
Therapeutic Targeting of NF-κB in Acute Lung Injury: A Double-Edged Sword



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Pulmonary Endothelium: A Key Orchestrator of Inflammatory Lung Injury
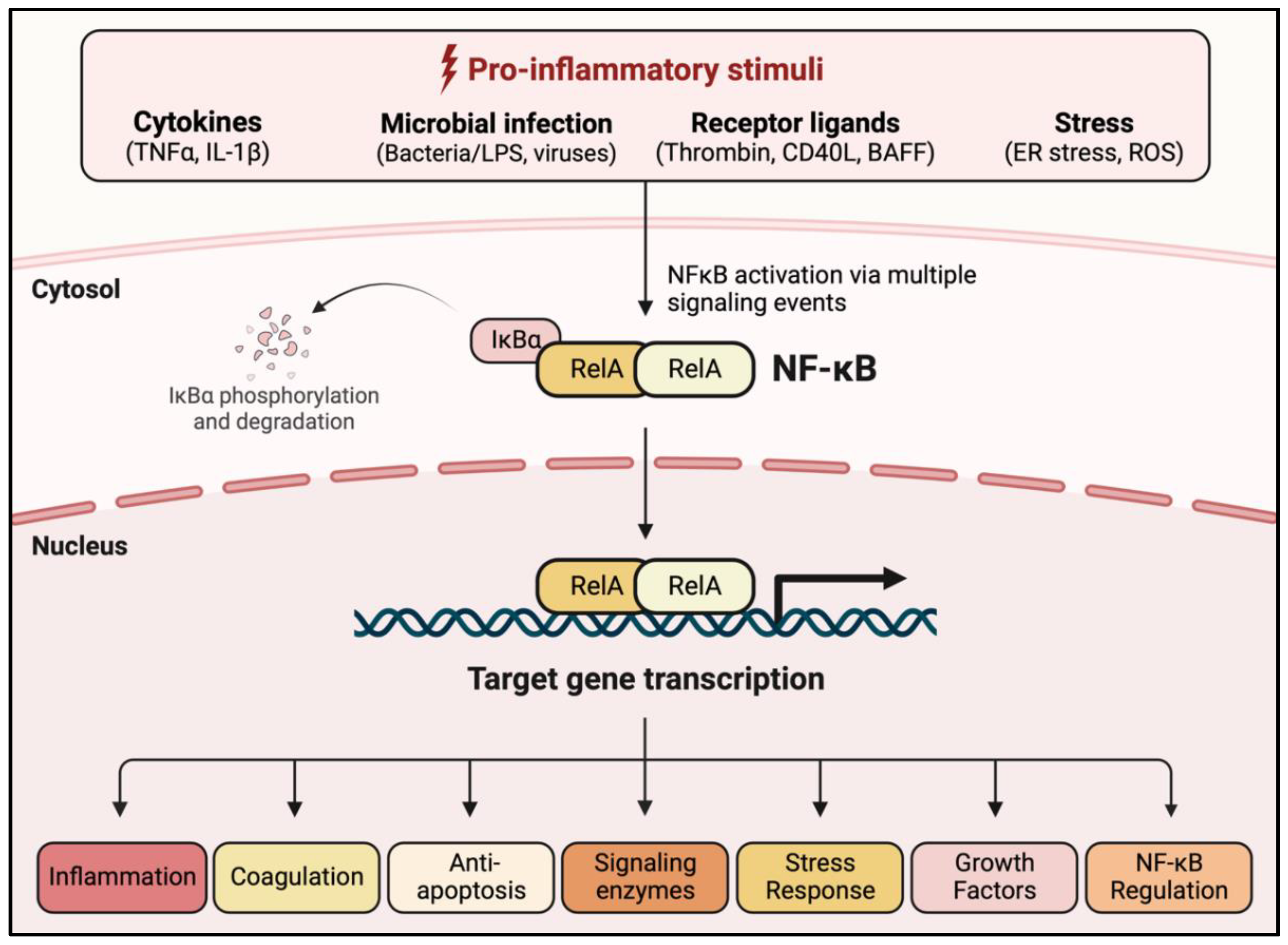
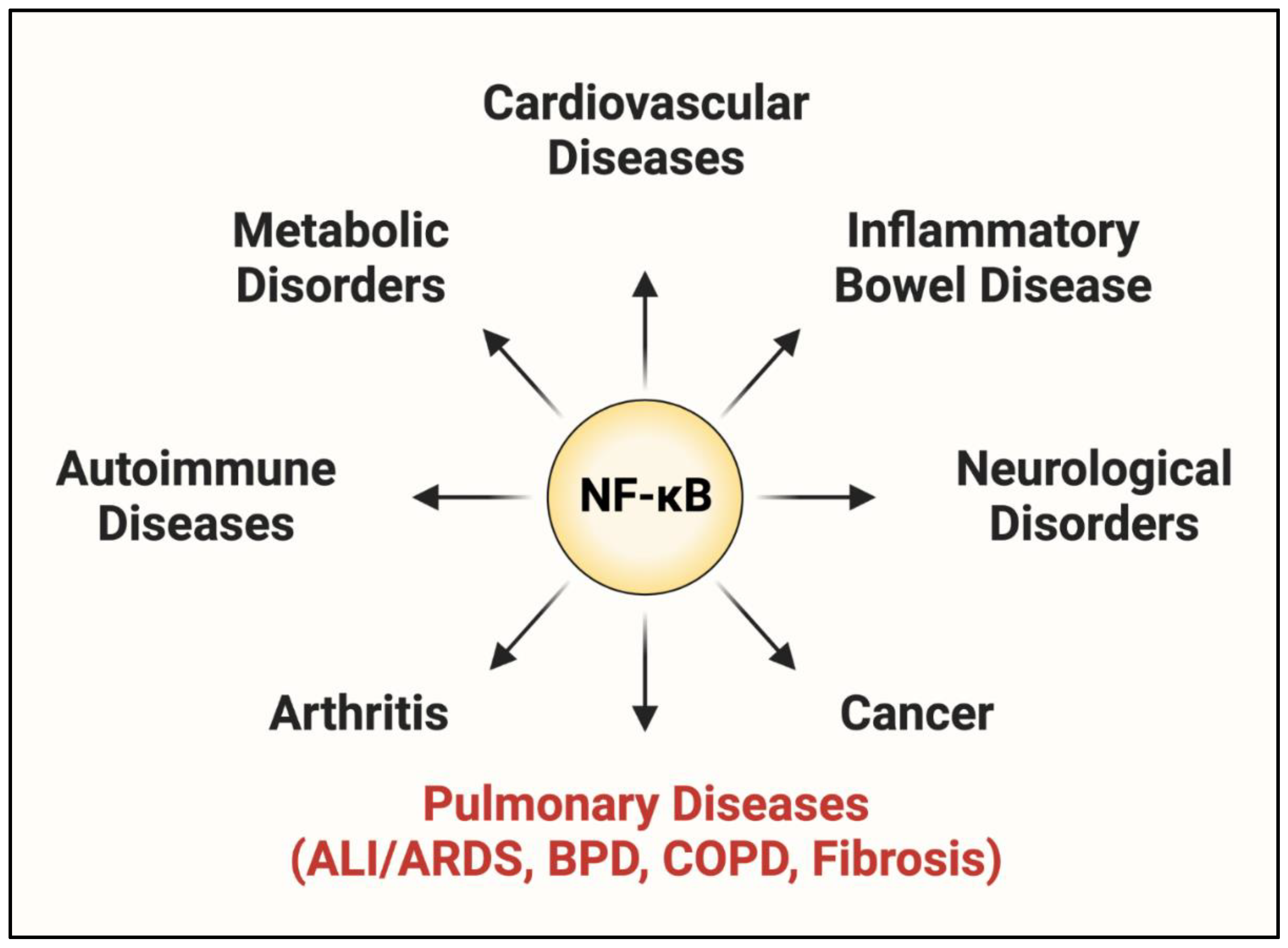
1.2. NF-κB: A Central Regulator of EC Dysfunction
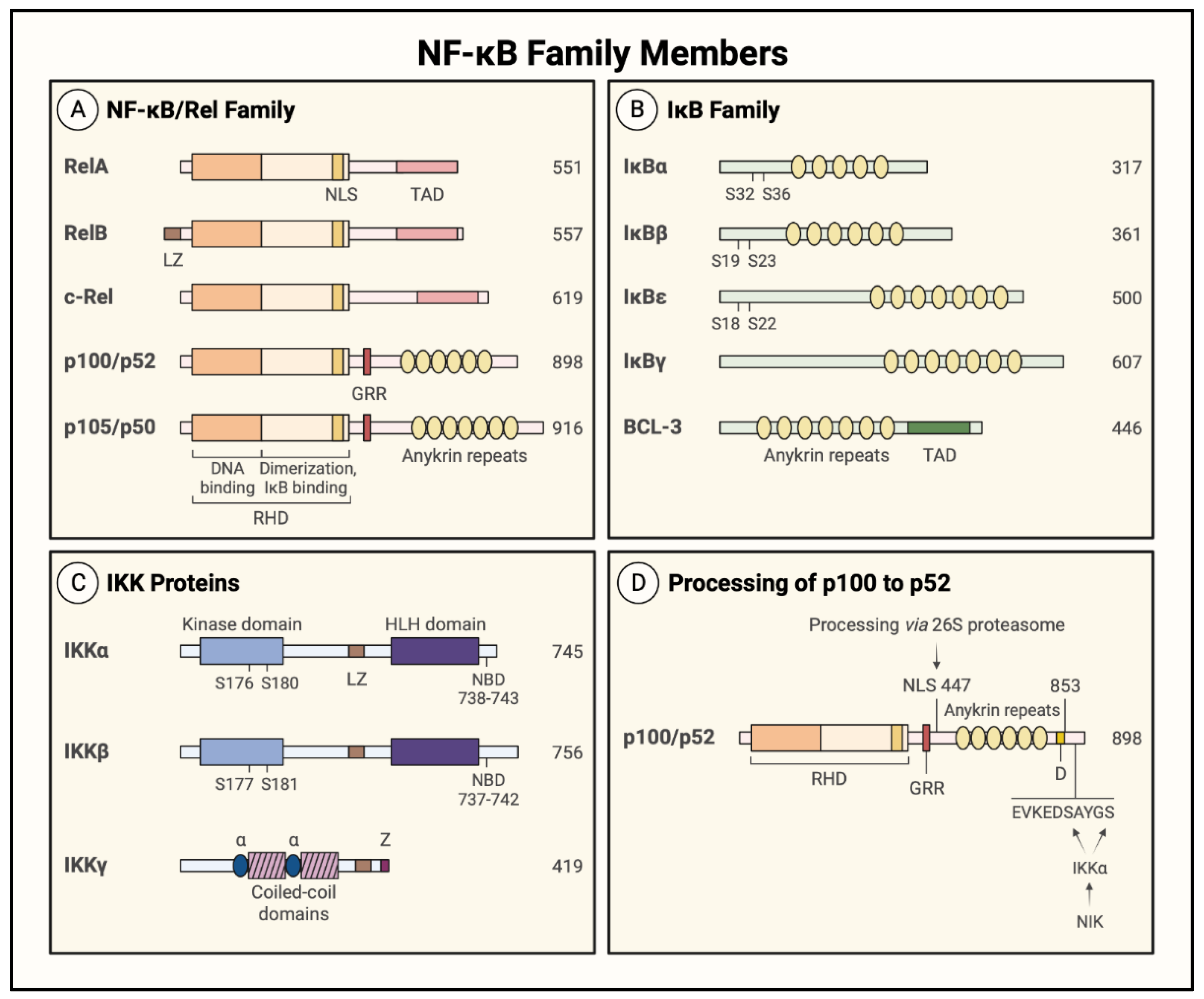
1.3. NF-κB Family Members and Inhibitors
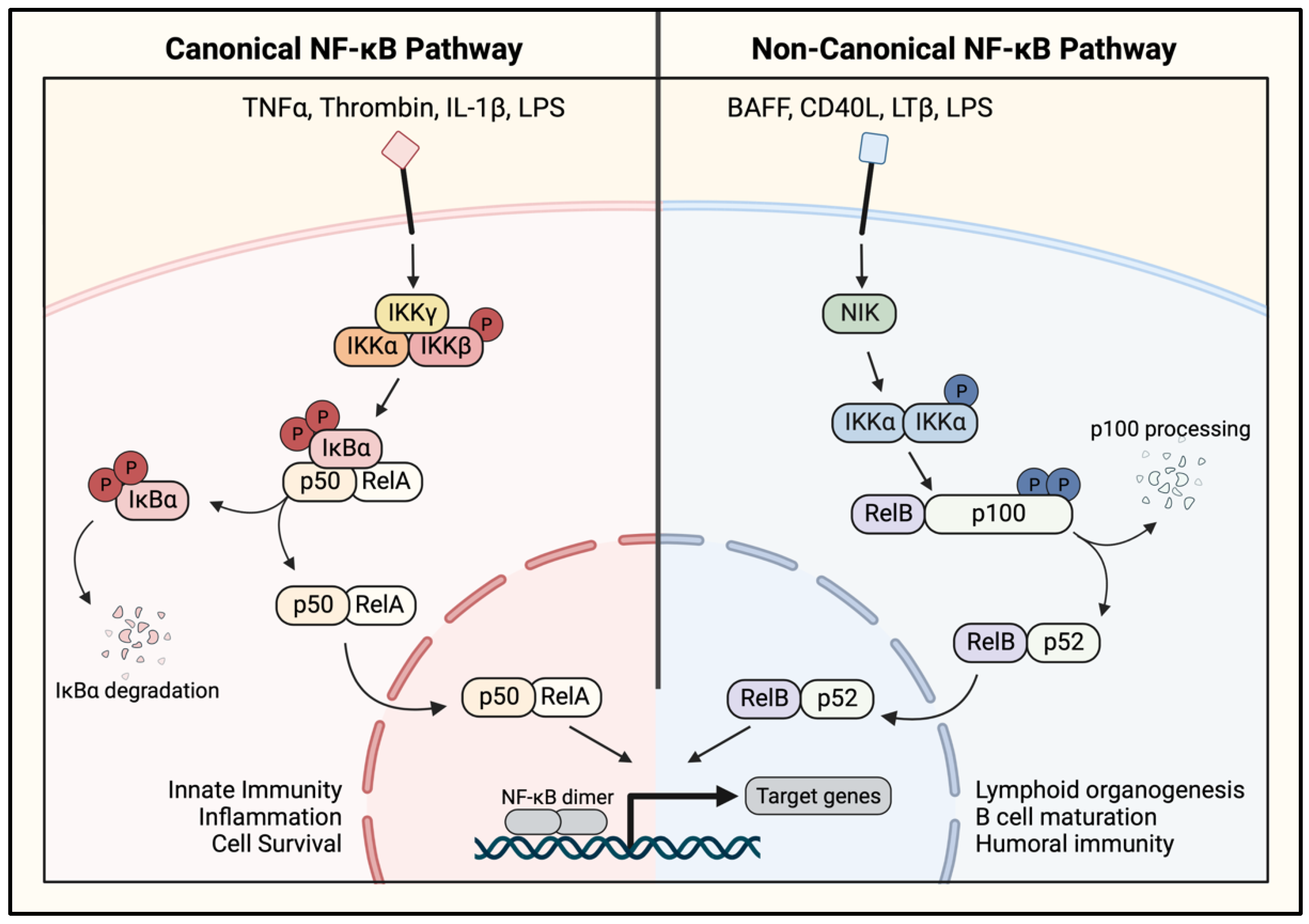
1.4. NF-κB Activation Pathways
2. Signaling to NF-κB in the Endothelium
2.1. PKC Signaling to IKK/NF-κB
2.2. PI3K/Akt Signaling to IKK/NF-κB
2.3. MAPK/p38 Signaling to IKK/NF-κB
2.4. Calcium Signaling to IKK/NF-κB
