
A Linkage between Angiogenesis and Inflammation in Neovascular Age-Related Macular Degeneration



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
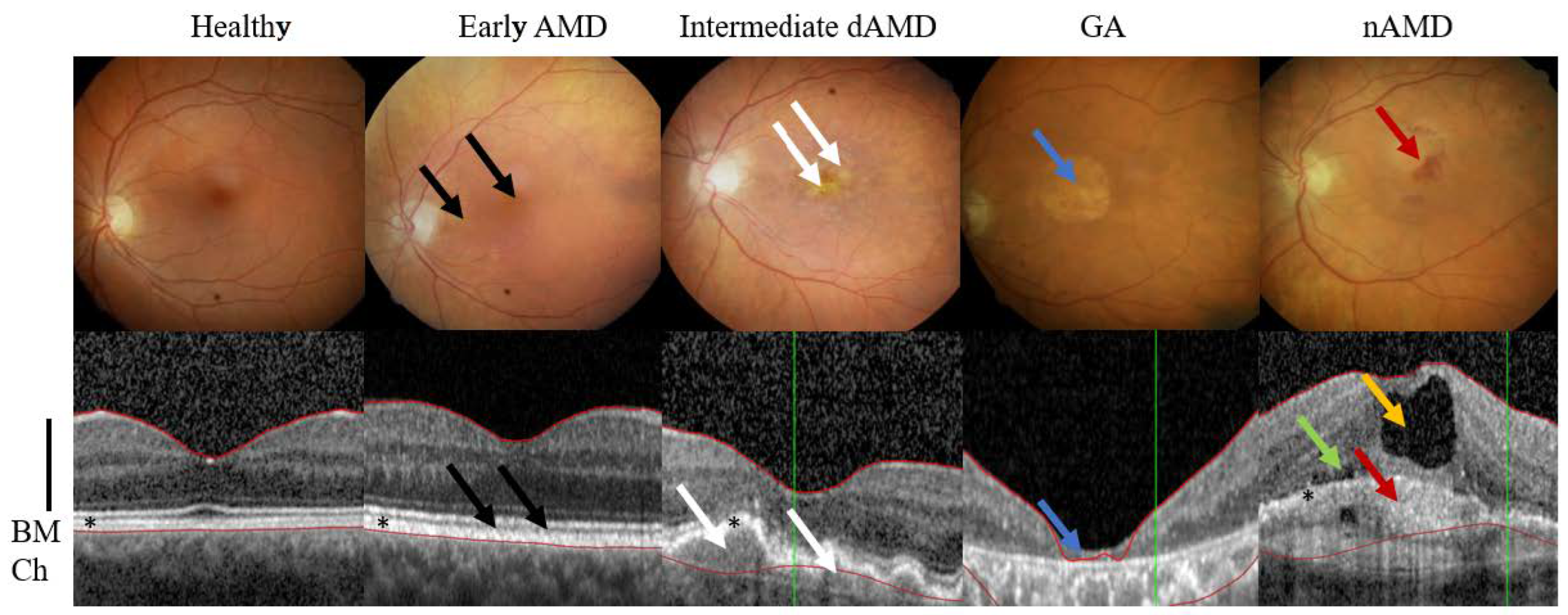
2. AMD Phenotypes
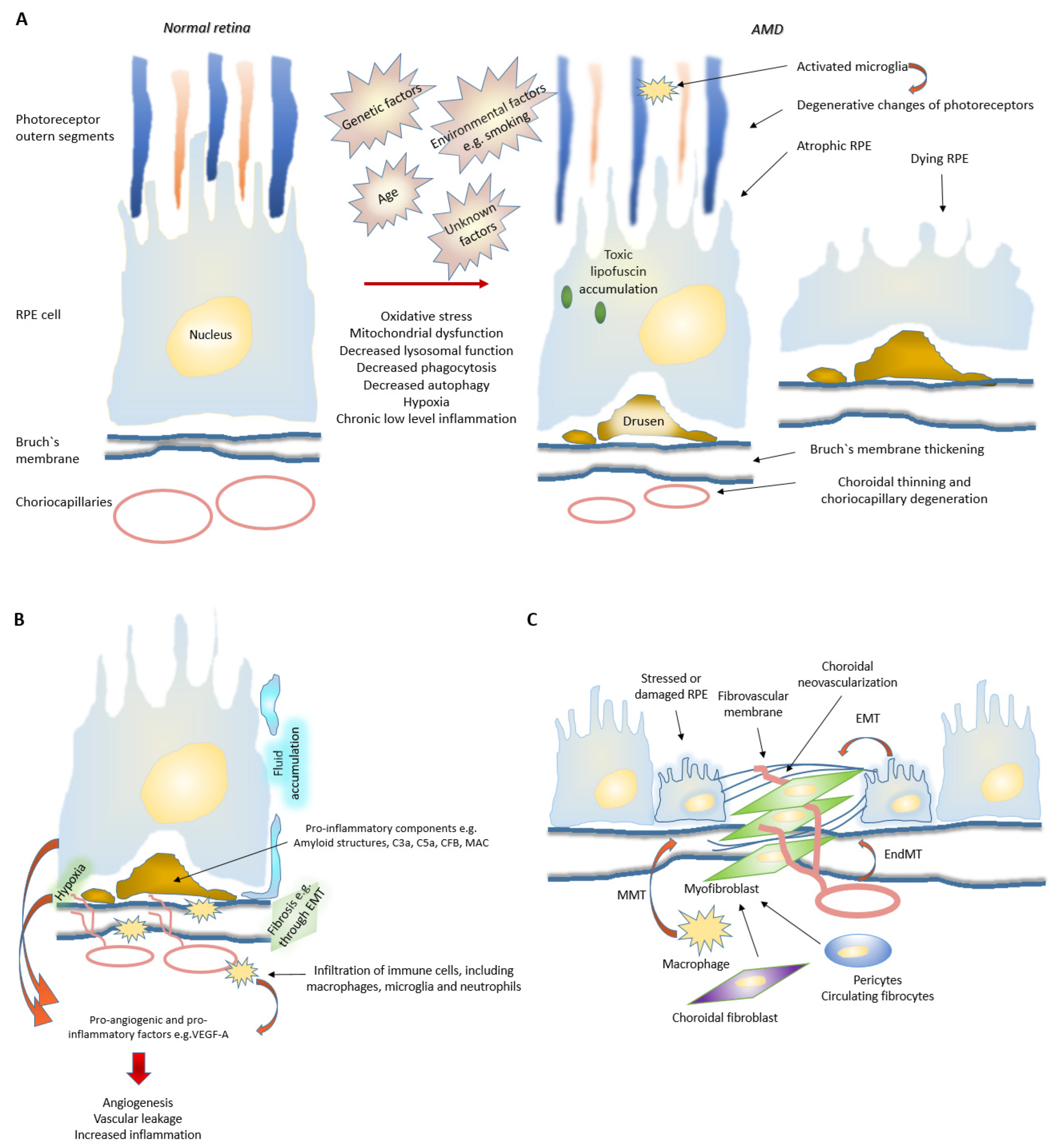
3. Evolution of AMD
3.1. Disturbed Proteostasis in AMD
3.2. Role of Choriocapillaris in nAMD Development
3.3. Senescence in AMD
3.4. Fibrosis in nAMD
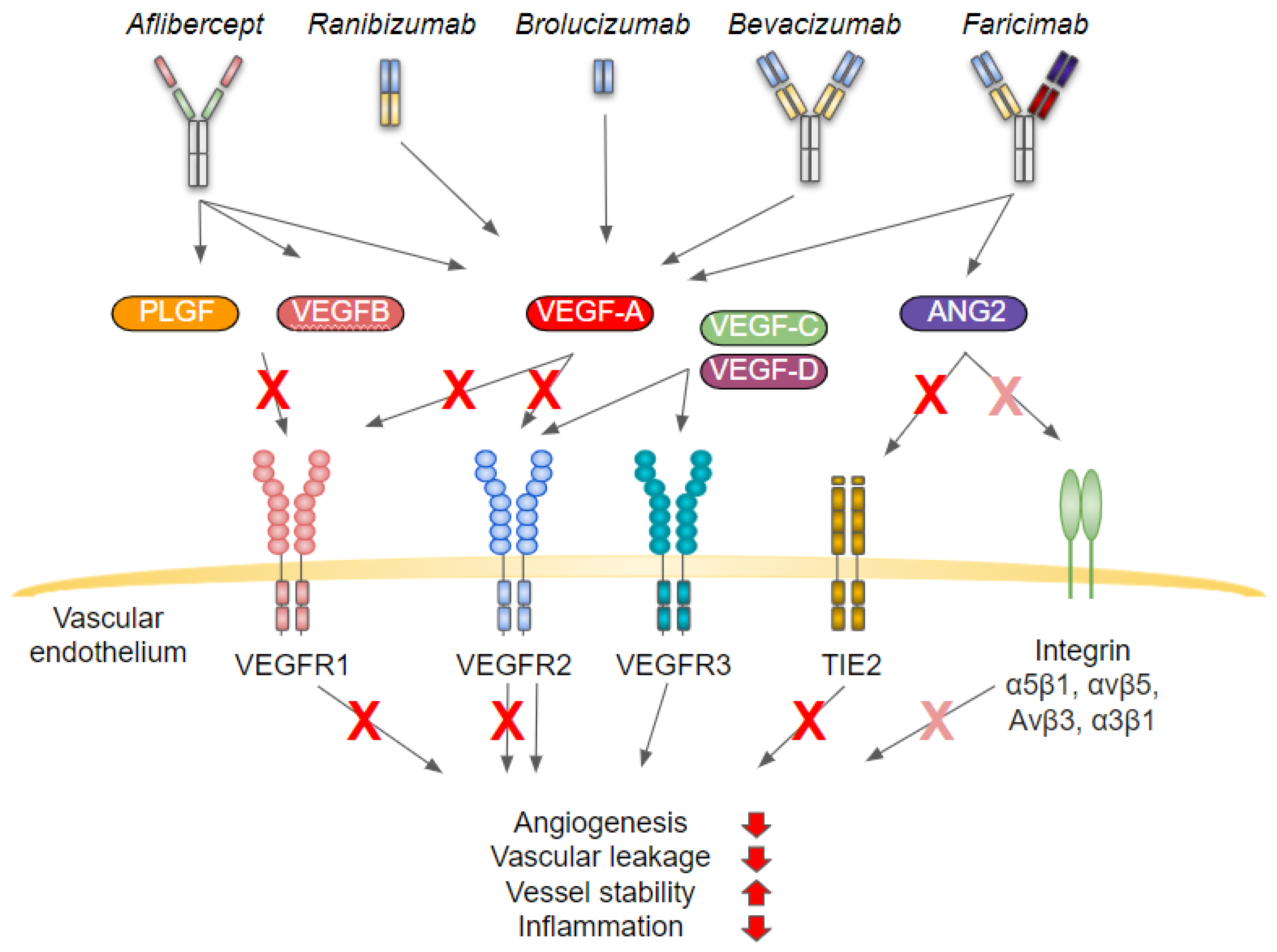
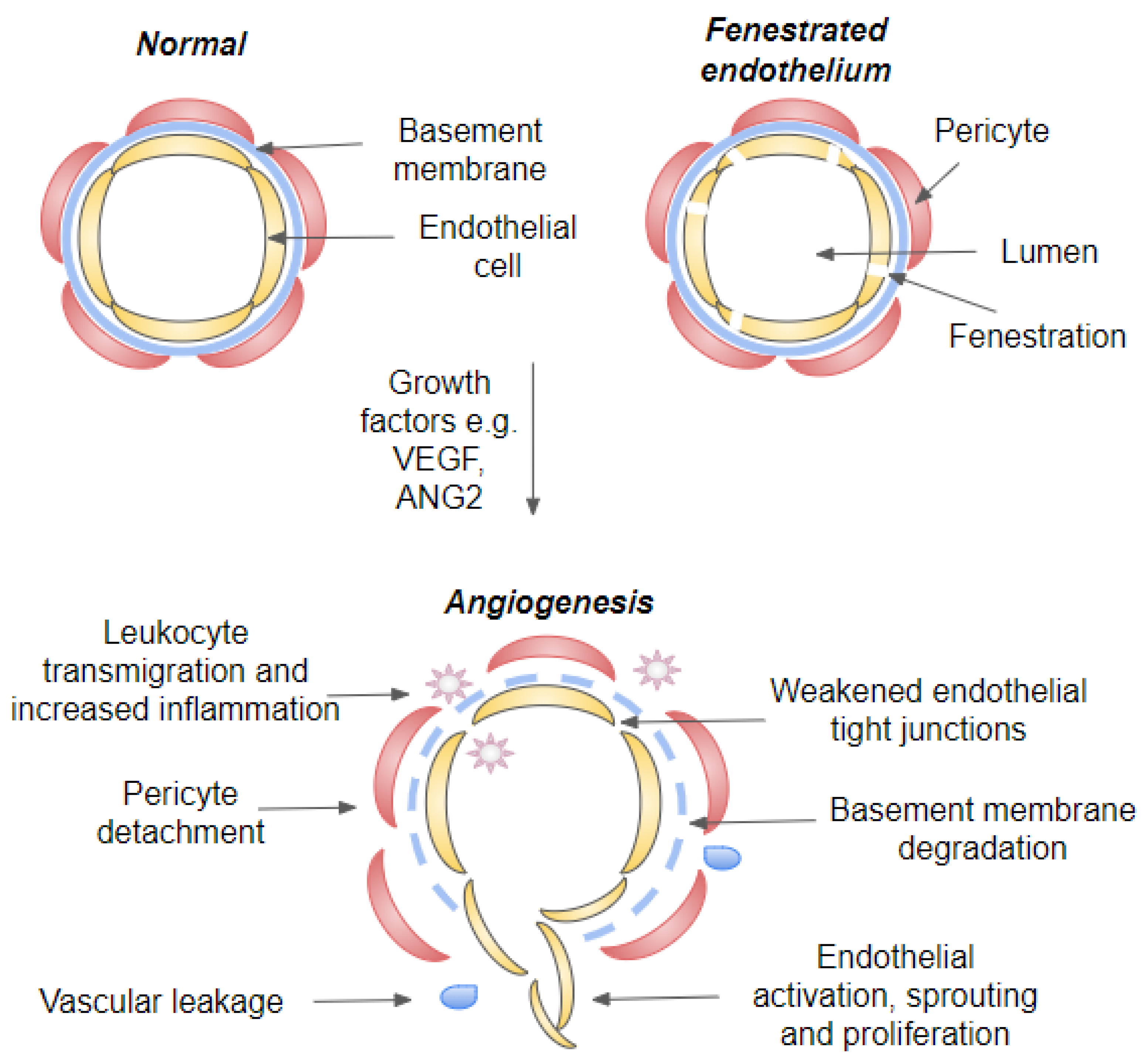
4. Angiogenesis and Inflammation
4.1. Inflammatory Signaling Cascades
4.2. Blood-Retinal-Barriers
4.3. Inflammatory Cell Recruitment
4.4. Effects of Hypoxia
5. Concluding Remarks
Funding
Data Availability Statement
Conflicts of Interest
References
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990-2020: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Q.; Welchowski, T.; Schmid, M.; Mauschitz, M.M.; Holz, F.G.; Finger, R.P. Prevalence and incidence of age-related macular degeneration in europe: A systematic review and meta-analysis. Br. J. Ophthalmol. 2020, 104, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Flaxel, C.J.; Adelman, R.A.; Bailey, S.T.; Fawzi, A.; Lim, J.I.; Vemulakonda, G.A.; Ying, G.S. Age-related macular degeneration preferred practice pattern(r). Ophthalmology 2020, 127, P1–P65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corazza, P.; D’Alterio, F.M.; Kabbani, J.; Alam, M.M.R.; Mercuri, S.; Orlans, H.O.; Younis, S. Long-term outcomes of intravitreal anti-vegf therapies in patients affected by neovascular age-related macular degeneration: A real-life study. BMC Ophthalmol. 2021, 21, 300. [Google Scholar] [CrossRef] [PubMed]
- Heier, J.S.; Brown, D.M.; Chong, V.; Korobelnik, J.F.; Kaiser, P.K.; Nguyen, Q.D.; Kirchhof, B.; Ho, A.; Ogura, Y.; Yancopoulos, G.D.; et al. Intravitreal aflibercept (vegf trap-eye) in wet age-related macular degeneration. Ophthalmology 2012, 119, 2537–2548. [Google Scholar] [CrossRef]
- Rosenfeld, P.J.; Brown, D.M.; Heier, J.S.; Boyer, D.S.; Kaiser, P.K.; Chung, C.Y.; Kim, R.Y.; Group, M.S. Ranibizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2006, 355, 1419–1431. [Google Scholar] [CrossRef] [Green Version]
- Dugel, P.U.; Koh, A.; Ogura, Y.; Jaffe, G.J.; Schmidt-Erfurth, U.; Brown, D.M.; Gomes, A.V.; Warburton, J.; Weichselberger, A.; Holz, F.G.; et al. Hawk and harrier: Phase 3, multicenter, randomized, double-masked trials of brolucizumab for neovascular age-related macular degeneration. Ophthalmology 2020, 127, 72–84. [Google Scholar] [CrossRef]
- Gragoudas, E.S.; Adamis, A.P.; Cunningham, E.T., Jr.; Feinsod, M.; Guyer, D.R. VEGF Inhibition Study in Ocular Neovascularization Clinical Trial Group Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 2004, 351, 2805–2816. [Google Scholar] [CrossRef] [Green Version]
- Heier, J.S.; Khanani, A.M.; Quezada Ruiz, C.; Basu, K.; Ferrone, P.J.; Brittain, C.; Figueroa, M.S.; Lin, H.; Holz, F.G.; Patel, V.; et al. Efficacy, durability, and safety of intravitreal faricimab up to every 16 weeks for neovascular age-related macular degeneration (tenaya and lucerne): Two randomised, double-masked, phase 3, non-inferiority trials. Lancet 2022, 399, 729–740. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Martin, J.; Ruan, Q.; Rafique, A.; Rosconi, M.P.; Shi, E.; Pyles, E.A.; Yancopoulos, G.D.; Stahl, N.; Wiegand, S.J. Binding and neutralization of vascular endothelial growth factor (vegf) and related ligands by vegf trap, ranibizumab and bevacizumab. Angiogenesis 2012, 15, 171–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avery, R.L.; Pieramici, D.J.; Rabena, M.D.; Castellarin, A.A.; Nasir, M.A.; Giust, M.J. Intravitreal bevacizumab (avastin) for neovascular age-related macular degeneration. Ophthalmology 2006, 113, 363–372 e365. [Google Scholar] [CrossRef] [PubMed]
