gao, J.; Boothman, D. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Iglesias-Figueroa, B.; Siqueiros-Cendón, T.; Gutierrez, D.A.; Aguilera, R.J.; Espinoza-Sánchez, E.; Arévalo-Gallegos, S.; Varela-Ramirez, A.; Rascón-Cruz, Q. Recombinant human lactoferrin induces apoptosis, disruption of F-actin structure and cell cycle arrest with selective cytotoxicity on human triple negative breast cancer cells. Apoptosis 2019, 27, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [Green Version]
- Jang, C.; Wong, J.; Coppinger, J.; Seki, A.; Yates, J.; Fang, G. DDA3 recruits microtubule depolymerase Kif2a to spindle poles and controls spindle dynamics and mitotic chromosome movement. J. Cell Biol. 2008, 181, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, C.; Fang, G. The N-terminal domain of DDA3 regulates the spindle-association of the microtubule depolymerase Kif2a and controls the mitotic function of DDA3. Cell Cycle 2009, 8, 3165–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Ongusaha, P.; Hong, Y.; Kurdistani, S.; Nakamura, M.; Lu, K.; Lee, S. Function of Drg1/Rit42 in p53-dependent mitotic spindle checkpoint. J. Biol. Chem. 2004, 279, 38597–38602. [Google Scholar] [CrossRef] [Green Version]
- WHO. Latest Global Cancer Data: Cancer Burden Rises to 18.1 Million New Cases and 9.6 Million Cancer Deaths in 2018. Available online: https://www.who.int/cancer/PRGlobocanFinal.pdf (accessed on 20 September 2018).
- Torre, L.; Islami, F.; Siegel, R.; Ward, E.; Jemal, A. Global Cancer in Women: Burden and Trends. Cancer Epidemiol. Biomarkers Prev. 2017, 26, 444–457. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Debatin, K. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [Green Version]
- Gorrini, C.; Harris, I.; Mak, T. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Wu, C.; Bratton, S. Regulation of the intrinsic apoptotic pathway by reactive oxygen species. Antiox. Redox Signal. 2013, 19, 546–548. [Google Scholar] [CrossRef] [Green Version]
- Oliver, F.; de la Rubia, G.; Rolli, V.; Ruiz-Ruiz, M.; de Murcia, G.; Murcia, J. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. J. Biol. Chem. 1998, 273, 33533–33539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, D.; Blais, V.; Denault, J. Caspase-7 uses an exosite to promote poly(ADP ribose) polymerase 1 proteolysis. Proc. Natl. Acad. Sci. USA 2012, 109, 5669–5674. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.; Meng, F.; Li, Q.; Ho, C.; Lam, T.; To, Y.; Lee, W.; Li, M.; Chu, K.; Toh, M. Cucurbitacin B induces apoptosis and S phase cell cycle arrest in BEL-7402 human hepatocellular carcinoma cells and is effective via oral administration. Cancer Lett. 2010, 294, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Czarnomysy, R.; Surażyński, A.; Muszynska, A.; Gornowicz, A.; Bielawska, A.; Bielawski, K. A novel series of pyrazole-platinum(II) complexes as potential anti-cancer agents that induce cell cycle arrest and apoptosis in breast cancer cells. J. Enzyme. Inhib. Med. Chem. 2018, 33, 1006–1023. [Google Scholar] [CrossRef]
- Trepat, X.; Chen, Z.; Jacobson, K. Cell Migration. Compr. Physiol. 2012, 2, 2369–2392. [Google Scholar] [PubMed] [Green Version]
- Martin, T.; Ye, L.; Sanders, A.; Lane, J.; Jiang, W. Cancer invasion and metastasis: Molecular and cellular perspective. In Metastatic Cancer Clinical and Biological Perspectives; Landes Bioscience: Austin, TX, USA, 2013; pp. 135–168. [Google Scholar]
