


Intracellular Metabolomics Identifies Efflux Transporter Inhibitors in a Routine Caco-2 Cell Permeability Assay—Biological Implications
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Transport Analysis
- Papp = (dQ/dt)/(C0 × Vr A)
- dQ/dt = slope of the linear portion of the permeated amount versus time curve (µmol/l/s)
- A = effective surface area of the Transwell insert (0.11 cm2)
- C0 = initial concentration of the substrate (µmol/L)
- Vr = the volume of the receiver chamber (mL).
- Papp (B to A)/Papp (A to B)
2.4. Knockout Cell Lines
2.5. Metabolite Extraction
2.6. LC-MS Sample Analysis for Targeted Metabolomics
2.7. Metabolite Linearity and Sensitivity
2.8. Data Analysis
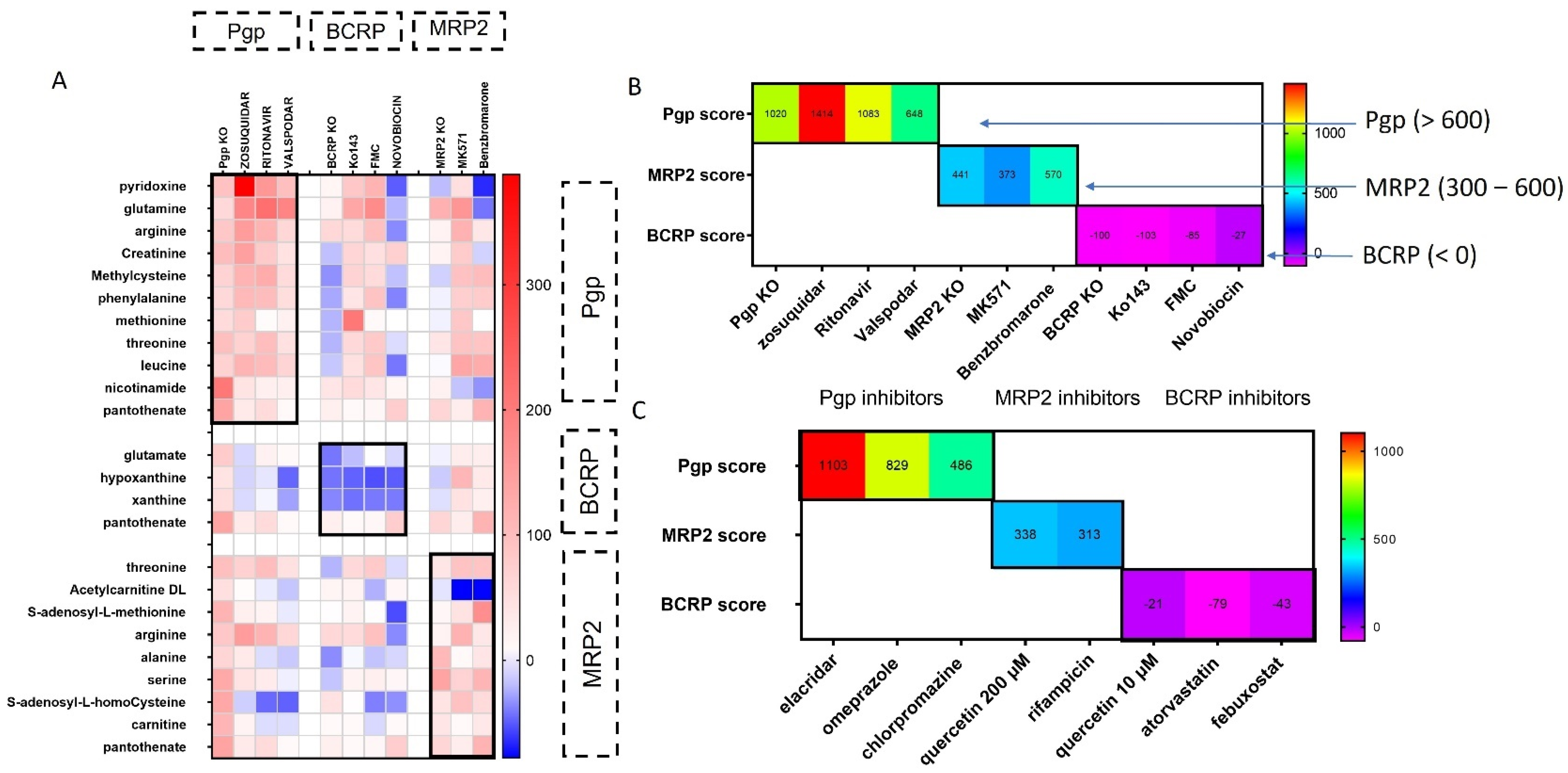