- Tadayoni, R.; Sararols, L.; Weissgerber, G.; Verma, R.; Clemens, A.; Holz, F.G. Brolucizumab: A newly developed anti-vegf molecule for the treatment of neovascular age-related macular degeneration. Ophthalmologica 2021, 244, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef]
- Stacker, S.A.; Achen, M.G. Emerging roles for vegf-d in human disease. Biomolecules 2018, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Tammela, T.; Zarkada, G.; Wallgard, E.; Murtomaki, A.; Suchting, S.; Wirzenius, M.; Waltari, M.; Hellstrom, M.; Schomber, T.; Peltonen, R.; et al. Blocking vegfr-3 suppresses angiogenic sprouting and vascular network formation. Nature 2008, 454, 656–660. [Google Scholar] [CrossRef]
- Singh, N.K.; Kotla, S.; Kumar, R.; Rao, G.N. Cyclic amp response element binding protein mediates pathological retinal neovascularization via modulating dll4-notch1 signaling. EBioMedicine 2015, 2, 1767–1784. [Google Scholar] [CrossRef] [Green Version]
- Felcht, M.; Luck, R.; Schering, A.; Seidel, P.; Srivastava, K.; Hu, J.; Bartol, A.; Kienast, Y.; Vettel, C.; Loos, E.K.; et al. Angiopoietin-2 differentially regulates angiogenesis through tie2 and integrin signaling. J. Clin. Invest. 2012, 122, 1991–2005. [Google Scholar] [CrossRef] [Green Version]
- Joussen, A.M.; Ricci, F.; Paris, L.P.; Korn, C.; Quezada-Ruiz, C.; Zarbin, M. Angiopoietin/tie2 signalling and its role in retinal and choroidal vascular diseases: A review of preclinical data. Eye 2021, 35, 1305–1316. [Google Scholar] [CrossRef]
- Park, S.W.; Yun, J.H.; Kim, J.H.; Kim, K.W.; Cho, C.H.; Kim, J.H. Angiopoietin 2 induces pericyte apoptosis via alpha3beta1 integrin signaling in diabetic retinopathy. Diabetes 2014, 63, 3057–3068. [Google Scholar] [CrossRef]
- Krzystolik, M.G.; Afshari, M.A.; Adamis, A.P.; Gaudreault, J.; Gragoudas, E.S.; Michaud, N.A.; Li, W.; Connolly, E.; O’Neill, C.A.; Miller, J.W. Prevention of experimental choroidal neovascularization with intravitreal anti-vascular endothelial growth factor antibody fragment. Arch. Ophthalmol. 2002, 120, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Damico, L.; Shams, N.; Lowman, H.; Kim, R. Development of ranibizumab, an anti-vascular endothelial growth factor antigen binding fragment, as therapy for neovascular age-related macular degeneration. Retina 2006, 26, 859–870. [Google Scholar] [CrossRef]
- Szabo, E.; Phillips, D.J.; Droste, M.; Marti, A.; Kretzschmar, T.; Shamshiev, A.; Weller, M. Antitumor activity of dlx1008, an anti-vegfa antibody fragment with low picomolar affinity, in human glioma models. J. Pharmacol. Exp. Ther. 2018, 365, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Presta, L.G.; Chen, H.; O’Connor, S.J.; Chisholm, V.; Meng, Y.G.; Krummen, L.; Winkler, M.; Ferrara, N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997, 57, 4593–4599. [Google Scholar] [PubMed]
- Regula, J.T.; Lundh von Leithner, P.; Foxton, R.; Barathi, V.A.; Cheung, C.M.; Bo Tun, S.B.; Wey, Y.S.; Iwata, D.; Dostalek, M.; Moelleken, J.; et al. Targeting key angiogenic pathways with a bispecific crossmab optimized for neovascular eye diseases. EMBO Mol. Med. 2016, 8, 1265–1288. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Rajendran, S.; Dowd, J.; Wilson, D.J. Rapid characterization of structural and functional similarity for a candidate bevacizumab (avastin) biosimilar using a multipronged mass-spectrometry-based approach. Drug. Test Anal. 2019, 11, 1207–1217. [Google Scholar] [CrossRef]
- Holash, J.; Davis, S.; Papadopoulos, N.; Croll, S.D.; Ho, L.; Russell, M.; Boland, P.; Leidich, R.; Hylton, D.; Burova, E.; et al. Vegf-trap: A vegf blocker with potent antitumor effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, Q.D.; Das, A.; Do, D.V.; Dugel, P.U.; Gomes, A.; Holz, F.G.; Koh, A.; Pan, C.K.; Sepah, Y.J.; Patel, N.; et al. Brolucizumab: Evolution through preclinical and clinical studies and the implications for the management of neovascular age-related macular degeneration. Ophthalmology 2020, 127, 963–976. [Google Scholar] [CrossRef]