- Liang, C.; Park, A.; Guan, J. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Hurtley, S. Cell Biology of the Cytoskeleton. Science 1998, 279, 459. [Google Scholar] [CrossRef]
- Alam, R.; Gorska, M. Mitogen-activated protein kinase signalling and ERK1/2 bistability in asthma. Clin. Exp. Allergy. 2011, 41, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Han, J. The p38 signal transduction pathway: Activation and function. Cell Signal. 2000, 12, 1–13. [Google Scholar] [CrossRef]
- Kralova, J.; Dvorak, M.; Koc, M.; Kral, V. p38 MAPK plays an essential role in apoptosis induced by photoactivation of a novel ethylene glycol porphyrin derivative. Oncogene 2008, 27, 3010–3020. [Google Scholar] [CrossRef] [Green Version]
- Koul, H.; Pal, M.; Koul, S. Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes Cancer 2013, 4, 342–359. [Google Scholar] [CrossRef]
- Cheung, P.; Campbell, D.; Nebreda, A.; Cohen, P. Feedback control of the protein kinase TAK1 by SAPK2a/p38alpha. EMBO J. 2003, 22, 5793–5805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdiguero, E.; Ruiz-Bonilla, V.; Serrano, A.; Muñoz-Cánoves, P. Genetic deficiency of p38alpha reveals its critical role in myoblast cell cycle exit: The p38alpha-JNK connection. Cell Cycle 2007, 6, 1298–1303. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebolt-Leopold, J.; Dudley, D.; Herrera, R.; Van Becelaere, K.; Wiland, A.; Gowan, R.; Tecle, H.; Barrett, S.; Bridges, A.; Przybranowski, S.; et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat. Med. 1999, 5, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Maemura, K.; Shiraishi, N.; Sakagami, K.; Kawakami, K.; Inoue, T.; Murano, M.; Watanabe, M.; Otsuki, Y. Proliferative effects of gamma-aminobutyric acid on the gastric cancer cell line are associated with extracellular signal-regulated kinase 1/2 activation. J. Gastroenterol. Hepatol. 2009, 24, 688–696. [Google Scholar] [CrossRef]
- Gauthier, R.; Harnois, C.; Drolet, J.; Reed, J.; Vézina, A.; Vachon, P. Human intestinal epithelial cell survival: Differentiation state specific control mechanisms. Am. J. Physiol. Cell Physiol. 2001, 280, C1540–C1554. [Google Scholar] [CrossRef] [Green Version]
- Abramovitch, R.; Tavor, E.; Jacob-Hirsch, J.; Zeira, E.; Amariglio, N.; Pappo, O.; Rechavi, G.; Galun, E.; Honigman, A. A pivotal role of cyclic AMP-responsive element binding protein in tumor progression. Cancer Res. 2004, 64, 1338–1346. [Google Scholar] [CrossRef] [Green Version]
- Mitton, B.; Chae, H.; Hsu, K.; Dutta, R.; Aldana-Masangkay, G.; Ferrari, R.; Davis, K.; Tiu, B.; Kaul, A.; Lacayo, N.; et al. Small molecule inhibition of cAMP response element binding protein in human acute myeloid leukemia cells. Leukemia 2016, 30, 2302–2311. [Google Scholar] [CrossRef]
- Chen, S.; Tao, J.; Zhong, F.; Jiao, Y.; Xu, J.; Shen, Q.; Wang, H.; Fan, S.; Zhang, Y. Polydatin down-regulates the phosphorylation level of Creb and induces apoptosis in human breast cancer cell. PLoS ONE 2017, 12, e0176501. [Google Scholar] [CrossRef]
- Lim, C.; Cao, X. Structure, function, and regulation of STAT proteins. Mol. Biosyst. 2006, 2, 536–550. [Google Scholar] [CrossRef]
- Wong, A.; Hirpara, J.; Pervaiz, S.; Eu, J.; Sethi, G.; Goh, B. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef] [Green Version]