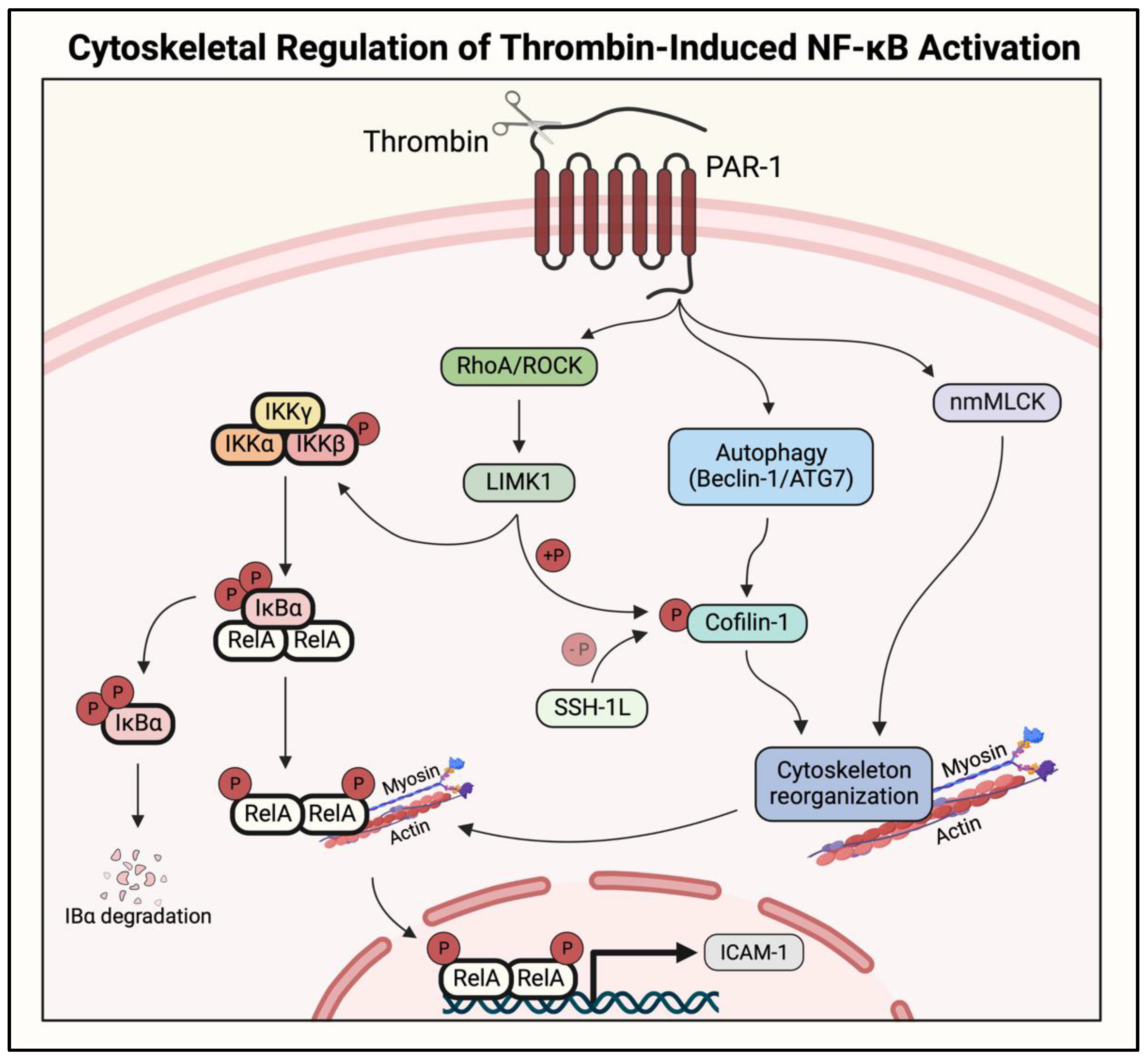
2.5. Actin–Myosin Interaction and Small GTPase Signaling to IKK/NF-κB
2.6. Oxidant Signaling to IKK/NF-κB
2.7. Tyrosine Kinase Signaling to IKK/NF-κB
2.8. ER and Mitochondrial Chaperone Signaling to IKK/NF-κB
2.9. Autophagy Protein Signaling to IKK/NF-κB
2.10. MTOR Signaling to IKK/NF-κB
3. Therapeutic Potential and Problems of Targeting NF-κB in ALI
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Prim. 2019, 5, 18. [Google Scholar] [CrossRef]
- Perl, M.; Lomas-Neira, J.; Venet, F.; Chung, C.S.; Ayala, A. Pathogenesis of indirect (secondary) acute lung injury. Expert. Rev. Respir. Med. 2011, 5, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Jagrosse, M.L.; Dean, D.A.; Rahman, A.; Nilsson, B.L. RNAi therapeutic strategies for acute respiratory distress syndrome. Transl. Res. 2019, 214, 30–49. [Google Scholar] [CrossRef] [PubMed]
- Tzotzos, S.J.; Fischer, B.; Fischer, H.; Zeitlinger, M. Incidence of ARDS and outcomes in hospitalized patients with COVID-19: A global literature survey. Crit. Care 2020, 24, 516. [Google Scholar] [CrossRef]
- Hasan, A.; Susanto, H.; Kasim, M.F.; Nuraini, N.; Lestari, B.; Triany, D.; Widyastuti, W. Superspreading in early transmissions of COVID-19 in Indonesia. Sci. Rep. 2020, 10, 22386. [Google Scholar] [CrossRef]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 22 September 2022).