2.9. Scoring System for Transporter Inhibitor Classification
2.10. Analysis of Protein Expression
2.11. Statistical Analysis
3. Results
3.1. Functional Assays to Evaluate Inhibition of Transporters
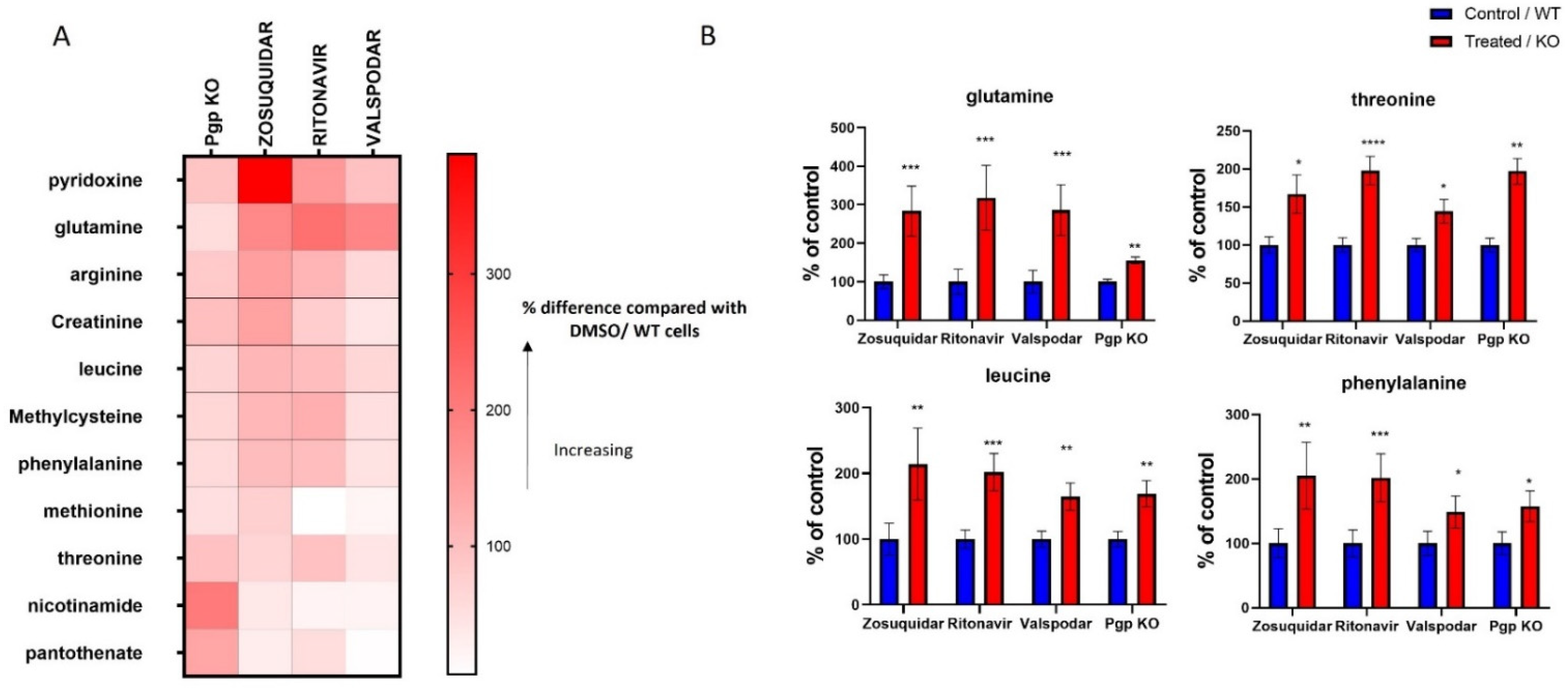
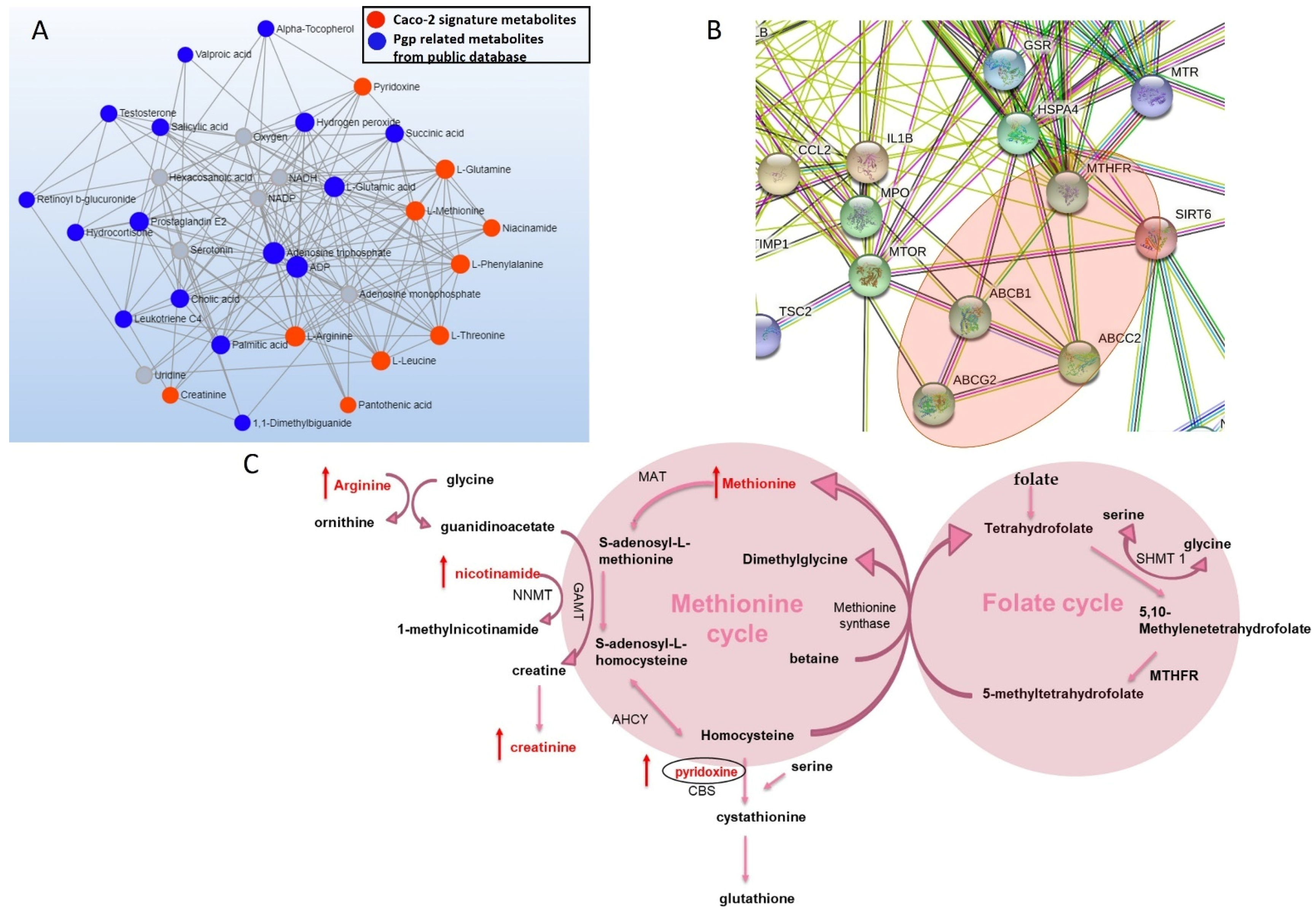
3.2. Metabolites Associated with Pgp Inhibition
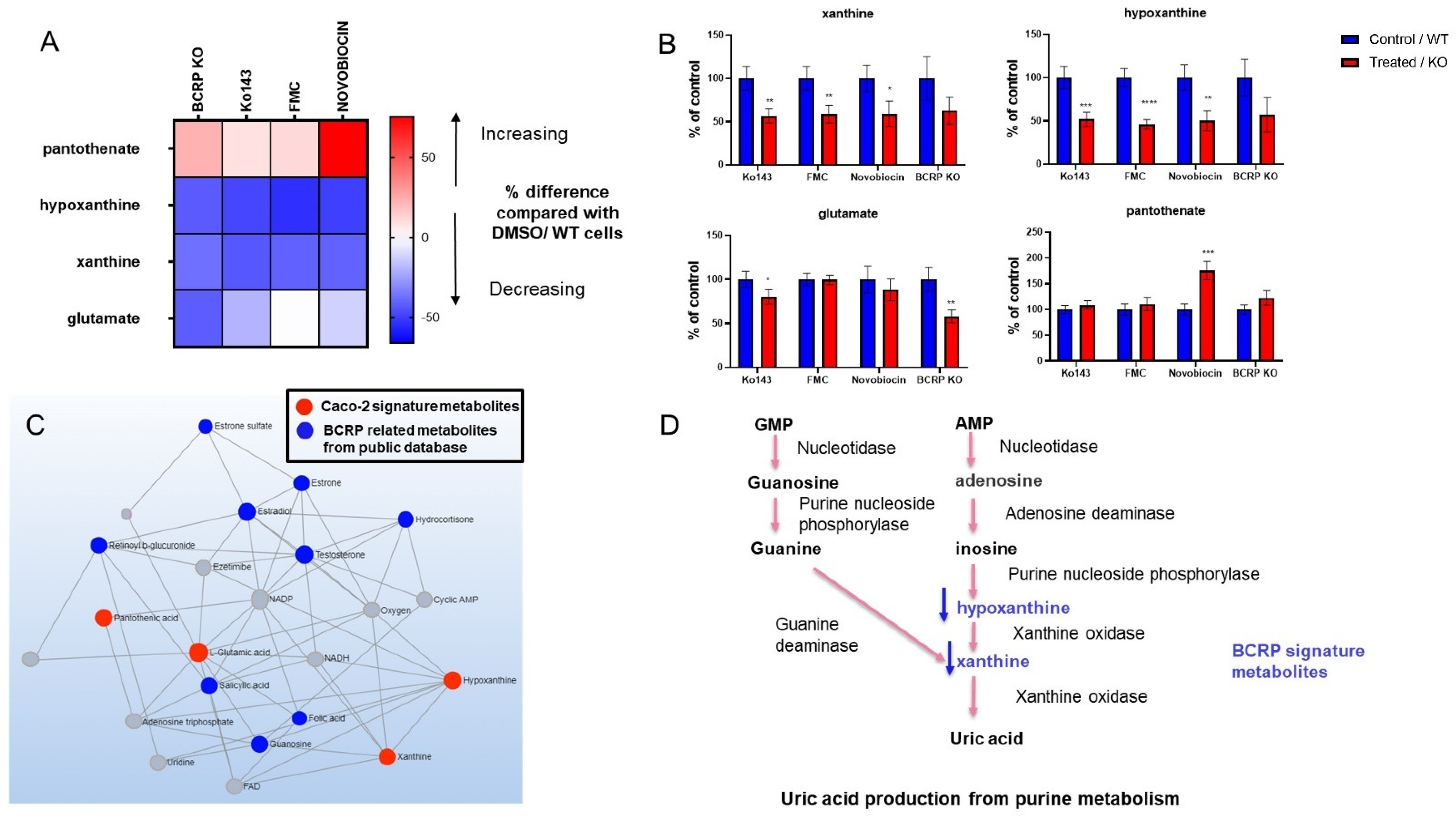
3.3. Metabolomic Changes Associated with BCRP Inhibition
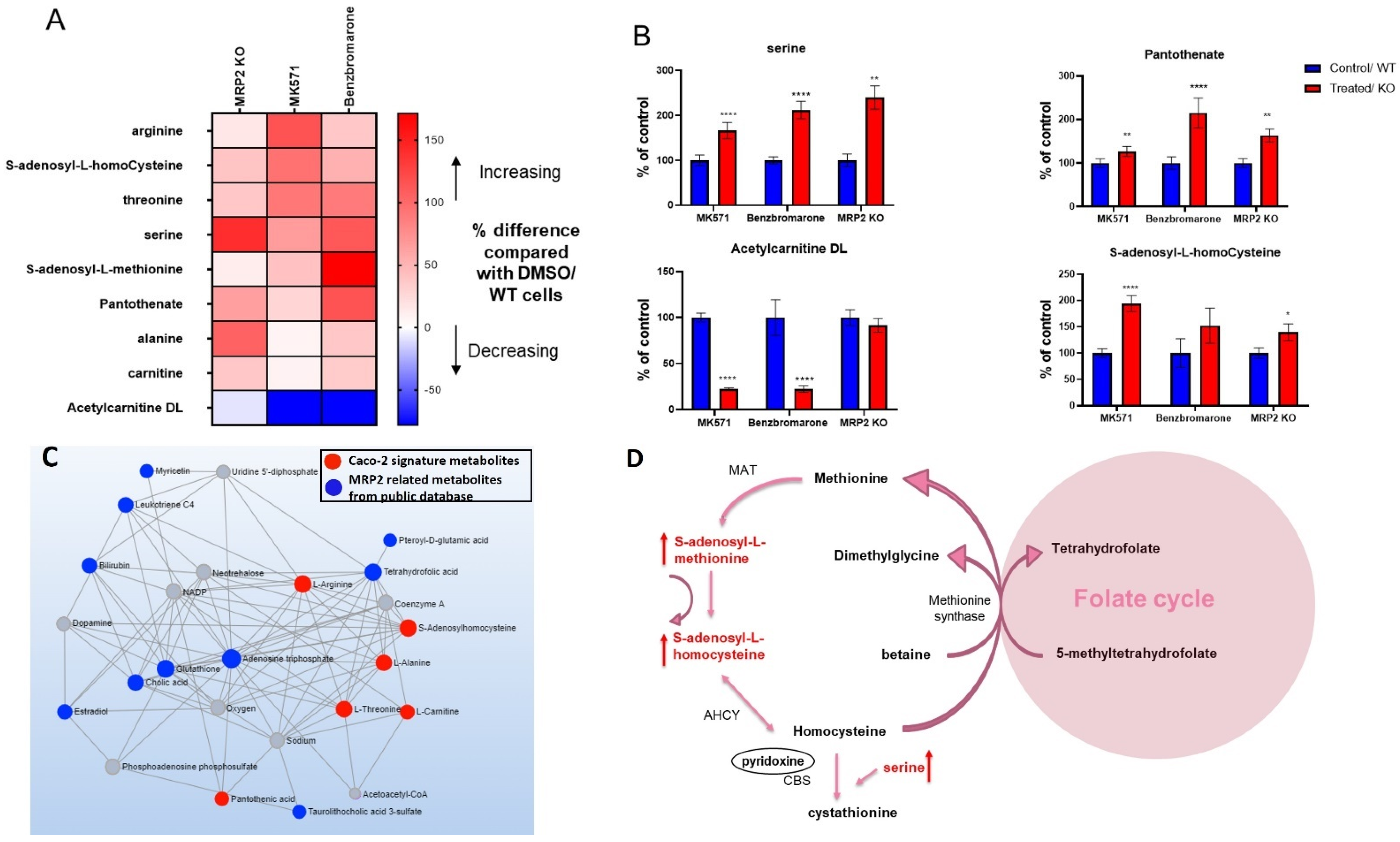
3.4. Alterations in Metabolites Induced by MRP2 Inhibition
3.5. Scoring System for Evaluating Transporter–Inhibitor Interactions
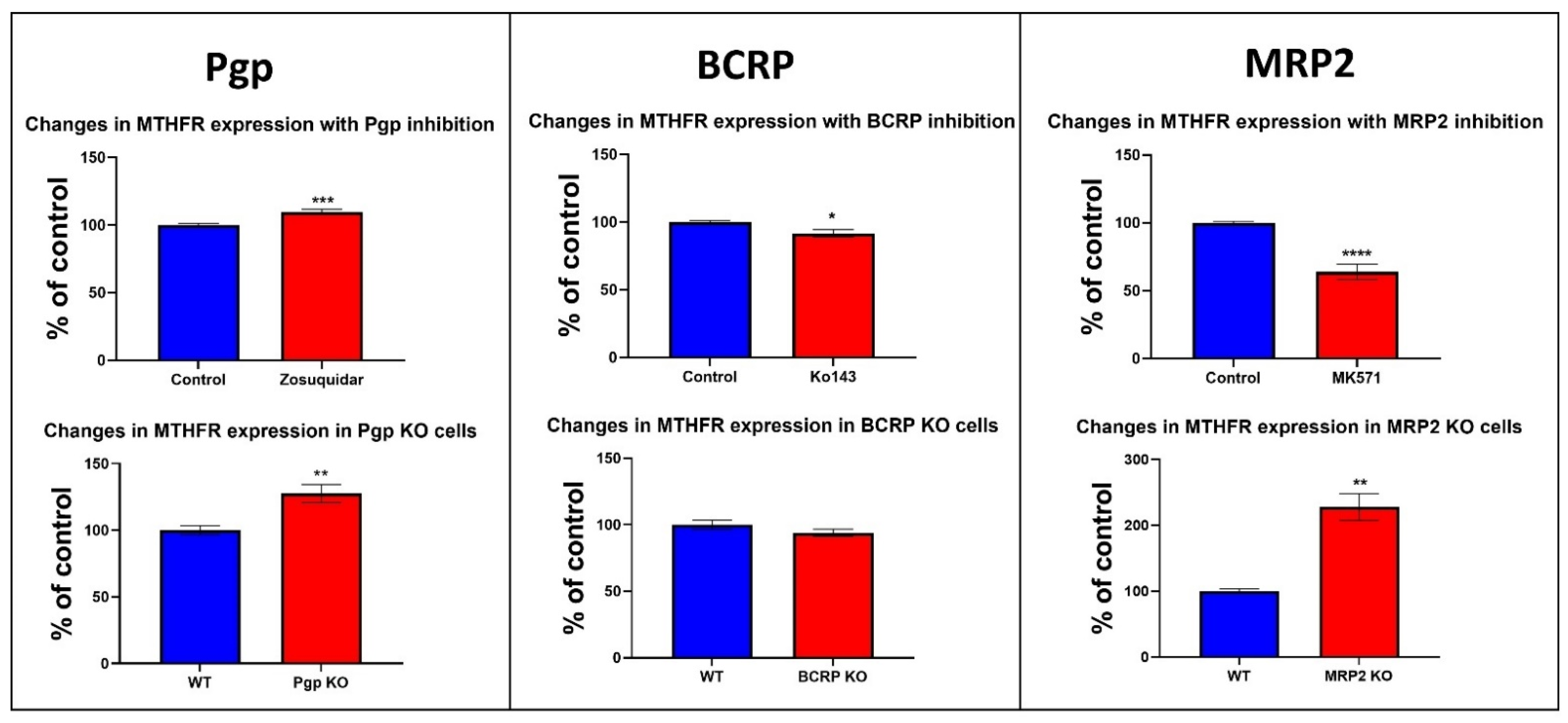
3.6. Changes in the Protein Expression of Enzymes Involved in Folate and Methionine Metabolism with Pgp, BCRP and MRP2 