- Groner, B.; von Manstein, V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell Endocrinol. 2017, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Greten, F.; Karin, M. The IKK/NF-κB activation pathway—A target for prevention and treatment of cancer. Cancer Lett. 2004, 206, 193–199. [Google Scholar] [CrossRef]
- Wang, W.; Nag, S.; Zhang, R. Targeting the NFκB signaling pathways for breast cancer prevention and therapy. Curr. Med. Chem. 2015, 22, 264–289. [Google Scholar] [CrossRef]
- Gkouveris, I.; Nikitakis, N.; Karanikou, M.; Rassidakis, G.; Sklavounou, A. JNK1/2 expression and modulation of STAT3 signaling in oral cancer. Oncol. Lett. 2016, 12, 699–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruman, D.; Chiu, H.; Hopkins, B.; Bagrodia, S.; Cantley, L.; Abraham, R. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Sunami, T.; Byrne, N.; Diehl, R.; Funabashi, K.; Hall, D.; Ikuta, M.; Patel, S.; Shipman, J.; Smith, R.; Takahashi, I.; et al. Structural basis of human p70 ribosomal S6 kinase-1 regulation by activation loop phosphorylation. J. Biol. Chem. 2010, 285, 4587–4591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieu, C.; Kopetz, S. The SRC family of protein tyrosine kinases: A new and promising target for colorectal cancer therapy. Clin. Colorectal. Cancer 2010, 9, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Palacios, E.; Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004, 23, 7990–8000. [Google Scholar] [CrossRef] [Green Version]
- Cabodi, S.; Calautti, E.; Talora, C.; Kuroki, T.; Stein, P.; Dotto, G. A PKC-eta/Fyn-dependent pathway leading to keratinocyte growth arrest and differentiation. Mol. Cell 2000, 6, 1121–1129. [Google Scholar] [CrossRef]
- Resh, M. Fyn, a Src family tyrosine kinase. Int. J. Biochem. Cell Biol. 1998, 39, 265–277. [Google Scholar] [CrossRef]
- Yuniati, L.; Scheijen, B.; van der Meer, L.; van Leeuwen, F. Tumor suppressors BTG1 and BTG2: Beyond growth control. J. Cell Physiol. 2019, 234, 5379–5389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Choi, Y.; Lim, I.; Park, T. BTG2 suppresses cancer cell migration through inhibition of Src-FAK signaling by downregulation of reactive oxygen species generation in mitochondria. Clin. Exp. Metastasis 2012, 29, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.; Wei, C.; Ku, B.; Jeon, T.; Chien, P.; Kim, J.; Park, S.; Hwang, H.; Ryu, S.; Park, H.; et al. The family-wide structure and function of human dual-specificity protein phosphatases. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 421–435. [Google Scholar] [CrossRef]
- Ryu, J.; Lee, D. Dual-specificity phosphatase 18 modulates the SUMOylation and aggregation of Ataxin-1. Biochem. Biophys. Res. Commun. 2018, 502, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Guo, P.; Takeda, K.; Fu, Q.; Fang, H.; Frucht, D. Erk1/2 inactivation promotes a rapid redistribution of COP1 and degradation of COP1 substrates. PNAS 2020, 117, 4078–4087. [Google Scholar] [CrossRef]
- Miura, H.; Kondo, Y.; Matsuda, M.; Aoki, K. Cell-to-Cell Heterogeneity in p38-Mediated Cross-Inhibition of JNK Causes Stochastic Cell Death. Cell Rep. 2018, 24, 2658–2668. [Google Scholar] [CrossRef] [Green Version]