- Ware, L.B.; Matthay, M.A. The Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef]
- Ashbaugh, D.G.; Bigelow, D.B.; Petty, T.L.; Levine, B.E. Acute respiratory distress in adults. Lancet 1967, 2, 319–323. [Google Scholar] [CrossRef]
- Maniatis, N.A.; Orfanos, S.E. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr. Opin. Crit. Care 2008, 14, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniatis, N.A.; Kotanidou, A.; Catravas, J.D.; Orfanos, S.E. Endothelial pathomechanisms in acute lung injury. Vascul. Pharmacol. 2008, 49, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Orfanos, S.E.; Mavrommati, I.; Korovesi, I.; Roussos, C. Pulmonary endothelium in acute lung injury: From basic science to the critically ill. Appl. Physiol. Intensive Care Med. 2004, 30, 1702–1714. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ye, R.D.; Malik, A.B. Transcriptional mechanisms of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L1037–L1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, R.B.; Strieter, R.M.; Martin, D.P.; Steinberg, K.P.; Milberg, J.A.; Maunder, R.J.; Kunkel, S.L.; Walz, A.; Hudson, L.D.; Martin, T.R. Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1996, 154, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C. Neutrophil kinetics and lung injury. Physiol. Rev. 1987, 67, 1249–1295. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Inoue, T.; Kyi, M.; Sawada, M.; Miyake, S.; Yoshizawa, Y. Effects of a neutrophil elastase inhibitor (ONO-5046) on acute pulmonary injury induced by tumor necrosis factor alpha (TNFalpha) and activated neutrophils in isolated perfused rabbit lungs. Am. J. Respir. Crit. Care Med. 1998, 157, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Uchiba, M.; Okajima, K.; Murakami, K.; Okabe, H.; Takatsuki, K. Endotoxin-induced pulmonary vascular injury is mainly mediated by activated neutrophils in rats. Thromb. Res. 1995, 78, 117–125. [Google Scholar] [CrossRef]
- Folz, R.J.; Abushamaa, A.M.; Suliman, H.B. Extracellular superoxide dismutase in the airways of transgenic mice reduces inflammation and attenuates lung toxicity following hyperoxia. J. Clin. Investig. 1999, 103, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Aird, W.C. Sepsis and coagulation. Crit. Care Clin. 2005, 21, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Fazal, F. Blocking NF-kappaB: An inflammatory issue. Proc. Am. Thorac. Soc. 2011, 8, 497–503. [Google Scholar] [CrossRef]
- Dudek, S.M.; Garcia, J.G. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 2001, 91, 1487–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A.; Fazal, F. Hug tightly and say goodbye: Role of endothelial ICAM-1 in leukocyte transmigration. Antioxid. Redox Signal. 2009, 11, 823–839. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Ye, X.; Xu, H.; Liu, S.F. Activation of endothelial intrinsic NF-{kappa}B pathway impairs protein C anticoagulation mechanism and promotes coagulation in endotoxemic mice. Blood 2009, 114, 2521–2529. [Google Scholar] [CrossRef] [Green Version]
- Sen, R.; Baltimore, D. Multiple Nuclear Factors Interact with the Immunoglobulin Enhancer Sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Tiruppathi, C.; Soni, D.; Wang, D.M.; Xue, J.; Singh, V.; Thippegowda, P.B.; Cheppudira, B.P.; Mishra, R.K.; Debroy, A.; Qian, Z.; et al. The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nat. Immunol. 2014, 15, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Hayden, M.S. New regulators of NF-kappaB in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [Green Version]
- Bijli, K.M.; Rahman, A. Nuclear Factor (NF)-κB Signaling in Endothelium. In Endothelial Biomedicine; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Takada, Y.; Boriek, A.M.; Aggarwal, B.B. Nuclear factor-kappaB: Its role in health and disease. J. Mol. Med. 2004, 82, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Ding, J.; Zhou, X.; Chen, G.; Liu, S.F. Divergent roles of endothelial NF-kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J. Exp. Med. 2008, 205, 1303–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen-Heininger, Y.M.; Poynter, M.E.; Aesif, S.W.; Pantano, C.; Ather, J.L.; Reynaert, N.L.; Ckless, K.; Anathy, V.; van der Velden, J.; Irvin, C.G.; et al. Nuclear factor kappaB, airway epithelium, and asthma: Avenues for redox control. Proc. Am. Thorac. Soc. 2009, 6, 249–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castranova, V. Signaling pathways controlling the production of inflammatory mediators in response to crystalline silica exposure: Role of reactive oxygen/nitrogen species. Free Radic. Biol. Med. 2004, 37, 916–925. [Google Scholar] [CrossRef]
- Rieger-Fackeldey, E.; Hentschel, R. Bronchopulmonary dysplasia and early prophylactic inhaled nitric oxide in preterm infants: Current concepts and future research strategies in animal models. J. Perinat. Med. 2008, 36, 442–447. [Google Scholar] [CrossRef]
- MacNee, W.; Tuder, R.M. New paradigms in the pathogenesis of chronic obstructive pulmonary disease I. Proc. Am. Thorac. Soc. 2009, 6, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Mett, I.; Bhunia, A.K.; Bowman, J.; Perez, M.; Zhang, L.; Gandjeva, A.; Zhen, L.; Chukwueke, U.; Mao, T.; et al. Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema. Nat. Med. 2010, 16, 767–773. [Google Scholar] [CrossRef]
- Liu, S.F.; Malik, A.B. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L622–L645. [Google Scholar] [CrossRef] [PubMed]
- Chousterman, B.G.; Swirski, F.K.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Robles, J.P.; Zamora, M.; Adan-Castro, E.; Siqueiros-Marquez, L.; Martinez de la Escalera, G.; Clapp, C. The spike protein of SARS-CoV-2 induces endothelial inflammation through integrin alpha5beta1 and NF-kappaB signaling. J. Biol. Chem. 2022, 298, 101695. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Tian, Y.; Nguyen, V.; Zhang, Y.; Gao, C.; Yin, R.; Carver, W.; Fan, D.; Albrecht, H.; Cui, T.; et al. Spike protein of SARS-CoV-2 activates macrophages and contributes to induction of acute lung inflammation in male mice. FASEB J. 2021, 35, e21801. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Lan, Z.; Ye, J.; Pang, L.; Liu, Y.; Wu, W.; Qin, X.; Guo, Y.; Zhang, P. Cytokine Storm: The Primary Determinant for the Pathophysiological Evolution of COVID-19 Deterioration. Front. Immunol. 2021, 12, 589095. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Karin, M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A.; Anwar, K.N.; True, A.L.; Malik, A.B. Thrombin-induced p65 homodimer binding to downstream NF-kappa B site of the promoter mediates endothelial ICAM-1 expression and neutrophil adhesion. J. Immunol. 1999, 162, 5466–5476. [Google Scholar]
- Chen, L.F.; Greene, W.C. Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell. Biol. 2004, 5, 392–394. [Google Scholar] [CrossRef]