- Rudge, J.S.; Holash, J.; Hylton, D.; Russell, M.; Jiang, S.; Leidich, R.; Papadopoulos, N.; Pyles, E.A.; Torri, A.; Wiegand, S.J.; et al. Vegf trap complex formation measures production rates of vegf, providing a biomarker for predicting efficacious angiogenic blockade. Proc. Natl. Acad. Sci. USA 2007, 104, 18363–18370. [Google Scholar] [CrossRef] [Green Version]
- Ruckman, J.; Green, L.S.; Beeson, J.; Waugh, S.; Gillette, W.L.; Henninger, D.D.; Claesson-Welsh, L.; Janjic, N. 2′-fluoropyrimidine rna-based aptamers to the 165-amino acid form of vascular endothelial growth factor (vegf165). Inhibition of receptor binding and vegf-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem. 1998, 273, 20556–20567. [Google Scholar] [CrossRef]
- Holekamp, N.M.; Campochiaro, P.A.; Chang, M.A.; Miller, D.; Pieramici, D.; Adamis, A.P.; Brittain, C.; Evans, E.; Kaufman, D.; Maass, K.F.; et al. Archway randomized phase 3 trial of the port delivery system with ranibizumab for neovascular age-related macular degeneration. Ophthalmology 2022, 129, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Gillies, M.C.; Campain, A.; Barthelmes, D.; Simpson, J.M.; Arnold, J.J.; Guymer, R.H.; McAllister, I.L.; Essex, R.W.; Morlet, N.; Hunyor, A.P.; et al. Long-term outcomes of treatment of neovascular age-related macular degeneration: Data from an observational study. Ophthalmology 2015, 122, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.T.; Eliott, D.; Sobrin, L. Inflammatory complications of intravitreal anti-vegf injections. J. Clin. Med. 2021, 10, 981. [Google Scholar] [CrossRef] [PubMed]
- Moon, B.G.; Joe, S.G.; Hwang, J.U.; Kim, H.K.; Choe, J.; Yoon, Y.H. Prevalence and risk factors of early-stage age-related macular degeneration in patients examined at a health promotion center in korea. J. Korean Med. Sci. 2012, 27, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.H.; Mullins, R.F.; Hageman, G.S.; Johnson, L.V. A role for local inflammation in the formation of drusen in the aging eye. Am. J. Ophthalmol. 2002, 134, 411–431. [Google Scholar] [CrossRef]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [Green Version]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the rpe is associated with increased susceptibility to oxidative stress and amd. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [Green Version]
- Hollyfield, J.G.; Bonilha, V.L.; Rayborn, M.E.; Yang, X.; Shadrach, K.G.; Lu, L.; Ufret, R.L.; Salomon, R.G.; Perez, V.L. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat. Med. 2008, 14, 194–198. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 2015, 35, 7304–7311. [Google Scholar] [CrossRef]
- Du, H.; Yang, W.; Chen, L.; Shen, B.; Peng, C.; Li, H.; Ann, D.K.; Yen, Y.; Qiu, W. Emerging role of autophagy during ischemia-hypoxia and reperfusion in hepatocellular carcinoma. Int. J. Oncol. 2012, 40, 2049–2057. [Google Scholar] [PubMed] [Green Version]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Vereb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Garcia, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An overview of the role of lipofuscin in age-related neurodegeneration. Front Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, K.N.; Mahroo, O.A.; Khan, R.S.; Mohamed, M.D.; McKibbin, M.; Bird, A.; Michaelides, M.; Tufail, A.; Moore, A.T. Differentiating drusen: Drusen and drusen-like appearances associated with ageing, age-related macular degeneration, inherited eye disease and other pathological processes. Prog. Retin. Eye Res. 2016, 53, 70–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.T.; Gao, J.; Cao, S.; Sandhu, N.; Cui, J.Z.; Chou, C.L.; Fang, E.; Matsubara, J.A. Inflammatory mediators induced by amyloid-beta in the retina and rpe in vivo: Implications for inflammasome activation in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2013, 54, 2225–2237. [Google Scholar] [CrossRef] [Green Version]
