
Tfeb-Mediated Transcriptional Regulation of Autophagy Induces Autosis during Ischemia/Reperfusion in the Heart

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ischemia/Reperfusion In Vivo
2.2. Measurement of Infarct Size
2.3. Cardiomyocyte Cultures
2.4. Viability of the Cells
2.5. Tat-Beclin 1
2.6. Electron Microscopy
2.7. Real-Time Quantitative Polymerase Chain Reaction (qPCR)
2.8. Immunoblot Analyses
2.9. Immunostaining Analysis
2.10. Statistical Analysis
3. Results
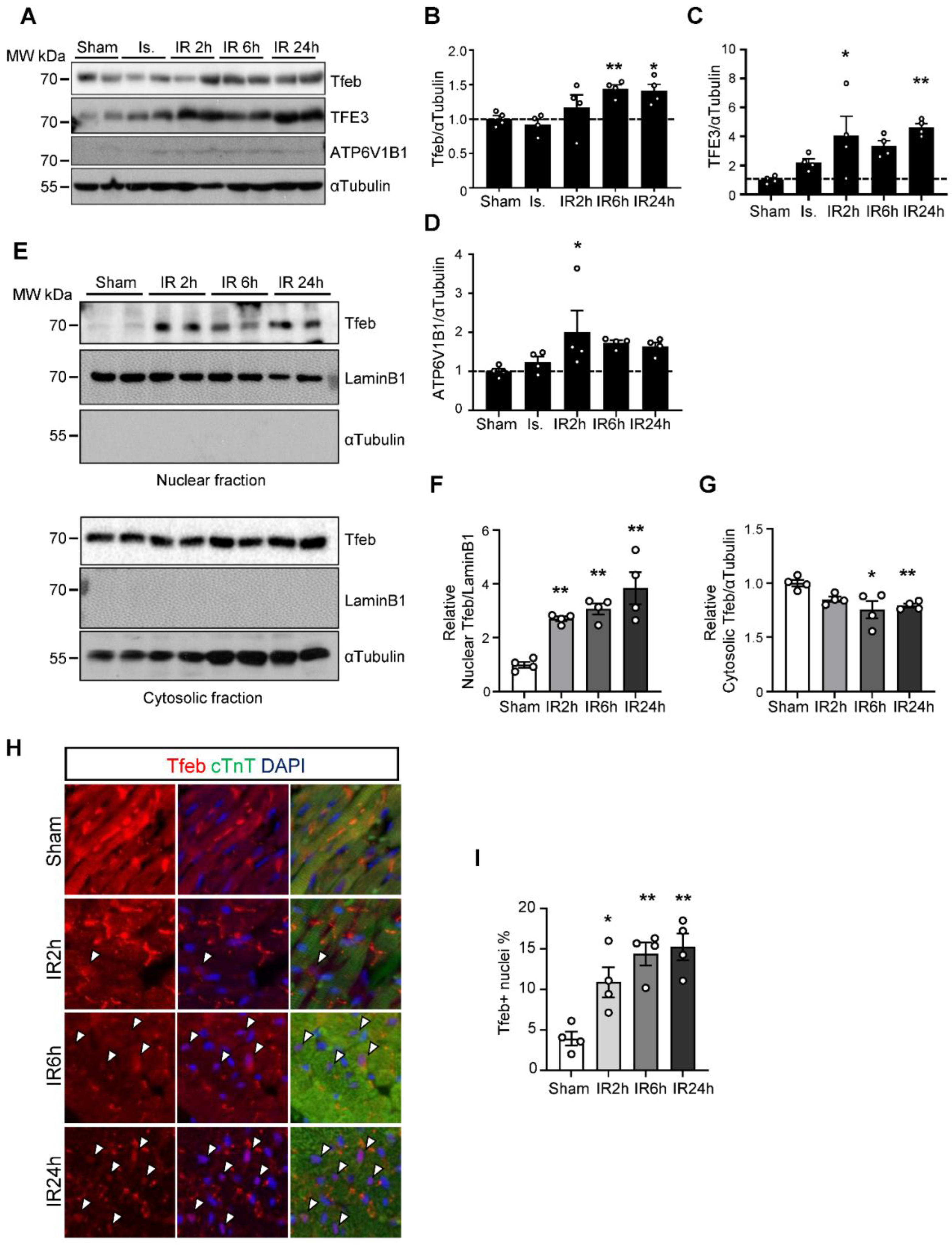
3.1. The Tfeb-Mediated Autophagy–Lysosome Pathway Is Persistently Upregulated during I/R
3.2. Tfeb Facilitates Tat-Beclin 1-Induced Autosis in Neonatal Rat Cardiomyocytes (NRCMs)
3.3. Persistent Activation of Tfeb Triggers Accumulation of Autophagic Vacuoles and Autosis during I/R
3.4. Acute Activation of Tfeb by 3,4-Dimethoxychalcone Aggravates Myocardial Infarction (MI) Caused by I/R
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef] [Green Version]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef]
- Nah, J.; Zablocki, D.; Sadoshima, J. Autosis: A New Target to Prevent Cell Death. JACC Basic Transl. Sci. 2020, 5, 857–869. [Google Scholar] [CrossRef]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, A.F.; Liu, Y.; Ginet, V.; Shi, M.; Nah, J.; Zou, Z.; Zhou, A.; Posner, B.A.; Xiao, G.; Tanguy, M.; et al. Interaction between the autophagy protein Beclin 1 and Na+,K+-ATPase during starvation, exercise, and ischemia. JCI Insight 2020, 5, e133282. [Google Scholar] [CrossRef]
- Garner, M.H. Na,K-ATPase in the nuclear envelope regulates Na+: K+ gradients in hepatocyte nuclei. J. Membr. Biol. 2002, 187, 97–115. [Google Scholar] [CrossRef]
- Nah, J.; Zhai, P.; Huang, C.Y.; Fernandez, A.F.; Mareedu, S.; Levine, B.; Sadoshima, J. Upregulation of Rubicon promotes autosis during myocardial ischemia/reperfusion injury. J. Clin. Investig. 2020, 130, 2978–2991. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef]
- Gatica, D.; Chiong, M.; Lavandero, S.; Klionsky, D.J. Molecular mechanisms of autophagy in the cardiovascular system. Circ. Res. 2015, 116, 456–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional Regulation of Autophagy: Mechanisms and Diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [Green Version]