- Huang, B.; Yang, X.D.; Lamb, A.; Chen, L.F. Posttranslational modifications of NF-kappaB: Another layer of regulation for NF-kappaB signaling pathway. Cell. Signal. 2010, 22, 1282–1290. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Westerheide, S.D.; Hanson, J.L.; Baldwin, A.S., Jr. Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J. Biol. Chem. 2000, 275, 32592–32597. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; May, M.J.; Jimi, E.; Ghosh, S. The Phosphorylation Status of Nuclear NF-ΚB Determines Its Association with CBP/p300 or HDAC-1. Mol. Cell 2002, 9, 625–636. [Google Scholar] [CrossRef]
- Ding, J.; Song, D.; Ye, X.; Liu, S.F. A pivotal role of endothelial-specific NF-kappaB signaling in the pathogenesis of septic shock and septic vascular dysfunction. J. Immunol. 2009, 183, 4031–4038. [Google Scholar] [CrossRef] [Green Version]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basilio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-kappaB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Gudowska-Sawczuk, M.; Mroczko, B. The Role of Nuclear Factor Kappa B (NF-kappaB) in Development and Treatment of COVID-19: Review. Int. J. Mol. Sci. 2022, 23, 5283. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Morigi, M.; Galbusera, M.; Pezzotta, A.; Gastoldi, S.; Imberti, B.; Perna, A.; Ruggenenti, P.; Donadelli, R.; Benigni, A.; et al. SARS-CoV-2 Spike Protein 1 Activates Microvascular Endothelial Cells and Complement System Leading to Platelet Aggregation. Front. Immunol. 2022, 13, 827146. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T. Two-way interactions between inflammation and coagulation. Trends Cardiovasc. Med. 2005, 15, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Batah, S.S.; Fabro, A.T. Pulmonary pathology of ARDS in COVID-19: A pathological review for clinicians. Respir. Med. 2021, 176, 106239. [Google Scholar] [CrossRef]
- Eisenhut, M.; Shin, J.I. Pathways in the Pathophysiology of Coronavirus 19 Lung Disease Accessible to Prevention and Treatment. Front. Physiol. 2020, 11, 872. [Google Scholar] [CrossRef]
- Mondrinos, M.J.; Kennedy, P.A.; Lyons, M.; Deutschman, C.S.; Kilpatrick, L.E. Protein kinase C and acute respiratory distress syndrome. Shock 2013, 39, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Black, A.R.; Black, J.D. Protein kinase C signaling and cell cycle regulation. Front. Immunol. 2012, 3, 423. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.; Anwar, K.N.; Uddin, S.; Xu, N.; Ye, R.D.; Platanias, L.C.; Malik, A.B. Protein kinase C-delta regulates thrombin-induced ICAM-1 gene expression in endothelial cells via activation of p38 mitogen-activated protein kinase. Mol. Cell. Biol. 2001, 21, 5554–5565. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Hu, C.; Fan, X.; Wang, Y.; Li, Q.; Su, Y.Q.; Zhang, D.M.; Yang, Q.; Passerini, A.G.; Sun, C. mTOR Inhibition Promotes Pneumonitis Through Inducing Endothelial Contraction and Hyperpermeability. Am. J. Respir. Cell Mol. Biol. 2021, 65, 646–657. [Google Scholar] [CrossRef]
- Paria, B.C.; Bair, A.M.; Xue, J.; Yu, Y.; Malik, A.B.; Tiruppathi, C. Ca2+ influx induced by protease-activated receptor-1 activates a feed-forward mechanism of TRPC1 expression via nuclear factor-kappaB activation in endothelial cells. J. Biol. Chem. 2006, 281, 20715–20727. [Google Scholar] [CrossRef]
- Bair, A.M.; Thippegowda, P.B.; Freichel, M.; Cheng, N.; Ye, R.D.; Vogel, S.M.; Yu, Y.; Flockerzi, V.; Malik, A.B.; Tiruppathi, C. Ca2+ entry via TRPC channels is necessary for thrombin-induced NF-kappaB activation in endothelial cells through AMP-activated protein kinase and protein kinase Cdelta. J. Biol. Chem. 2009, 284, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.; Anwar, K.N.; Malik, A.B. Protein kinase C-zeta mediates TNF-alpha-induced ICAM-1 gene transcription in endothelial cells. Am. J. Physiol. Cell Physiol. 2000, 279, C906–C914. [Google Scholar] [CrossRef] [Green Version]
- Anrather, J.; Csizmadia, V.; Soares, M.P.; Winkler, H. Regulation of NF-kappaB RelA phosphorylation and transcriptional activity by p21(ras) and protein kinase Czeta in primary endothelial cells. J. Biol. Chem. 1999, 274, 13594–13603. [Google Scholar] [CrossRef] [Green Version]
- Alleboina, S.; Wong, T.; Singh, M.V.; Dokun, A.O. Inhibition of protein kinase C beta phosphorylation activates nuclear factor-kappa B and improves postischemic recovery in type 1 diabetes. Exp. Biol. Med. 2020, 245, 785–796. [Google Scholar] [CrossRef]
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. Biosyst. 2015, 11, 1946–1954. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Graupera, M.; Guillermet-Guibert, J.; Foukas, L.C.; Phng, L.K.; Cain, R.J.; Salpekar, A.; Pearce, W.; Meek, S.; Millan, J.; Cutillas, P.R.; et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 2008, 453, 662–666. [Google Scholar] [CrossRef]
- Minhajuddin, M.; Bijli, K.M.; Fazal, F.; Sassano, A.; Nakayama, K.I.; Hay, N.; Platanias, L.C.; Rahman, A. Protein kinase C-delta and phosphatidylinositol 3-kinase/Akt activate mammalian target of rapamycin to modulate NF-kappaB activation and intercellular adhesion molecule-1 (ICAM-1) expression in endothelial cells. J. Biol. Chem. 2009, 284, 4052–4061. [Google Scholar] [CrossRef] [Green Version]
- Frey, R.S.; Gao, X.; Javaid, K.; Siddiqui, S.S.; Rahman, A.; Malik, A.B. Phosphatidylinositol 3-kinase gamma signaling through protein kinase Czeta induces NADPH oxidase-mediated oxidant generation and NF-kappaB activation in endothelial cells. J. Biol. Chem. 2006, 281, 16128–16138. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Gengaro, P.; Wang, W.; Wang, X.Q.; Li, C.; Faubel, S.; Rivard, C.; Schrier, R.W. Role of NF-kappaB and PI 3-kinase/Akt in TNF-alpha-induced cytotoxicity in microvascular endothelial cells. Am. J. Physiol. Renal. Physiol. 2008, 295, F932–F941. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, J.Q.; Jin, X.H.; Dong, M.P.; Zheng, J.F. Up-regulation of miR-146b-3p protects septic mice with acute respiratory distress syndrome by inhibiting PI3K/AKT signaling pathway. J. Bioenerg. Biomembr. 2020, 52, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ge, S.; He, W.; Chen, Q.; Xu, C.; Zeng, M. Ghrelin protects against lipopolysaccharide-induced acute respiratory distress syndrome through the PI3K/AKT pathway. J. Biol. Chem. 2021, 297, 101111. [Google Scholar] [CrossRef]
- Ji, S.; Wang, L. mu-Opioid receptor signalling via PI3K/Akt pathway ameliorates lipopolysaccharide-induced acute respiratory distress syndrome. Exp. Physiol. 2019, 104, 1555–1561. [Google Scholar] [CrossRef]
- Huang, X.; Dai, Z.; Cai, L.; Sun, K.; Cho, J.; Albertine, K.H.; Malik, A.B.; Schraufnagel, D.E.; Zhao, Y.Y. Endothelial p110gammaPI3K Mediates Endothelial Regeneration and Vascular Repair After Inflammatory Vascular Injury. Circulation 2016, 133, 1093–1103. [Google Scholar] [CrossRef] [Green Version]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Rajan, S.; Ye, J.; Bai, S.; Huang, F.; Guo, Y.L. NF-kappaB, but not p38 MAP kinase, is required for TNF-alpha-induced expression of cell adhesion molecules in endothelial cells. J. Cell Biochem. 2008, 105, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Kuldo, J.M.; Westra, J.; Asgeirsdottir, S.A.; Kok, R.J.; Oosterhuis, K.; Rots, M.G.; Schouten, J.P.; Limburg, P.C.; Molema, G. Differential effects of NF-κB and p38 MAPK inhibitors and combinations thereof on TNF-α- and IL-1β-induced proinflammatory status of endothelial cells in vitro. Am. J. Physiol. Cell Physiol. 2005, 289, C1229–C1239. [Google Scholar] [CrossRef]