Inhibition and KO in Caco-2 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oosterhuis, B.; Grindstaff, K.; Kurunczi, A.; Toth, B.; Temesszentandrasi-Ambrus, C.; Novak, E.; Kovacs, E.; Kis, E.; Nagy, I.; Sahi, J.; et al. The Transporter Book, 3rd ed.; SOLVO Biotechnology: Budaörs, Hungary, 2017; pp. 28–29, 150–192. [Google Scholar]
- Liang, Y.; Li, S.; Ligong, C. The physiological role of drug transporters. Protein Cell 2015, 6, 334–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomini, K.M.; Huang, S.-M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toomula, N.; Sathish Kumar, D.; Kumar, A.; Phaneendra, M. Role of Pharmacokinetic Studies in Drug Discovery. J. Bioequiv. Bioavailab. 2011, 3, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Strong, J.M.; Qiu, W.; Lesko, L.J.; Huang, S.-M. Scientific Perspectives on Drug Transporters and Their Role in Drug Interactions. Mol. Pharm. 2006, 3, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Kivisto, K.T.; Niemi, M.; Fromm, M.F. Functional interaction of intestinal CYP3A4 and P-glycoprotein. Fundam. Clin. Pharmacol. 2004, 18, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Lan, L.-b.; Sanglard, D.; Furuya, K.; Schuetz, J.D.; Schuetz, E.G. Interaction of Cytochrome P450 3A Inhibitors with P-Glycoprotein. J. Pharmacol. Exp. Ther. 2002, 303, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Guo, X.; Lin, E.T.; Benet, L.Z. Overlapping Substrate Specificities of Cytochrome P450 3A and P-Glycoprotein for a Novel Cysteine Protease Inhibitor. Drug Metab. Dispos. 1998, 26, 360–366. [Google Scholar] [PubMed]
- Shugarts, S.; Benet, L.Z. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm. Res. 2009, 26, 2039–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, K.; Crowe, A.; Wang, X.; Zhang, P.; Ding, K.; Li, L.; Yue, W. Regulation of Organic Anion Transporting Polypeptides (OATP) 1B1- and OATP1B3-Mediated Transport: An Updated Review in the Context of OATP-Mediated Drug-Drug Interactions. Int. J. Mol. Sci. 2018, 19, 855. [Google Scholar] [CrossRef]
- In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry; U.S. Food and Drug Administration, 2020. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions (accessed on 1 August 2022).