- Lipecz, A.; Csipo, T.; Tarantini, S.; Hand, R.A.; Ngo, B.N.; Conley, S.; Nemeth, G.; Tsorbatzoglou, A.; Courtney, D.L.; Yabluchanska, V.; et al. Age-related impairment of neurovascular coupling responses: A dynamic vessel analysis (dva)-based approach to measure decreased flicker light stimulus-induced retinal arteriolar dilation in healthy older adults. Geroscience 2019, 41, 341–349. [Google Scholar] [CrossRef]
- Long, Q.; Cao, X.; Bian, A.; Li, Y. C3a increases vegf and decreases pedf mrna levels in human retinal pigment epithelial cells. Biomed. Res. Int. 2016, 2016, 6958752. [Google Scholar] [CrossRef] [Green Version]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components c3a and c5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef] [Green Version]
- Murray, H.; Qiu, B.; Ho, S.Y.; Wang, X. Complement factor b mediates ocular angiogenesis through regulating the vegf signaling pathway. Int. J. Mol. Sci. 2021, 22, 9580. [Google Scholar] [CrossRef]
- Kunchithapautham, K.; Rohrer, B. Sublytic membrane-attack-complex (mac) activation alters regulated rather than constitutive vascular endothelial growth factor (vegf) secretion in retinal pigment epithelium monolayers. J. Biol. Chem. 2011, 286, 23717–23724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, B.; Merriam, J.E.; Zernant, J.; Hancox, L.S.; Taiber, A.J.; Gehrs, K.; Cramer, K.; Neel, J.; Bergeron, J.; Barile, G.R.; et al. Variation in factor b (bf) and complement component 2 (c2) genes is associated with age-related macular degeneration. Nat. Genet. 2006, 38, 458–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, E.H.; Wang, K.; Thompson, S.; Riker, M.J.; Hoffmann, J.M.; Stone, E.M.; Mullins, R.F. Comparison of drusen and modifying genes in autosomal dominant radial drusen and age-related macular degeneration. Retina 2015, 35, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefansson, E.; Geirsdottir, A.; Sigurdsson, H. Metabolic physiology in age related macular degeneration. Prog. Retin. Eye Res. 2011, 30, 72–80. [Google Scholar] [CrossRef]
- McHugh, K.J.; Li, D.; Wang, J.C.; Kwark, L.; Loo, J.; Macha, V.; Farsiu, S.; Kim, L.A.; Saint-Geniez, M. Computational modeling of retinal hypoxia and photoreceptor degeneration in patients with age-related macular degeneration. PLoS ONE 2019, 14, e0216215. [Google Scholar] [CrossRef]
- Wang, S.; Wang, X.; Cheng, Y.; Ouyang, W.; Sang, X.; Liu, J.; Su, Y.; Liu, Y.; Li, C.; Yang, L.; et al. Autophagy dysfunction, cellular senescence, and abnormal immune-inflammatory responses in amd: From mechanisms to therapeutic potential. Oxid. Med. Cell Longev. 2019, 2019, 3632169. [Google Scholar] [CrossRef] [Green Version]
- Kaarniranta, K.; Blasiak, J.; Liton, P.; Boulton, M.; Klionsky, D.J.; Sinha, D. Autophagy in age-related macular degeneration. Autophagy 2022, 1–13. [Google Scholar] [CrossRef]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and its hallmarks: How to oppose aging strategically? A review of potential options for therapeutic intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.S.; Lin, S.; Copland, D.A.; Dick, A.D.; Liu, J. Cellular senescence in the aging retina and developments of senotherapies for age-related macular degeneration. J. Neuroinflammation 2021, 18, 32. [Google Scholar] [CrossRef]