- Javaheri, A.; Bajpai, G.; Picataggi, A.; Mani, S.; Foroughi, L.; Evie, H.; Kovacs, A.; Weinheimer, C.J.; Hyrc, K.; Xiao, Q.; et al. TFEB activation in macrophages attenuates postmyocardial infarction ventricular dysfunction independently of ATG5-mediated autophagy. JCI Insight 2019, 4, e127312. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Huang, H.; Jin, Y.; Shen, K.; Chen, X.; Xu, Z.; Jin, B.; Pan, H. Role of TFEB in autophagic modulation of ischemia reperfusion injury in mice kidney and protection by urolithin A. Food Chem. Toxicol. 2019, 131, 110591. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Del Re, D.P.; Zablocki, D.K.; Hsu, C.P.; et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Xie, W.; Nah, J.; Sauvat, A.; Liu, P.; Pietrocola, F.; Sica, V.; Carmona-Gutierrez, D.; Zimmermann, A.; Pendl, T.; et al. 3,4-Dimethoxychalcone induces autophagy through activation of the transcription factors TFE3 and TFEB. EMBO Mol. Med. 2019, 11, e10469. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Sense (5′→3′) | Antisense (5′→3′) |
|---|---|---|
| Akt1 | GCTTCTATGGTGCGGAGATT | CTTGTCCAGCATGAGGTTCT |
| Ambra1 | ATACTACGCCCAGAGGATGA | GAAGAAGAGGAGGTGGAAGAAC |
| App | TCTCTGTCCCTGCTCTACAA | CAAGACATCGTCGGAGTAGTTC |
| Atg10 | CGAGCGGGTTCTCATTAACA | TCCTTGGCTGTTCTCCATTC |
| Atg12 | AGCTCTTCAGTCCTGTCATTTC | ACTCCTGGTTCACTCTTCCT |
| Atg16l1 | TGTGTGTGTGTGAGGAAAGG | GCAGTGATGCAGGGTAAAGA |
| Atg16l2 | CGAGACAACACACTCAAGGTTA | CAGCTTTGGTCCAGTCAGAA |
| Atg3 | CCGGTCCTCAAGGAATCAAA | GTTGGACAGTGGTGGACTAAG |
| Atg4a | GAAACGAGGGCATTTGGAAAG | ACCTGAAAGGGTGGAGTAGTA |
| Atg4b | CAGATGCAGCAGCTCTAAGT | GGGACAAGATATGGGAGAGAAAG |
| Atg4c | GCGGAGTCTTCTATGAGGAATG | GGAAGGAAGGAAGGAAGGAAAG |
| Atg4d | CCTGGTCTTGTTGTCTCACTAC | TTCCGCTGGTCCAACTAATC |
| Atg5 | CAGGTGATGATTCACGGGATAG | GGAGGACACACTCTTTCAATCT |
| Atg7 | TCCTGAGAGCATCCCTCTAAT | GGCTCGACACAGATCATCATAG |
| Atg9a | GACATCTACCATCGCATCCTAC | GGGTGAAGAAGACAACCTCTC |
| Atg9b | GATGACAGGCCCAGGATATTT | GCCAGGCTAGTGTTCTCTTATC |
| Bcl2 | GAGCAGGTGCCTACAAGAAA | CTTTGTCCTCTGACTGGGTATG |
| Bcl2l1 | CTGGTTGAGCCCATCTCTATT | CTGACTCCAGCTGTATCCTTTC |