- Brault, L.; Menter, T.; Obermann, E.; Knapp, S.; Thommen, S.; Schwaller, J.; Tzankov, A. PIM kinases are progression markers and emerging therapeutic targets in diffuse large B-cell lymphoma. Br. J. Cancer 2012, 107, 491–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemskova, M.; Sahakian, E.; Bashkirova, S.; Lilly, M. The PIM1 kinase is a critical component of a survival pathway activated by docetaxel and promotes survival of docetaxel-treated prostate cancer cells. J. Biol. Chem. 2008, 283, 20635–20644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ononye, O.; Sausen, C.; Bochman, M.; Balakrishnan, L. Dynamic regulation of Pif1 acetylation is crucial to the maintenance of genome stability. Curr. Genet 2021, 67, 85–92. [Google Scholar] [CrossRef]
- Degli-Esposti, M.; Dougall, W.; Smolak, P.; Waugh, J.; Smith, C.; Goodwin, R. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 1997, 7, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Lalaoui, N.; Morlé, A.; Mérino, D.; Jacquemin, G.; Iessi, E.; Morizot, A.; Shirley, S.; Robert, B.; Solary, E.; Garrido, C.; et al. TRAIL-R4 promotes tumor growth and resistance to apoptosis in cervical carcinoma HeLa cells through AKT. PLoS ONE 2011, 6, 19679. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Thomas, E.; Herzog, B.; Westfall, M.; Rocheleau, J.; Jackson, R.; Wang, M.; Liang, P. NDRG1 is necessary for p53-dependent apoptosis. J. Biol. Chem. 2004, 279, 48930–48940. [Google Scholar] [CrossRef] [Green Version]
- Kovacevic, Z.; Menezes, S.; Sahni, S.; Kalinowski, D.; Bae, D.; Lane, D.; Richardson, D. The Metastasis Suppressor, N-MYC Downstream-regulated Gene-1 (NDRG1), Down-regulates the ErbB Family of Receptors to Inhibit Downstream Oncogenic Signaling Pathways. J. Biol. Chem. 2016, 291, 1029–1052. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Tissue Origin | CC50 (µM) * | SCI ** | p-Value ‡ |
|---|---|---|---|---|
| 72 h | ||||
| Hs27 | Normal foreskin epithelial | 3.47 +/− 0.45 | 1 | n/a |
| MDA-MB-231 | Breast adenocarcinoma | 0.49 +/− 0.12 | 7.1 | 0.0003 |
| MDA-MB-468 | 0.25 +/− 0.02 | 13.9 | 0.0002 | |
| HCC70 | Breast carcinoma | 5.40 +/− 0.084 | 0.6 | 0.001 |
| HCC1419 | 6.27 +/− 0.14 | 0.5 | 0.0005 | |
| MCF-7 | 0.45 +/− 0.014 | 7.7 | 0.0003 | |
| T47D | 0.78 +/− 0.07 | 4.5 | 0.0005 | |
| OVCAR-8 | Ovarian carcinoma | 0.44 +/− 0.07 | 7.9 | 0.0003 |
| OVCAR-5 | 0.60 +/− 0.008 | 5.8 | 0.0003 | |
| OVCAR-3 | Ovarian adenocarcinoma | 0.30 +/− 0.03 | 11.6 | 0.0002 |
| OV-90 | Metastatic ovarian adenocarcinoma | 0.65 +/− 0.19 | 5.3 | 0.0005 |
| NCI-H358 | Non-small cell lung cancer | 0.19 +/− 0.11 | 18.3 | 0.000254 |
| NCI-H460 | Large cell lung cancer | 0.67 +/− 0.17 | 5.2 | 0.000545 |
| A549 | Lung carcinoma | 0.27 +/− 0.03 | 12.9 | 0.000262 |
| PC-3 | Prostatic adenocarcinoma | 1.41 +/− 0.09 | 2.5 | 0.00147 |
| LnCap | Prostatic carcinoma | 0.79 +/− 0.09 | 4.5 | 0.000538 |
| PANC-1 | Pancreatic carcinoma | 0.76 +/− 0.10 | 4.6 | 0.000524 |
| A375 | Melanoma | 0.40 +/− 0.03 | 8.7 | 0.000296 |
| WM-115 | 0.38 +/− 0.05 | 41.3 | 0.000292 | |
| 48 h | ||||
| Hs27 | Normal foreskin epithelial | 5.76 +/− 0.36 | 1 | n/a |
| CEM | T lymphoblastic leukemia | 0.48 +/− 0.02 | 12 | 0.000014 |
| MOLT-3 | Acute lymphoblastic leukemia | 6.54 +/− 0.36 | 0.9 | 0.05676 |
| HL-60 | Acute promyelocytic leukemia | 0.58 +/− 0.05 | 9.9 | 0.000016 |
| JURKAT | Acute T cell leukemia | 0.37 +/− 0.01 | 15.6 | 0.000013 |
| NALM-6 | Lymphoblastic leukemia | 4.91 +/− 0.33 | 1.2 | 0.03937 |
| RAMOS | Burkitt’s lymphoma | 0.31 +/− 0.009 | 18.6 | 0.000013 |
| RPMI-8226 | Myeloma | 4.45 +/− 0.091 | 1.3 | 0.003631 |
| MM.1S | 0.96 +/− 0.014 | 6 | 0.000021 | |