- Fang, W.; Cai, S.X.; Wang, C.L.; Sun, X.X.; Li, K.; Yan, X.W.; Sun, Y.B.; Sun, X.Z.; Gu, C.K.; Dai, M.Y.; et al. Modulation of mitogenactivated protein kinase attenuates sepsisinduced acute lung injury in acute respiratory distress syndrome rats. Mol. Med. Rep. 2017, 16, 9652–9658. [Google Scholar] [CrossRef] [Green Version]
- Kania, E.; Roest, G.; Vervliet, T.; Parys, J.B.; Bultynck, G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 2017, 7, 140. [Google Scholar] [CrossRef] [PubMed]
- Tiruppathi, C.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 2006, 13, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.W.; Han, F.; Tauseef, M.; Birnbaumer, L.; Mehta, D.; Muller, W.A. TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J. Exp. Med. 2015, 212, 1883–1899. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Ahmmed, G.U.; Paria, B.C.; Holinstat, M.; Voyno-Yasenetskaya, T.; Tiruppathi, C.; Minshall, R.D.; Malik, A.B. RhoA interaction with inositol 1,4,5-trisphosphate receptor and transient receptor potential channel-1 regulates Ca2+ entry. Role in signaling increased endothelial permeability. J. Biol. Chem. 2003, 278, 33492–33500. [Google Scholar] [CrossRef] [Green Version]
- Tauseef, M.; Knezevic, N.; Chava, K.R.; Smith, M.; Sukriti, S.; Gianaris, N.; Obukhov, A.G.; Vogel, S.M.; Schraufnagel, D.E.; Dietrich, A.; et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J. Exp. Med. 2012, 209, 1953–1968. [Google Scholar] [CrossRef]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Sundivakkam, P.C.; Natarajan, V.; Malik, A.B.; Tiruppathi, C. Store-operated Ca2+ entry (SOCE) induced by protease-activated receptor-1 mediates STIM1 protein phosphorylation to inhibit SOCE in endothelial cells through AMP-activated protein kinase and p38beta mitogen-activated protein kinase. J. Biol. Chem. 2013, 288, 17030–17041. [Google Scholar] [CrossRef] [Green Version]
- Paria, B.C.; Malik, A.B.; Kwiatek, A.M.; Rahman, A.; May, M.J.; Ghosh, S.; Tiruppathi, C. Tumor necrosis factor-alpha induces nuclear factor-kappaB-dependent TRPC1 expression in endothelial cells. J. Biol. Chem. 2003, 278, 37195–37203. [Google Scholar] [CrossRef] [Green Version]
- Tiruppathi, C.; Naqvi, T.; Sandoval, R.; Mehta, D.; Malik, A.B. Synergistic effects of tumor necrosis factor-alpha and thrombin in increasing endothelial permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L958–L968. [Google Scholar] [CrossRef]
- Anwar, K.N.; Fazal, F.; Malik, A.B.; Rahman, A. RhoA/Rho-associated kinase pathway selectively regulates thrombin-induced intercellular adhesion molecule-1 expression in endothelial cells via activation of I kappa B kinase beta and phosphorylation of RelA/p65. J. Immunol. 2004, 173, 6965–6972. [Google Scholar] [CrossRef] [Green Version]
- Cernuda-Morollon, E.; Ridley, A.J. Rho GTPases and leukocyte adhesion receptor expression and function in endothelial cells. Circ. Res. 2006, 98, 757–767. [Google Scholar] [CrossRef]
- Chen, X.L.; Zhang, Q.; Zhao, R.; Ding, X.; Tummala, P.E.; Medford, R.M. Rac1 and superoxide are required for the expression of cell adhesion molecules induced by tumor necrosis factor-alpha in endothelial cells. J. Pharmacol. Exp. Ther. 2003, 305, 573–580. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Q.; Zhao, R.; Medford, R.M. Superoxide, H2O2, and iron are required for TNF-α-induced MCP-1 gene expression in endothelial cells: Role of Rac1 and NADPH oxidase. Am. J. Physiol. Heart Circ. Physiol. 2003, 186, H1001–H1007. [Google Scholar] [CrossRef] [Green Version]
- Spindler, V.; Schlegel, N.; Waschke, J. Role of GTPases in control of microvascular permeability. Cardiovasc. Res. 2010, 87, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazal, F.; Minhajuddin, M.; Bijli, K.M.; McGrath, J.L.; Rahman, A. Evidence for actin cytoskeleton-dependent and -independent pathways for RelA/p65 nuclear translocation in endothelial cells. J. Biol. Chem. 2007, 282, 3940–3950. [Google Scholar] [CrossRef] [Green Version]
- Fazal, F.; Bijli, K.M.; Minhajuddin, M.; Rein, T.; Finkelstein, J.N.; Rahman, A. Essential role of cofilin-1 in regulating thrombin-induced RelA/p65 nuclear translocation and intercellular adhesion molecule 1 (ICAM-1) expression in endothelial cells. J. Biol. Chem. 2009, 284, 21047–21056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, A.; Marando, C.; Rahman, A.; Fazal, F. Thrombin selectively engages LIM kinase 1 and slingshot-1L phosphatase to regulate NF-kappaB activation and endothelial cell inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L651–L664. [Google Scholar] [CrossRef] [Green Version]
- Fazal, F.; Bijli, K.M.; Murrill, M.; Leonard, A.; Minhajuddin, M.; Anwar, K.N.; Finkelstein, J.N.; Watterson, D.M.; Rahman, A. Critical role of non-muscle myosin light chain kinase in thrombin-induced endothelial cell inflammation and lung PMN infiltration. PLoS ONE 2013, 8, e59965. [Google Scholar] [CrossRef] [Green Version]
- Burridge, K.; Guilluy, C. Focal adhesions, stress fibers and mechanical tension. Exp. Cell. Res. 2016, 343, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagnoli, T.L.; da Silva, J.S.; Sudo, S.Z.; Santos, A.D.; Gomide, G.F.; de Sa, M.P.L.; Zapata-Sudo, G. ROCK Inhibition as Potential Target for Treatment of Pulmonary Hypertension. Cells 2021, 10, 1648. [Google Scholar] [CrossRef]
- Frey, R.S.; Rahman, A.; Kefer, J.C.; Minshall, R.D.; Malik, A.B. PKCzeta regulates TNF-alpha-induced activation of NADPH oxidase in endothelial cells. Circ. Res. 2002, 90, 1012–1019. [Google Scholar] [CrossRef]
- Pan, W.; Yu, H.; Huang, S.; Zhu, P. Resveratrol Protects against TNF-alpha-Induced Injury in Human Umbilical Endothelial Cells through Promoting Sirtuin-1-Induced Repression of NF-KB and p38 MAPK. PLoS ONE 2016, 11, e0147034. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, B.J.; Solt, L.A.; Chowdhury, I.; Kazi, A.S.; Abid, M.R.; Aird, W.C.; May, M.J.; Foskett, J.K.; Madesh, M. G protein-coupled receptor Ca2+-linked mitochondrial reactive oxygen species are essential for endothelial/leukocyte adherence. Mol. Cell. Biol. 2007, 27, 7582–7593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, J.D.; McArdle, P.J.; O’Reilly, P.J.; Matalon, S. Oxidant-Antioxidant Balance in Acute Lung Injury. Chest 2002, 122, 314S–320S. [Google Scholar] [CrossRef] [Green Version]
- von Knethen, A.; Heinicke, U.; Laux, V.; Parnham, M.J.; Steinbicker, A.U.; Zacharowski, K. Antioxidants as Therapeutic Agents in Acute Respiratory Distress Syndrome (ARDS) Treatment-From Mice to Men. Biomedicines 2022, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Sittipunt, C.; Steinberg, K.P.; Ruzinski, J.T.; Myles, C.; Zhu, S.; Goodman, R.B.; Hudson, L.D.; Matalon, S.; Martin, T.R. Nitric Oxide and Nitrotyrosine in the Lungs of Patients with Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2001, 163, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Galvão, A.M.; de Andrade, A.D.; de Souza Maia, M.B.; da Silva, K.E.R.; Bezerra, A.D.; de Melo, J.F.; de Morais, N.G.; da Costa, T.B.; de Castro, C.M.M.B. Antioxidant supplementation for the treatment of acute lung injury: A meta-analysis. Rev. Bras. Ter. Intensiv. 2011, 23, 41–48. [Google Scholar]