| Becn1 | CAGGAACTCACAGCTCCATTAC | CCATCCTGGCGAGTTTCAATA |
| Bnip3 | TCCAGCCTCCGTCTCTATTT | CTGTCACAGTGAGAACTCTTGG |
| Cdkn1b | CCTTCCGCCTGCAGAAAT | CTGACTCGCTTCTTCCATATCC |
| Cdkn2a | CATGTTGTTGAGGCTAGAGAGG | CACCGTAGTTGAGCAGAAGAG |
| Cln3 | CCACCTGTCCCAGACTTTAATC | CCGCAGAACTTCCCATAACA |
| Ctsb | CATGCACTGGAGAAGGAGATAC | CTGTTAGACACGCTGTAGGAAG |
| Ctsd | CCTGGGCGATGTCTTCATT | GTGGAGAAGGAGCAAGTTAGAG |
| Ctss | GGGAGACATGACCAATGAAGAA | TGTCAGGCAATGTCCGATTAG |
| Cxcr4 | CTGCCCACCATCTACTTCATC | CGTCATGCTCCTTAGCTTCTT |
| Dapk1 | CAACAGGGCTTCTAGGGATTT | AGCTCCCGAAGTTTGGATATAAG |
| Dram1 | CAGATCCAAGTCAGGGAGAAAG | CAGGCATTGATAGGAGCAGATAA |
| Dram2 | TGGTAGTGCACGCCTTTAAT | CTGTCCTGGAACTCATTCTGTAG |
| Eif2ak3 | CCCAGGCATTGTGAGGTATT | CCAGTCTGTGCTTTCGTCTT |
| Eif4g1 | GCTACAAGCACTCTATGCTCTC | CTTCACCACGTCCTCATCATATAG |
| Esr1 | CTTGGAAGGCCGAAATGAAATG | GGCAGGGCTATTCTTCTTAGTG |
| Gaa | CCAATTCCTCTCCACACACTAC | TTGAGCGGGAGATCACAAAG |
| Gabarap | CTCCTTCCTTGACATCCAGTTC | GGAGAGACAGCCTCAAACATTA |
| Gabarapl1 | CCTAACCTCTGTCTCCATACCT | GAATGTCTCCTGCCACAACT |
| Gabarapl2 | GCACTGTCCTCATGGCTATT | CACACCTACCTCCCTTCATTAC |
| Hdac1 | GCTGCTCAACTATGGTCTCTAC | CACTGTGGTACTTGGTCATCTC |
| Hdac6 | GACCTGTCTACCTGGTGTTATG | GCAGTGTGGTCTGGGATTTA |
| Hgs | TGAGGAGAAAGAGAGGATGAGA | GTTCACAGGCGAGGAATACA |
| Hsp90aa1 | GACGAGATGGTTTCTCTGAAGG | TTCCACAAAGGCGGAGTTAG |
| Hspa8 | ACTCCTCTTTCCCTTGGTATTG | GTCAGAGTAGGTGGTGAAAGTC |
| Ifng | CTCTTCCTCATGGCTGTTTCT | TTCTTCCACATCTATGCCACTT |
| Igf1 | CAGAAGAGGGAGAGAGAGAGAA | TAGCAAGCAGAAGAGGGATTTAG |
| Ins2 | CCCTAAGTGATCCGCTACAATC | CAGGGCCATGTTGAAACAATAA |
| Irgm1 | CGAAGACCAGAAGCTGAAAGA | CCGGATGTTGAGAGATGAGATG |
| Lamp1 | GACCCTGAAAGTGGAGAACAA | GGGCATCAGGAAGAGTCATATT |
| Map1lc3a | GTCTACGCCTCCCAAGAAAC | GGTGTCACATCTCTGCCTAATC |
| Map1lc3b | CGCATGCCTTTAGCCTTTAATC | CCTGGAACTCACTTTGTAGACC |
| Mapk14 | GAAAGCAGGGACCTTCTCATAG | GTGCTCAGGACTCCATTTCTT |
| Mapk8 | TCCAGCACCCATACATCAAC | CTATTGTGTGCTCCCTCTCATC |
| Mtor | CGGGACTACAGAGAGAAGAAGA | CATCAACGTCAGGTGGTCATAG |
| Nfkb1 | GGGATTTCGATTCCGCTATGT | GACCTGTGGGTAGGATTTCTTG |
| Npc1 | GAGCAGATCCTGTCTGCATTAT | CTGGTGTATCCGTGAAGTGTT |
| Pik3c3 | CCTGATGCCTGGCTCAATAA | TAACTTCTGGACACCCTCTCT |