- Little, K.; Ma, J.H.; Yang, N.; Chen, M.; Xu, H. Myofibroblasts in macular fibrosis secondary to neovascular age-related macular degeneration-the potential sources and molecular cues for their recruitment and activation. EBioMedicine 2018, 38, 283–291. [Google Scholar] [CrossRef]
- Tenbrock, L.; Wolf, J.; Boneva, S.; Schlecht, A.; Agostini, H.; Wieghofer, P.; Schlunck, G.; Lange, C. Subretinal fibrosis in neovascular age-related macular degeneration: Current concepts, therapeutic avenues, and future perspectives. Cell Tissue Res. 2022, 387, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M. Fibrosis and diseases of the eye. J. Clin. Investig. 2007, 117, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Yelle, N.; Venugopal, C.; Singh, S.K. Emt: Mechanisms and therapeutic implications. Pharmacol. Ther. 2018, 182, 80–94. [Google Scholar] [CrossRef]
- Li, H. Angiogenesis in the progression from liver fibrosis to cirrhosis and hepatocelluar carcinoma. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 217–233. [Google Scholar] [CrossRef]
- Heier, J.S.; Singh, R.P.; Wykoff, C.C.; Csaky, K.G.; Lai, T.Y.Y.; Loewenstein, A.; Schlottmann, P.G.; Paris, L.P.; Westenskow, P.D.; Quezada-Ruiz, C. The angiopoietin/tie pathway in retinal vascular diseases: A review. Retina 2021, 41, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Ojha, U.; Lee, Y.M. Pathological angiogenesis and inflammation in tissues. Arch. Pharm. Res. 2021, 44, 1–15. [Google Scholar] [CrossRef]
- DiPietro, L.A. Angiogenesis and wound repair: When enough is enough. J. Leukoc. Biol. 2016, 100, 979–984. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N. Vascular endothelial growth factor and age-related macular degeneration: From basic science to therapy. Nat. Med. 2010, 16, 1107–1111. [Google Scholar] [CrossRef]
- Marazita, M.C.; Dugour, A.; Marquioni-Ramella, M.D.; Figueroa, J.M.; Suburo, A.M. Oxidative stress-induced premature senescence dysregulates vegf and cfh expression in retinal pigment epithelial cells: Implications for age-related macular degeneration. Redox Biol. 2016, 7, 78–87. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, F.; Campbell, M. The blood-retina barrier in health and disease. FEBS J. 2021. Published Online Ahead of Print. [Google Scholar] [CrossRef]
- Eelen, G.; Treps, L.; Li, X.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis updated. Circ. Res. 2020, 127, 310–329. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Cazares, D.; Chavez-Dominguez, R.; Carlos-Reyes, A.; Lopez-Camarillo, C.; Hernadez de la Cruz, O.N.; Lopez-Gonzalez, J.S. Contribution of angiogenesis to inflammation and cancer. Front. Oncol. 2019, 9, 1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Patel, N.; Ohbayashi, M.; Nugent, A.K.; Ramchand, K.; Toda, M.; Chau, K.Y.; Bunce, C.; Webster, A.; Bird, A.C.; Ono, S.J.; et al. Circulating anti-retinal antibodies as immune markers in age-related macular degeneration. Immunology 2005, 115, 422–430. [Google Scholar] [CrossRef]
- Penfold, P.L.; Provis, J.M.; Furby, J.H.; Gatenby, P.A.; Billson, F.A. Autoantibodies to retinal astrocytes associated with age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 1990, 228, 270–274. [Google Scholar] [CrossRef]
- Joachim, S.C.; Bruns, K.; Lackner, K.J.; Pfeiffer, N.; Grus, F.H. Analysis of igg antibody patterns against retinal antigens and antibodies to alpha-crystallin, gfap, and alpha-enolase in sera of patients with “wet” age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2007, 245, 619–626. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the endothelial barrier: Identifying and reconciling controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef]