| MM.1R | Multiple Myeloma | 0.76 +/− 0.01 | 7.6 | 0.000018 |
| Ingenuity Canonical Pathways | Genes | p-Values |
|---|---|---|
| SAPK/JNK Signaling | DUSP10,DUSP4 | 6.46 × 10−3 |
| Cell Cycle Regulation | BTG2 | 4.27 × 10−2 |
| p38 MAPK Signaling | DUSP10 | 8.13 × 10−2 |
| STAT3 Pathway | PIM1 | 1.31 × 10−1 |
| ERK/MAPK Signaling | DUSP4 | 1.48 × 10−1 |
| RNA-Seq | Gene Function | Supporting In Vitro Assay | Pathway |
|---|---|---|---|
| ↑BTG2 | Cell cycle regulation and Antiproliferation | Arrest in S & G2/M phases during cell cycle by FC * | BTG2 Cell cycle and apoptosis |
| Anti-metastasis and Anti-invasive | Scratch wound assay by CM ‡ | ||
| Mitochondrial depolarization | JC-1 assay via FC | ||
| ↑DUSP4 ↑DUSP10 | Dephosphorylation of ERK1/2, SAPK/JNK, and p38 MAP Kinases | Inactivation of ERK1/2 and p38 during multiplex cell signaling assays | ERK1/2, SAPK/JNK and p38 MAPK |
| ↑PIM1 | Cell proliferation | Cell cycle arrest found by FC | JAK/STAT |
| Apoptosis | Apoptosis assay via FC | ||
| Migration | Scratch wound invasive assay by CM analysis | ||
| ↓PIF1 | Maintenance of nuclear and mitochondrial DNA | Cellular DNA fragmentation detected during cell cycle FC analysis | PIF1 DNA repair |
| ↑TNFRSF10D | Apoptosis | Apoptosis assay via FC | NF-κB |
| Signals NF-κB activation | Activation of NF-κB found via Multiplex cell signaling assays | ||
| ↑NDRG1 | Caspase activation | Caspase 3/7 and 8 assays by FC | NDRG1 cell cycle, apoptosis |
| Antiproliferation and Apoptosis | Cell cycle and apoptosis assays via FC | ||
| Anti-metastasis and Anti-invasive | Scratch wound assay by CM ‡ | ||
| ↓PSRC1 | Microtubule maintenance and stability, including regulation of spindle during mitosis | Disturbance of microtubules and a deformed spindle unable to undergo mitosis found by immunohistochemistry and CM | PSRC1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutierrez, D.A.; Contreras, L.; Villanueva, P.J.; Borrego, E.A.; Morán-Santibañez, K.; Hess, J.D.; DeJesus, R.; Larragoity, M.; Betancourt, A.P.; Mohl, J.E.; et al. Identification of a Potent Cytotoxic Pyrazole with Anti-Breast Cancer Activity That Alters Multiple Pathways. Cells 2022, 11, 254. https://doi.org/10.3390/cells11020254
Gutierrez DA, Contreras L, Villanueva PJ, Borrego EA, Morán-Santibañez K, Hess JD, DeJesus R, Larragoity M, Betancourt AP, Mohl JE, et al. Identification of a Potent Cytotoxic Pyrazole with Anti-Breast Cancer Activity That Alters Multiple Pathways. Cells. 2022; 11(2):254. https://doi.org/10.3390/cells11020254
Chicago/Turabian StyleGutierrez, Denisse A., Lisett Contreras, Paulina J. Villanueva, Edgar A. Borrego, Karla Morán-Santibañez, Jessica D. Hess, Rebecca DeJesus, Manuel Larragoity, Ana P. Betancourt, Jonathon E. Mohl, and et al. 2022. "Identification of a Potent Cytotoxic Pyrazole with Anti-Breast Cancer Activity That Alters Multiple Pathways" Cells 11, no. 2: 254. https://doi.org/10.3390/cells11020254
APA StyleGutierrez, D. A., Contreras, L., Villanueva, P. J., Borrego, E. A., Morán-Santibañez, K., Hess, J. D., DeJesus, R., Larragoity, M., Betancourt, A. P., Mohl, J. E., Robles-Escajeda, E., Begum, K., Roy, S., Kirken, R. A., Varela-Ramirez, A., & Aguilera, R. J. (2022). Identification of a Potent Cytotoxic Pyrazole with Anti-Breast Cancer Activity That Alters Multiple Pathways. Cells, 11(2), 254. https://doi.org/10.3390/cells11020254