- Wessler, J.D.; Grip, L.T.; Mendell, J.; Giugliano, R.P. The P-Glycoprotein Transport System and Cardiovascular Drugs. J. Am. Coll. Cardiol. 2013, 61, 2495–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.; Zahir, H.; Matsushima, N.; Noveck, R.; Lee, F.; Chen, S.; Zhang, G.; Shi, M. Drug-drug interaction studies of cardiovascular drugs involving P-glycoprotein, an efflux transporter, on the pharmacokinetics of edoxaban, an oral factor Xa inhibitor. Am. J. Cardiovasc. Drugs 2013, 13, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsby, R.; Martin, P.D.; Surry, D.; Sharma, P.; Fenner, K. Solitary Inhibition of the Breast Cancer Resistance Protein (BCRP) Efflux Transporter Results in a Clinically Significant Drug-Drug Interaction with Rosuvastatin by Causing Up To a Two-Fold Increase in Statin Exposure. Drug Metab. Dispos. 2015, 44, 398–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Liu, K.; Liu, Z.; Liu, Q.; Wang, C.; Meng, Q.; Huo, X.; Sun, P.; Sun, H.; Yang, X.; et al. P-gp, MRP2 and OAT1/OAT3 mediate the drug-drug interaction between resveratrol and methotrexate. Toxicol. Appl. Pharmacol. 2016, 306, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.T.; Wu, W.; Lun, C.; Nigam, S.K. The drug transporter OAT3 (SLC22A8) and endogenous metabolite communication via the gut-liver-kidney axis. J. Biol. Chem. 2017, 292, 15789–15803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köck, K.; Ferslew, B.C.; Netterberg, I.; Yang, K.; Urban, T.J.; Swaan, P.W.; Stewart, P.W.; Brouwer, K.L.R. Risk factors for development of cholestatic drug-induced liver injury: Inhibition of hepatic basolateral bile acid transporters multidrug resistance-associated proteins 3 and 4. Drug Metab. Dispos. 2014, 42, 665–674. [Google Scholar] [CrossRef]
- MacLean, C.; Moenning, U.; Reichel, A.; Fricker, G. Closing the gaps: A full scan of the intestinal expression of p-glycoprotein, breast cancer resistance protein, and multidrug resistance-associated protein 2 in male and female rats. Drug Metab. Dispos. 2008, 36, 1249–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, I.J.; Raub, T.J.; Borchardt, R.T. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989, 96, 736–749. [Google Scholar] [CrossRef]
- Sun, H.; Chow, E.C.Y.; Liu, S.; Du, Y.; Pang, K.S. The Caco-2 cell monolayer: Usefulness and limitations. Expert Opin. Drug Metab. Toxicol. 2008, 4, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.C.; Curi, R.; Genvigir, F.D.; Hirata, M.H.; Hirata, R.D. The expression of efflux and uptake transporters are regulated by statins in Caco-2 and HepG2 cells. Acta Pharmacol. Sin. 2009, 30, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Siissalo, S.; Hannukainen, J.; Kolehmainen, J.; Hirvonen, J.; Kaukonen, A.M. A Caco-2 cell based screening method for compounds interacting with MRP2 efflux protein. Eur. J. Pharm. Biopharm. 2009, 71, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Sampson, K.E.; Brinker, A.; Pratt, J.; Venkatraman, N.; Xiao, Y.; Blasberg, J.; Steiner, T.; Bourner, M.; Thompson, D.C. Zinc Finger Nuclease–Mediated Gene Knockout Results in Loss of Transport Activity for P-Glycoprotein, BCRP, and MRP2 in Caco-2 Cells. Drug Metab. Dispos. 2015, 43, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, M.; Breitkopf, S.B.; Yang, X.; Asara, J.M. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 2012, 7, 872–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepard, R.L.; Cao, J.; Starling, J.J.; Dantzig, A.H. Modulation of P-glycoprotein but not MRP1- or BCRP-mediated drug resistance by LY335979. Int. J. Cancer 2003, 103, 121–125. [Google Scholar] [CrossRef] [PubMed]
- SOLVO. SOLVO Introducing PSC 833 the Specific P-gp Inhibitor and New Drug Transporter Services. SOLVO, 4 June 2009. [Google Scholar]
- Drewe, J.; Gutmann, H.; Fricker, G.; Török, M.; Beglinger, C.; Huwyler, J. HIV protease inhibitor ritonavir: A more potent inhibitor of P-glycoprotein than the cyclosporine analog SDZ PSC 833. Biochem. Pharmacol. 1999, 57, 1147–1152. [Google Scholar] [CrossRef]
- Vidmar Golja, M.; Šmid, A.; Karas Kuželički, N.; Trontelj, J.; Geršak, K.; Mlinarič-Raščan, I. Folate Insufficiency Due to MTHFR Deficiency Is Bypassed by 5-Methyltetrahydrofolate. J. Clin. Med. 2020, 9, 2836. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine Oxidoreductase in Drug Metabolism: Beyond a Role as a Detoxifying Enzyme. Curr. Med. Chem. 2016, 23, 4027–4036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosomi, A.; Nakanishi, T.; Fujita, T.; Tamai, I. Extra-renal elimination of uric acid via intestinal efflux transporter BCRP/ABCG2. PLoS ONE 2012, 7, e30456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Wang, Z.; Zhang, K.; Chi, Z.; Xu, T.; Jiang, D.; Chen, S.; Li, W.; Yang, X.; Zhang, X.; et al. One-Carbon Metabolism Supports S-Adenosylmethionine and Histone Methylation to Drive Inflammatory Macrophages. Mol. Cell 2019, 75, 1147–1160.e5. [Google Scholar] [CrossRef] [PubMed]