- Chanchal, S.; Mishra, A.; Singh, M.K.; Ashraf, M.Z. Understanding inflammatory responses in the manifestation of prothrombotic phenotypes. Front Cell Dev. Biol. 2020, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Subhi, Y.; Krogh Nielsen, M.; Molbech, C.R.; Kruger Falk, M.; Singh, A.; Hviid, T.V.F.; Nissen, M.H.; Sorensen, T.L. Association of cd11b+ monocytes and anti-vascular endothelial growth factor injections in treatment of neovascular age-related macular degeneration and polypoidal choroidal vasculopathy. JAMA Ophthalmol. 2019, 137, 515–522. [Google Scholar] [CrossRef]
- Tisi, A.; Feligioni, M.; Passacantando, M.; Ciancaglini, M.; Maccarone, R. The impact of oxidative stress on blood-retinal barrier physiology in age-related macular degeneration. Cells 2021, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Marneros, A.G.; Fan, J.; Yokoyama, Y.; Gerber, H.P.; Ferrara, N.; Crouch, R.K.; Olsen, B.R. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am. J. Pathol. 2005, 167, 1451–1459. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.A.; Kim, S.J.; Choi, Y.A.; Yoon, H.J.; Kim, A.; Lee, J. Retinal vegfa maintains the ultrastructure and function of choriocapillaris by preserving the endothelial plvap. Biochem. Biophys. Res. Commun. 2020, 522, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Kamba, T.; Tam, B.Y.; Hashizume, H.; Haskell, A.; Sennino, B.; Mancuso, M.R.; Norberg, S.M.; O’Brien, S.M.; Davis, R.B.; Gowen, L.C.; et al. Vegf-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H560–H576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzedine, H.; Escudier, B.; Lhomme, C.; Pautier, P.; Rouvier, P.; Gueutin, V.; Baumelou, A.; Derosa, L.; Bahleda, R.; Hollebecque, A.; et al. Kidney diseases associated with anti-vascular endothelial growth factor (vegf): An 8-year observational study at a single center. Medicine 2014, 93, 333–339. [Google Scholar] [CrossRef]
- Ollero, M.; Sahali, D. Inhibition of the vegf signalling pathway and glomerular disorders. Nephrol. Dial. Transpl. 2015, 30, 1449–1455. [Google Scholar] [CrossRef]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-specific alterations of vegf-a expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig. 2003, 111, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Hikichi, T.; Agarie, M. Reduced vessel density of the choriocapillaris during anti-vascular endothelial growth factor therapy for neovascular age-related macular degeneration. Investig. Opthalmology Vis. Sci. 2019, 60, 1088–1095. [Google Scholar] [CrossRef] [Green Version]
- Yi, X.; Ogata, N.; Komada, M.; Yamamoto, C.; Takahashi, K.; Omori, K.; Uyama, M. Vascular endothelial growth factor expression in choroidal neovascularization in rats. Graefes Arch. Clin. Exp. Ophthalmol. 1997, 235, 313–319. [Google Scholar] [CrossRef]
- Hudson, N.; Cahill, M.; Campbell, M. Inner blood-retina barrier involvement in dry age-related macular degeneration (amd) pathology. Neural Regen. Res. 2020, 15, 1656–1657. [Google Scholar] [CrossRef]
- Penfold, P.L.; Killingsworth, M.C.; Sarks, S.H. Senile macular degeneration: The involvement of immunocompetent cells. Graefes Arch. Clin. Exp. Ophthalmol. 1985, 223, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Ronning, K.E.; Karlen, S.J.; Miller, E.B.; Burns, M.E. Molecular profiling of resident and infiltrating mononuclear phagocytes during rapid adult retinal degeneration using single-cell rna sequencing. Sci. Rep. 2019, 9, 4858. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Padmanabhan, A.; Vaidya, T.; Watson, A.M.; Bhutto, I.A.; Hose, S.; Shang, P.; Stepicheva, N.; Yazdankhah, M.; Weiss, J.; et al. Neutrophils homing into the retina trigger pathology in early age-related macular degeneration. Commun. Biol. 2019, 2, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsumi-Miyahara, C.; Sonoda, K.H.; Egashira, K.; Ishibashi, M.; Qiao, H.; Oshima, T.; Murata, T.; Miyazaki, M.; Charo, I.F.; Hamano, S.; et al. The relative contributions of each subset of ocular infiltrated cells in experimental choroidal neovascularisation. Br. J. Ophthalmol. 2004, 88, 1217–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buschini, E.; Piras, A.; Nuzzi, R.; Vercelli, A. Age related macular degeneration and drusen: Neuroinflammation in the retina. Prog. Neurobiol. 2011, 95, 14–25. [Google Scholar] [CrossRef] [PubMed]