- Weber, C.; Negrescu, E.; Erl, W.; Pietsch, A.; Frankenberger, M.; Ziegler-Heitbrock, H.W.L.; Siess, W.; Weber, P.C. Inhibitors of Protein Tyrosine Kinase Suppress TNF-Stimulated Induction of Endothelial Cell Adhesion Molecules. J. Immunol. 1995, 155, 445–451. [Google Scholar] [PubMed]
- Bijli, K.M.; Minhajuddin, M.; Fazal, F.; O’Reilly, M.A.; Platanias, L.C.; Rahman, A. c-Src interacts with and phosphorylates RelA/p65 to promote thrombin-induced ICAM-1 expression in endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L396–L404. [Google Scholar] [CrossRef] [Green Version]
- Bijli, K.M.; Fazal, F.; Minhajuddin, M.; Rahman, A. Activation of Syk by protein kinase C-delta regulates thrombin-induced intercellular adhesion molecule-1 expression in endothelial cells via tyrosine phosphorylation of RelA/p65. J. Biol. Chem. 2008, 283, 14674–14684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijli, K.M.; Fazal, F.; Rahman, A. Regulation of Rela/p65 and endothelial cell inflammation by proline-rich tyrosine kinase 2. Am. J. Respir. Cell. Mol. Biol. 2012, 47, 660–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMullen, M.; Keller, R.; Sussman, M.; Pumiglia, K. Vascular endothelial growth factor-mediated activation of p38 is dependent upon Src and RAFTK/Pyk2. Oncogene 2004, 23, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.R.; Bradley, R.; Ganju, R.K. LPS-induced MCP-1 expression in human microvascular endothelial cells is mediated by the tyrosine kinase, Pyk2 via the p38 MAPK/NF-kappaB-dependent pathway. Mol. Immunol. 2009, 46, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell. Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornmann, B. The molecular hug between the ER and the mitochondria. Curr. Opin. Cell Biol. 2013, 25, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell. Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Moro, F.; Okamoto, K.; Donzeau, M.; Neupert, W.; Brunner, M. Mitochondrial protein import: Molecular basis of the ATP-dependent interaction of MtHsp70 with Tim44. J. Biol. Chem. 2002, 277, 6874–6880. [Google Scholar] [CrossRef] [Green Version]
- Yaguchi, T.; Aida, S.; Kaul, S.C.; Wadhwa, R. Involvement of mortalin in cellular senescence from the perspective of its mitochondrial import, chaperone, and oxidative stress management functions. Ann. N. Y. Acad. Sci. 2007, 1100, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Garg, A.D.; Kaczmarek, A.; Krysko, O.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. ER stress-induced inflammation: Does it aid or impede disease progression? Trends Mol. Med. 2012, 18, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S.C.; Deocaris, C.C.; Wadhwa, R. Three faces of mortalin: A housekeeper, guardian and killer. Exp. Gerontol. 2007, 42, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Schelling, C.; Kato, H.; Rapaport, D.; Woitalla, D.; Schiesling, C.; Schulte, C.; Sharma, M.; Illig, T.; Bauer, P.; et al. Dissecting the role of the mitochondrial chaperone mortalin in Parkinson’s disease: Functional impact of disease-related variants on mitochondrial homeostasis. Hum. Mol. Genet. 2010, 19, 4437–4452. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.O.; Bhargava, P.; Na, Y.; Lee, J.S.; Ryu, J.; Kaul, S.C.; Wadhwa, R. Relevance of mortalin to cancer cell stemness and cancer therapy. Sci. Rep. 2017, 7, 42016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, A.; Grose, V.; Paton, A.W.; Paton, J.C.; Yule, D.I.; Rahman, A.; Fazal, F. Selective Inactivation of Intracellular BiP/GRP78 Attenuates Endothelial Inflammation and Permeability in Acute Lung Injury. Sci. Rep. 2019, 9, 2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, A.; Su, P.Y.; Yule, D.I.; Rahman, A.; Fazal, F. Critical Role of Mortalin/GRP75 in Endothelial Cell Dysfunction Associated with Acute Lung Injury. Shock 2020, 54, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Paton, A.W.; Beddoe, T.; Thorpe, C.M.; Whisstock, J.C.; Wilce, M.C.; Rossjohn, J.; Talbot, U.M.; Paton, J.C. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 2006, 443, 548–552. [Google Scholar] [CrossRef]
- Leonard, A.; Paton, A.W.; El-Quadi, M.; Paton, J.C.; Fazal, F. Preconditioning with endoplasmic reticulum stress ameliorates endothelial cell inflammation. PLoS ONE 2014, 9, e110949. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Moscat, J.; Diaz-Meco, M.T. Feedback on fat: p62-mTORC1-autophagy connections. Cell 2011, 147, 724–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, A.; Millar, M.W.; Slavin, S.A.; Bijli, K.M.; Dionisio Santos, D.A.; Dean, D.A.; Fazal, F.; Rahman, A. Critical role of autophagy regulator Beclin1 in endothelial cell inflammation and barrier disruption. Cell. Signal. 2019, 61, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Shadab, M.; Millar, M.W.; Slavin, S.A.; Leonard, A.; Fazal, F.; Rahman, A. Autophagy protein ATG7 is a critical regulator of endothelial cell inflammation and permeability. Sci. Rep. 2020, 10, 13708. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.H.; Chen, H.R.; Chuang, Y.C.; Yeh, T.M. Macrophage Migration Inhibitory Factor-Induced Autophagy Contributes to Thrombin-Triggered Endothelial Hyperpermeability in Sepsis. Shock 2018, 50, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Slavin, S.A.; Leonard, A.; Grose, V.; Fazal, F.; Rahman, A. Autophagy inhibitor 3-methyladenine protects against endothelial cell barrier dysfunction in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L388–L396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Li, H.; Gong, Y.; Zheng, H.; Zhao, D. Hydrogen sulfide ameliorated lipopolysaccharide-induced acute lung injury by inhibiting autophagy through PI3K/Akt/mTOR pathway in mice. Biochem. Biophys. Res. Commun. 2018, 507, 514–518. [Google Scholar] [CrossRef]
- Zeng, M.; Sang, W.; Chen, S.; Chen, R.; Zhang, H.; Xue, F.; Li, Z.; Liu, Y.; Gong, Y.; Zhang, H.; et al. 4-PBA inhibits LPS-induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol. Lett. 2017, 271, 26–37. [Google Scholar] [CrossRef]
- Sun, Y.; Li, C.; Shu, Y.; Ju, X.; Zou, Z.; Wang, H.; Rao, S.; Guo, F.; Liu, H.; Nan, W.; et al. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci. Signal. 2012, 5, ra16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, G.; Dull, R.O.; Schwartz, D.E.; Hu, G. Autophagy in pulmonary macrophages mediates lung inflammatory injury via NLRP3 inflammasome activation during mechanical ventilation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L173–L185. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhou, K.; Liao, L.; Zhang, T.; Yang, M.; Sun, C. Lipoxin A4 receptor agonist BML-111 induces autophagy in alveolar macrophages and protects from acute lung injury by activating MAPK signaling. Respir. Res. 2018, 19, 243. [Google Scholar] [CrossRef] [PubMed]