- Caudill, M.A.; Wang, J.C.; Melnyk, S.; Pogribny, I.P.; Jernigan, S.; Collins, M.D.; Santos-Guzman, J.; Swendseid, M.E.; Cogger, E.A.; James, S.J. Intracellular S-Adenosylhomocysteine Concentrations Predict Global DNA Hypomethylation in Tissues of Methyl-Deficient Cystathionine β-Synthase Heterozygous Mice. J. Nutr. 2001, 131, 2811–2818. [Google Scholar] [CrossRef] [PubMed]
- Sugie, M.; Asakura, E.; Zhao, Y.L.; Torita, S.; Nadai, M.; Baba, K.; Kitaichi, K.; Takagi, K.; Takagi, K.; Hasegawa, T. Possible involvement of the drug transporters P glycoprotein and multidrug resistance-associated protein Mrp2 in disposition of azithromycin. Antimicrob. Agents Chemother. 2004, 48, 809–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igari, S.; Ohtaki, A.; Yamanaka, Y.; Sato, Y.; Yohda, M.; Odaka, M.; Noguchi, K.; Yamada, K. Properties and crystal structure of methylenetetrahydrofolate reductase from Thermus thermophilus HB8. PLoS ONE 2011, 6, e23716. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, M.; Thakur, J.; Suyal, S.; Oniel, R.; Chakraborty, R.; Pradhan, S.; Sharma, M.; Sengupta, S.; Laxman, S.; Masakapalli, S.K.; et al. Allosteric inhibition of MTHFR prevents futile SAM cycling and maintains nucleotide pools in one-carbon metabolism. J. Biol. Chem. 2020, 295, 16037–16057. [Google Scholar] [CrossRef] [PubMed]
- McFadden, J.; Girard, C.; Tao, S.; Zhou, Z.; Bernard, J.; Duplessis, M.; White, H. Symposium review: One-carbon metabolism and methyl donor nutrition in the dairy cow. J. Dairy Sci. 2020, 103, 5668–5683. [Google Scholar] [CrossRef]
- Ganguly, S.; Finkelstein, D.; Shaw, T.I.; Michalek, R.D.; Zorn, K.M.; Ekins, S.; Yasuda, K.; Fukuda, Y.; Schuetz, J.D.; Mukherjee, K.; et al. Metabolomic and transcriptomic analysis reveals endogenous substrates and metabolic adaptation in rats lacking Abcg2 and Abcb1a transporters. PLoS ONE 2021, 16, e0253852. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.; Lee, J.; Park, J.-C.; Kim, K.H.; Ko, J.M.; Park, S.-H.; Kim, S.-K.; Mook-Jung, I.; Lee, J.Y. Neurotoxicity of phenylalanine on human iPSC-derived cerebral organoids. Mol. Genet. Metab. 2022, 136, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Rojas, C.; Lindsay, C.B.; Montecinos-Oliva, C.; Arrazola, M.S.; Retamales, R.M.; Bunout, D.; Hirsch, S.; Inestrosa, N.C. Is L-methionine a trigger factor for Alzheimer’s-like neurodegeneration?: Changes in Aβ oligomers, tau phosphorylation, synaptic proteins, Wnt signaling and behavioral impairment in wild-type mice. Mol. Neurodegener. 2015, 10, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, J.-I.; Tseng, Y.-J.; Chen, M.-H.; Huang, C.-Y.F.; Chang, P.M.-H. Clinical Perspective of FDA Approved Drugs with P-Glycoprotein Inhibition Activities for Potential Cancer Therapeutics. Front. Oncol. 2020, 10, 561936. [Google Scholar] [CrossRef] [PubMed]
- Ogura, J.; Kuwayama, K.; Sasaki, S.; Kaneko, C.; Koizumi, T.; Yabe, K.; Tsujimoto, T.; Takeno, R.; Takaya, A.; Kobayashi, M.; et al. Reactive oxygen species derived from xanthine oxidase interrupt dimerization of breast cancer resistance protein, resulting in suppression of uric acid excretion to the intestinal lumen. Biochem. Pharmacol. 2015, 97, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Bobulescu, I.A.; Moe, O.W. Renal Transport of Uric Acid: Evolving Concepts and Uncertainties. Adv. Chronic Kidney Dis. 2012, 19, 358–371. [Google Scholar] [CrossRef]
- Tátrai, P.; Erdő, F.; Dörnyei, G.; Krajcsi, P. Modulation of Urate Transport by Drugs. Pharmaceutics 2021, 13, 899. [Google Scholar] [CrossRef] [PubMed]
- Komori, H.; Yamada, K.; Tamai, I. Hyperuricemia enhances intracellular urate accumulation via down-regulation of cell-surface BCRP/ABCG2 expression in vascular endothelial cells. Biochim. Biophys. Acta Biomembr. 2018, 1860, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G. The role of multidrug resistance efflux transporters in antifolate resistance and folate homeostasis. Drug Resist. Updat. 2006, 9, 227–246. [Google Scholar] [CrossRef] [PubMed]
- Lemos, C.; Kathmann, I.; Giovannetti, E.; Belien, J.A.; Scheffer, G.L.; Calhau, C.; Jansen, G.; Peters, G.J. Cellular folate status modulates the expression of BCRP and MRP multidrug transporters in cancer cell lines from different origins. Mol. Cancer Ther. 2009, 8, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Ifergan, I.; Shafran, A.; Jansen, G.; Hooijberg, J.H.; Scheffer, G.L.; Assaraf, Y.G. Folate deprivation results in the loss of breast cancer resistance protein (BCRP/ABCG2) expression. A role for BCRP in cellular folate homeostasis. J. Biol. Chem. 2004, 279, 25527–25534. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Guo, W.; Yang, Y.; Ji, T.; Xu, J. More severe toxicity of genetic polymorphisms on MTHFR activity in osteosarcoma patients treated with high-dose methotrexate. Oncotarget 2017, 9, 11465–11476. [Google Scholar] [CrossRef] [Green Version]