- McLeod, D.S.; Bhutto, I.; Edwards, M.M.; Silver, R.E.; Seddon, J.M.; Lutty, G.A. Distribution and quantification of choroidal macrophages in human eyes with age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5843–5855. [Google Scholar] [CrossRef]
- Cherepanoff, S.; McMenamin, P.; Gillies, M.C.; Kettle, E.; Sarks, S.H. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br. J. Ophthalmol. 2010, 94, 918–925. [Google Scholar] [CrossRef]
- Hagbi-Levi, S.; Grunin, M.; Jaouni, T.; Tiosano, L.; Rinsky, B.; Elbaz-Hayoun, S.; Peled, A.; Chowers, I. Proangiogenic characteristics of activated macrophages from patients with age-related macular degeneration. Neurobiol. Aging 2017, 51, 71–82. [Google Scholar] [CrossRef]
- Oh, H.; Takagi, H.; Takagi, C.; Suzuma, K.; Otani, A.; Ishida, K.; Matsumura, M.; Ogura, Y.; Honda, Y. The potential angiogenic role of macrophages in the formation of choroidal neovascular membranes. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1891–1898. [Google Scholar]
- Grossniklaus, H.E.; Ling, J.X.; Wallace, T.M.; Dithmar, S.; Lawson, D.H.; Cohen, C.; Elner, V.M.; Elner, S.G.; Sternberg, P., Jr. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol. Vis. 2002, 8, 119–126. [Google Scholar]
- Wang, Z.; Koenig, A.L.; Lavine, K.J.; Apte, R.S. Macrophage plasticity and function in the eye and heart. Trends Immunol. 2019, 40, 825–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lis-Lopez, L.; Bauset, C.; Seco-Cervera, M.; Cosin-Roger, J. Is the macrophage phenotype determinant for fibrosis development? Biomedicines 2021, 9, 1747. [Google Scholar] [CrossRef]
- Combadiere, C.; Feumi, C.; Raoul, W.; Keller, N.; Rodero, M.; Pezard, A.; Lavalette, S.; Houssier, M.; Jonet, L.; Picard, E.; et al. Cx3cr1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J. Clin. Investig. 2007, 117, 2920–2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Brown, K.E.; Milam, A.H. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp. Eye Res. 2003, 76, 463–471. [Google Scholar] [CrossRef]
- Droho, S.; Cuda, C.M.; Perlman, H.; Lavine, J.A. Macrophage-derived interleukin-6 is necessary and sufficient for choroidal angiogenesis. Sci. Rep. 2021, 11, 18084. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Yao, L.C.; Feng, J.; Romano, T.; Jung, S.S.; Schreiter, J.L.; Yan, L.; Shealy, D.J.; McDonald, D.M. Tnf-alpha drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice. J. Clin. Investig. 2009, 119, 2954–2964. [Google Scholar]
- Fahey, E.; Doyle, S.L. Il-1 family cytokine regulation of vascular permeability and angiogenesis. Front. Immunol. 2019, 10, 1426. [Google Scholar] [CrossRef] [Green Version]
- Heidemann, J.; Ogawa, H.; Dwinell, M.B.; Rafiee, P.; Maaser, C.; Gockel, H.R.; Otterson, M.F.; Ota, D.M.; Lugering, N.; Domschke, W.; et al. Angiogenic effects of interleukin 8 (cxcl8) in human intestinal microvascular endothelial cells are mediated by cxcr2. J. Biol. Chem. 2003, 278, 8508–8515. [Google Scholar] [CrossRef] [Green Version]
- Nahavandipour, A.; Krogh Nielsen, M.; Sorensen, T.L.; Subhi, Y. Systemic levels of interleukin-6 in patients with age-related macular degeneration: A systematic review and meta-analysis. Acta Ophthalmol. 2020, 98, 434–444. [Google Scholar] [CrossRef]
- Chernykh, V.; Shevchenko, A.; Konenkov, V.; Prokofiev, V.; Eremina, A.; Trunov, A. Tnf-alpha gene polymorphisms: Association with age-related macular degeneration in russian population. Int. J. Ophthalmol. 2019, 12, 25–29. [Google Scholar]