- Nosaka, N.; Martinon, D.; Moreira, D.; Crother, T.R.; Arditi, M.; Shimada, K. Autophagy Protects Against Developing Increased Lung Permeability and Hypoxemia by Down Regulating Inflammasome Activity and IL-1beta in LPS Plus Mechanical Ventilation-Induced Acute Lung Injury. Front. Immunol. 2020, 11, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Chen, Y.; Zhang, P.; Lin, P.; Xie, N.; Wu, M. Protective Features of Autophagy in Pulmonary Infection and Inflammatory Diseases. Cells 2019, 8, 123. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; He, B.; Qian, H.; Liu, Q.; Wang, D.; Li, J.; Wei, Z.; Wang, Z.; Xu, Z.; Wu, G.; et al. RAB26-dependent autophagy protects adherens junctional integrity in acute lung injury. Autophagy 2018, 14, 1677–1692. [Google Scholar] [CrossRef]
- Ye, Y.; Tan, S.; Zhou, X.; Li, X.; Jundt, M.C.; Lichter, N.; Hidebrand, A.; Dhasarathy, A.; Wu, M. Inhibition of p-IkappaBalpha Ubiquitylation by Autophagy-Related Gene 7 to Regulate Inflammatory Responses to Bacterial Infection. J. Infect. Dis. 2015, 212, 1816–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryter, S.W.; Nakahira, K.; Haspel, J.A.; Choi, A.M. Autophagy in pulmonary diseases. Annu. Rev. Physiol. 2012, 74, 377–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, P.T.; Hay, N. The two TORCs and Akt. Dev. Cell. 2007, 12, 487–502. [Google Scholar] [CrossRef] [Green Version]
- Huang, S. mTOR Signaling in Metabolism and Cancer. Cells 2020, 9, 2278. [Google Scholar] [CrossRef]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Minhajuddin, M.; Fazal, F.; Bijli, K.M.; Amin, M.R.; Rahman, A. Inhibition of mammalian target of rapamycin potentiates thrombin-induced intercellular adhesion molecule-1 expression by accelerating and stabilizing NF-kappa B activation in endothelial cells. J. Immunol. 2005, 174, 5823–5829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A.; True, A.L.; Anwar, K.N.; Ye, R.D.; Voyno-Yasenetskaya, T.A.; Malik, A.B. Galpha(q) and Gbetagamma regulate PAR-1 signaling of thrombin-induced NF-kappaB activation and ICAM-1 transcription in endothelial cells. Circ. Res. 2002, 91, 398–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Weichhart, T.; Costantino, G.; Poglitsch, M.; Rosner, M.; Zeyda, M.; Stuhlmeier, K.M.; Kolbe, T.; Stulnig, T.M.; Horl, W.H.; Hengstschlager, M.; et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 2008, 29, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorne, E.; Zhao, X.; Zmijewski, J.W.; Liu, G.; Park, Y.J.; Tsuruta, Y.; Abraham, E. Participation of mammalian target of rapamycin complex 1 in Toll-like receptor 2- and 4-induced neutrophil activation and acute lung injury. Am. J. Respir. Cell. Mol. Biol. 2009, 41, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Bermudez, T.; Sammani, S.; Song, J.H.; Hernon, V.R.; Kempf, C.L.; Garcia, A.N.; Burt, J.; Hufford, M.; Camp, S.M.; Cress, A.E.; et al. eNAMPT neutralization reduces preclinical ARDS severity via rectified NFkB and Akt/mTORC2 signaling. Sci. Rep. 2022, 12, 696. [Google Scholar] [CrossRef]
- Lee, H.; Fei, Q.; Streicher, A.; Zhang, W.; Isabelle, C.; Patel, P.; Lam, H.C.; Arciniegas-Rubio, A.; Pinilla-Vera, M.; Amador-Munoz, D.P.; et al. mTORC1 is a mechanosensor that regulates surfactant function and lung compliance during ventilator-induced lung injury. JCI Insight. 2021, 6, e137708. [Google Scholar] [CrossRef]
- Ballesteros-Alvarez, J.; Andersen, J.K. mTORC2: The other mTOR in autophagy regulation. Aging Cell 2021, 20, e13431. [Google Scholar] [CrossRef]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-kappaB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M. Regulation of tissue homeostasis by NF-kappaB signalling: Implications for inflammatory diseases. Nat. Rev. Immunol. 2009, 9, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Gilroy, D.W.; Colville-Nash, P.R.; Willoughby, D.A. Possible new role for NF-κB in the resolution of inflammation. Nat. Med. 2001, 7, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Ye, X.; Miller, E.J.; Liu, S.F. NF-κB-to-AP-1 Switch: A Mechanism Regulating Transition from Endothelial Barrier Injury to Repair in Endotoxemic Mice. Sci. Rep. 2014, 4, 5543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Zhang, H.; Luo, L.; Lin, J.; Li, D.; Zheng, S.; Huang, H.; Yan, S.; Yang, J.; Hao, Y.; et al. Resolvin D1 Improves the Resolution of Inflammation via Activating NF-kappaB p50/p50-Mediated Cyclooxygenase-2 Expression in Acute Respiratory Distress Syndrome. J. Immunol. 2017, 199, 2043–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boone, D.L.; Turer, E.E.; Lee, E.G.; Ahmad, R.C.; Wheeler, M.T.; Tsui, C.; Hurley, P.; Chien, M.; Chai, S.; Hitotsumatsu, O.; et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 2004, 5, 1052–1060. [Google Scholar] [CrossRef]
- Song, H.Y.; Rothe, M.; Goeddel, D.V. The tumor necrosis factor-inducible zinc finger protein A20 interacts withTRAF1/TRAF2 and inhibits NF-κB activation. Proc. Natl. Acad. Sci. USA 1996, 93, 6721–6725. [Google Scholar] [CrossRef] [Green Version]
- Kisseleva, T.; Song, L.; Vorontchikhina, M.; Feirt, N.; Kitajewski, J.; Schindler, C. NF-κB regulation of endothelial cell function during LPS-induced toxemia and cancer. J. Clin. Investig. 2006, 116, 2955–2963. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.W.; Egan, L.; Li, Z.W.; Greten, F.R.; Kagnoff, M.F.; Karin, M. The two faces of IKK and NF-kappaB inhibition: Prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat. Med. 2003, 9, 575–581. [Google Scholar] [CrossRef]
- Dorrington, M.G.; Fraser, I.D.C. NF-kappaB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Scott, O.; Roifman, C.M. NF-kappaB pathway and the Goldilocks principle: Lessons from human disorders of immunity and inflammation. J. Allergy. Clin. Immunol. 2019, 143, 1688–1701. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Su, J.; Cui, X.; Li, Y.; Barochia, A.; Eichacker, P.Q. Can we predict the effects of NF-kappaB inhibition in sepsis? Studies with parthenolide and ethyl pyruvate. Expert. Opin. Investig. Drugs 2009, 18, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Iosef, C.; Alastalo, T.P.; Hou, Y.; Chen, C.; Adams, E.S.; Lyu, S.C.; Cornfield, D.N.; Alvira, C.M. Inhibiting NF-kappaB in the developing lung disrupts angiogenesis and alveolarization. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L1023–L1036. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millar, M.W.; Fazal, F.; Rahman, A. Therapeutic Targeting of NF-κB in Acute Lung Injury: A Double-Edged Sword. Cells 2022, 11, 3317. https://doi.org/10.3390/cells11203317
Millar MW, Fazal F, Rahman A. Therapeutic Targeting of NF-κB in Acute Lung Injury: A Double-Edged Sword. Cells. 2022; 11(20):3317. https://doi.org/10.3390/cells11203317
Chicago/Turabian StyleMillar, Michelle Warren, Fabeha Fazal, and Arshad Rahman. 2022. "Therapeutic Targeting of NF-κB in Acute Lung Injury: A Double-Edged Sword" Cells 11, no. 20: 3317. https://doi.org/10.3390/cells11203317
APA StyleMillar, M. W., Fazal, F., & Rahman, A. (2022). Therapeutic Targeting of NF-κB in Acute Lung Injury: A Double-Edged Sword. Cells, 11(20), 3317. https://doi.org/10.3390/cells11203317