- Calderon-Larranaga, A.; Saadeh, M.; Hooshmand, B.; Refsum, H.; Smith, A.D.; Marengoni, A.; Vetrano, D.L. Association of Homocysteine, Methionine, and MTHFR 677C>T Polymorphism with Rate of Cardiovascular Multimorbidity Development in Older Adults in Sweden. JAMA Netw. Open 2020, 3, e205316. [Google Scholar] [CrossRef]
- Raghubeer, S.; Matsha, T.E. Methylenetetrahydrofolate (MTHFR), the One-Carbon Cycle, and Cardiovascular Risks. Nutrients 2021, 13, 4562. [Google Scholar] [CrossRef]
- Bhatia, P.; Singh, N. Homocysteine excess: Delineating the possible mechanism of neurotoxicity and depression. Fundam. Clin. Pharmacol. 2015, 29, 522–528. [Google Scholar] [CrossRef]
- Clark, R.; Kerr, I.D.; Callaghan, R. Multiple drugbinding sites on the R482G isoform of the ABCG2 transporter. Br. J. Pharmacol. 2006, 149, 506–515. [Google Scholar] [CrossRef]
- Matsson, P.; Englund, G.; Ahlin, G.; Bergstrom, C.A.; Norinder, U.; Artursson, P. A global drug inhibition pattern for the human ATP-binding cassette transporter breast cancer resistance protein (ABCG2). J. Pharmacol. Exp. Ther. 2007, 323, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Gollavilli, P.N.; Ramesh, V.; Parma, B.; Schwab, A.; Vazakidou, M.E.; Natesan, R.; Saatci, O.; Rapa, I.; Bironzo, P.; et al. Thymidylate synthase drives the phenotypes of epithelial-to-mesenchymal transition in non-small cell lung cancer. Br. J. Cancer 2021, 124, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Berger, F.G.; Berger, S.H. Thymidylate synthase as a chemotherapeutic drug target: Where are we after fifty years? Cancer Biol. Ther. 2006, 5, 1238–1241. [Google Scholar] [CrossRef]
- Parkhitko, A.A.; Jouandin, P.; Mohr, S.E.; Perrimon, N. Methionine metabolism and methyltransferases in the regulation of aging and lifespan extension across species. Aging Cell 2019, 18, e13034. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.O.; Wang, L.; Kuo, Y.M.; Andrews, A.J.; Gupta, S.; Kruger, W.D. S-adenosylhomocysteine hydrolase over-expression does not alter S-adenosylmethionine or S-adenosylhomocysteine levels in CBS deficient mice. Mol. Genet. Metab. Rep. 2018, 15, 15–21. [Google Scholar] [CrossRef]
- Herbig, K.; Chiang, E.-P.; Lee, L.-R.; Hills, J.; Shane, B.; Stover, P.J. Cytoplasmic Serine Hydroxymethyltransferase Mediates Competition between Folate-dependent Deoxyribonucleotide andS-Adenosylmethionine Biosyntheses. J. Biol. Chem. 2002, 277, 38381–38389. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, I.; Sohn, K.-J.; Stempak, J.M.; Croxford, R.; Kim, Y.-I. Folate Deficiency Induces Cell-Specific Changes in the Steady-State Transcript Levels of Genes Involved in Folate Metabolism and 1-Carbon Transfer Reactions in Human Colonic Epithelial Cells. J. Nutr. 2007, 137, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Chen, X.; Li, J.; Sun, S. Serine and Metabolism Regulation: A Novel Mechanism in Antitumor Immunity and Senescence. Aging Dis. 2020, 11, 1640–1653. [Google Scholar] [CrossRef]
- Labuschagne, C.F.; Van Den Broek, N.J.F.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D.K. Serine, but Not Glycine, Supports One-Carbon Metabolism and Proliferation of Cancer Cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [Green Version]
- Reid, M.A.; Allen, A.E.; Liu, S.; Liberti, M.V.; Liu, P.; Liu, X.; Dai, Z.; Gao, X.; Wang, Q.; Liu, Y.; et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat. Commun. 2018, 9, 5442. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naseem, A.; Pal, A.; Gowan, S.; Asad, Y.; Donovan, A.; Temesszentandrási-Ambrus, C.; Kis, E.; Gaborik, Z.; Bhalay, G.; Raynaud, F. Intracellular Metabolomics Identifies Efflux Transporter Inhibitors in a Routine Caco-2 Cell Permeability Assay—Biological Implications. Cells 2022, 11, 3286. https://doi.org/10.3390/cells11203286
Naseem A, Pal A, Gowan S, Asad Y, Donovan A, Temesszentandrási-Ambrus C, Kis E, Gaborik Z, Bhalay G, Raynaud F. Intracellular Metabolomics Identifies Efflux Transporter Inhibitors in a Routine Caco-2 Cell Permeability Assay—Biological Implications. Cells. 2022; 11(20):3286. https://doi.org/10.3390/cells11203286
Chicago/Turabian StyleNaseem, Afia, Akos Pal, Sharon Gowan, Yasmin Asad, Adam Donovan, Csilla Temesszentandrási-Ambrus, Emese Kis, Zsuzsanna Gaborik, Gurdip Bhalay, and Florence Raynaud. 2022. "Intracellular Metabolomics Identifies Efflux Transporter Inhibitors in a Routine Caco-2 Cell Permeability Assay—Biological Implications" Cells 11, no. 20: 3286. https://doi.org/10.3390/cells11203286
APA StyleNaseem, A., Pal, A., Gowan, S., Asad, Y., Donovan, A., Temesszentandrási-Ambrus, C., Kis, E., Gaborik, Z., Bhalay, G., & Raynaud, F. (2022). Intracellular Metabolomics Identifies Efflux Transporter Inhibitors in a Routine Caco-2 Cell Permeability Assay—Biological Implications. Cells, 11(20), 3286. https://doi.org/10.3390/cells11203286