| Pik3cg | GTACCCTGGTGATCGAGAAATG | ATGGTTTCGTTGGATAGGACTG |
| Pik3r4 | AGGTTCTGGGACTTGGTTTC | GACCTCGGTGCCTTCTATTATC |
| Prkaa1 | TGGCTGGGTGTGTAAAGATATG | CATGTCAGAGCCCACAATGA |
| Pten | CAGTGAATGCCATCACCATTTC | CTGGGCTTAAGGTCTGATTCTC |
| Rab24 | ATCTGGCCCAGTGGAATTAG | GTGAGAGGCACCTGGAATAG |
| Rb1 | CCTCCTACCTTGTCACCAATAC | GTTACCTCCAGGAATCCGTAAG |
| Rgs19 | TGCGAGAAGGCATCAATAGG | GGAGTAATAGAGAGCGGTAGGT |
| Rps6kb1 | AGAGATGGGTCAGGGTTAAGA | TTCAGGTTCCCAGGAAGTAAAG |
| Snca | TGACAACAGTGGCTGAGAAG | CCAGTGGCAGCAGCTATATT |
| Sqstm1 | CTCTGGACACGATCCAGTATTC | CTGCTCTACGTGATGCAACTA |
| Tgfb1 | GGTGGTATACTGAGACACCTTG | CCCAAGGAAAGGTAGGTGATAG |
| Tgm2 | CATCACCAGCACTCTGTATCTC | GGTTCCTTCGGTTCCTTCAT |
| Tmem74 | TGCTCCAAGACAGTGCTATTC | GTTCCTGGCTGCAACAATTAC |
| Tnf | TTGTCTACTCCCAGGTTCTCT | GAGGTTGACTTTCTCCTGGTATG |
| Tnfsf10 | CAGCCCTAAAGTACCCAGTAATC | CACATCTGTCCTGAGGTTTCTAC |
| Trp53 | CAGTCTACTTCCCGCCATAAA | GTCTCAGCCCTGAAGTCATAAG |
| Ulk1 | CAGGGTGGACACATGCTAATAC | CAGCTTGTGGACACTCAGATAC |
| Ulk2 | CAACATCTCGTCAGACCACTC | CACTGCCCTCCACACATAAA |
| Uvrag | GACCACGAGACAGTTGAGATAG | GCAGGGACAATGGACTTAGAA |
| Wipi1 | GTGTGTCTAGACGACGAGAATG | GACTTCTGAGGTAGGCTTCTTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nah, J.; Sung, E.-A.; Zhai, P.; Zablocki, D.; Sadoshima, J. Tfeb-Mediated Transcriptional Regulation of Autophagy Induces Autosis during Ischemia/Reperfusion in the Heart. Cells 2022, 11, 258. https://doi.org/10.3390/cells11020258
Nah J, Sung E-A, Zhai P, Zablocki D, Sadoshima J. Tfeb-Mediated Transcriptional Regulation of Autophagy Induces Autosis during Ischemia/Reperfusion in the Heart. Cells. 2022; 11(2):258. https://doi.org/10.3390/cells11020258
Chicago/Turabian StyleNah, Jihoon, Eun-Ah Sung, Peiyong Zhai, Daniela Zablocki, and Junichi Sadoshima. 2022. "Tfeb-Mediated Transcriptional Regulation of Autophagy Induces Autosis during Ischemia/Reperfusion in the Heart" Cells 11, no. 2: 258. https://doi.org/10.3390/cells11020258
APA StyleNah, J., Sung, E. -A., Zhai, P., Zablocki, D., & Sadoshima, J. (2022). Tfeb-Mediated Transcriptional Regulation of Autophagy Induces Autosis during Ischemia/Reperfusion in the Heart. Cells, 11(2), 258. https://doi.org/10.3390/cells11020258