- Zhao, M.; Bai, Y.; Xie, W.; Shi, X.; Li, F.; Yang, F.; Sun, Y.; Huang, L.; Li, X. Interleukin-1beta level is increased in vitreous of patients with neovascular age-related macular degeneration (namd) and polypoidal choroidal vasculopathy (pcv). PLoS ONE 2015, 10, e0125150. [Google Scholar]
- Roshanipour, N.; Shahriyari, E.; Ghaffari Laleh, M.; Vahedi, L.; Mirjand Gerami, S.; Khamaneh, A. Associations of tlr4 and il-8 genes polymorphisms with age-related macular degeneration (amd): A systematic review and meta-analysis. Ophthalmic Genet. 2021, 42, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Liukkonen, M.P.K.; Paterno, J.J.; Kivinen, N.; Siintamo, L.; Koskela, A.K.J.; Kaarniranta, K. Epithelial-mesenchymal transition-related serum markers et-1, il-8 and tgf-beta2 are elevated in a finnish wet age-related macular degeneration cohort. Acta Ophthalmol. 2021, 100, e1153–e1162. [Google Scholar] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Mammadzada, P.; Corredoira, P.M.; Andre, H. The role of hypoxia-inducible factors in neovascular age-related macular degeneration: A gene therapy perspective. Cell Mol. Life Sci. 2020, 77, 819–833. [Google Scholar] [CrossRef]
- Sheridan, C.M.; Pate, S.; Hiscott, P.; Wong, D.; Pattwell, D.M.; Kent, D. Expression of hypoxia-inducible factor-1alpha and -2alpha in human choroidal neovascular membranes. Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 1361–1367. [Google Scholar] [CrossRef]
- Inoue, Y.; Yanagi, Y.; Matsuura, K.; Takahashi, H.; Tamaki, Y.; Araie, M. Expression of hypoxia-inducible factor 1alpha and 2alpha in choroidal neovascular membranes associated with age-related macular degeneration. Br. J. Ophthalmol. 2007, 91, 1720–1721. [Google Scholar] [CrossRef] [Green Version]
- Caprara, C.; Thiersch, M.; Lange, C.; Joly, S.; Samardzija, M.; Grimm, C. Hif1a is essential for the development of the intermediate plexus of the retinal vasculature. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Usui, Y.; Westenskow, P.D.; Kurihara, T.; Aguilar, E.; Sakimoto, S.; Paris, L.P.; Wittgrove, C.; Feitelberg, D.; Friedlander, M.S.; Moreno, S.K.; et al. Neurovascular crosstalk between interneurons and capillaries is required for vision. J. Clin. Investig. 2015, 125, 2335–2346. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Cui, X.; Han, Y.; Park, K.S.; Gao, X.; Zhang, X.; Yuan, Z.; Hu, Y.; Hsu, C.W.; Li, X.; et al. Hypoxic drive caused type 3 neovascularization in a preclinical model of exudative age-related macular degeneration. Hum. Mol. Genet. 2019, 28, 3475–3485. [Google Scholar] [CrossRef]
- Suzuki, S.; Sato, T.; Watanabe, M.; Higashide, M.; Tsugeno, Y.; Umetsu, A.; Furuhashi, M.; Ida, Y.; Hikage, F.; Ohguro, H. Hypoxia differently affects tgf-beta2-induced epithelial mesenchymal transitions in the 2d and 3d culture of the human retinal pigment epithelium cells. Int. J. Mol. Sci. 2022, 23, 5473. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heloterä, H.; Kaarniranta, K. A Linkage between Angiogenesis and Inflammation in Neovascular Age-Related Macular Degeneration. Cells 2022, 11, 3453. https://doi.org/10.3390/cells11213453
Heloterä H, Kaarniranta K. A Linkage between Angiogenesis and Inflammation in Neovascular Age-Related Macular Degeneration. Cells. 2022; 11(21):3453. https://doi.org/10.3390/cells11213453
Chicago/Turabian StyleHeloterä, Hanna, and Kai Kaarniranta. 2022. "A Linkage between Angiogenesis and Inflammation in Neovascular Age-Related Macular Degeneration" Cells 11, no. 21: 3453. https://doi.org/10.3390/cells11213453
APA StyleHeloterä, H., & Kaarniranta, K. (2022). A Linkage between Angiogenesis and Inflammation in Neovascular Age-Related Macular Degeneration. Cells, 11(21), 3453. https://doi.org/10.3390/cells11213453