
Only Acute but Not Chronic Thrombocytopenia Protects Mice against Left Ventricular Dysfunction after Acute Myocardial Infarction
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Animals
2.2. Antibody Induced Thrombocytopenia in Mice
2.3. Experimental Model of Acute Myocardial Infarction (AMI) and Reperfusion in Mice
2.4. Experiments with Human Blood
2.5. Isolation of Cardiac Fibroblasts and Incubation with Platelet Supernatant
2.6. Flow Cytometric Analysis of Blood Cells
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Immunohistochemistry of Cardiac Sections
2.9. Collagen-Staining of Cardiac Sections
2.10. Immunofluorescence (IF)-Staining of Heart Sections
2.11. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.12. Study Population and Light Transmission Aggregometry (LTA)
2.13. Statistical Analysis
3. Results
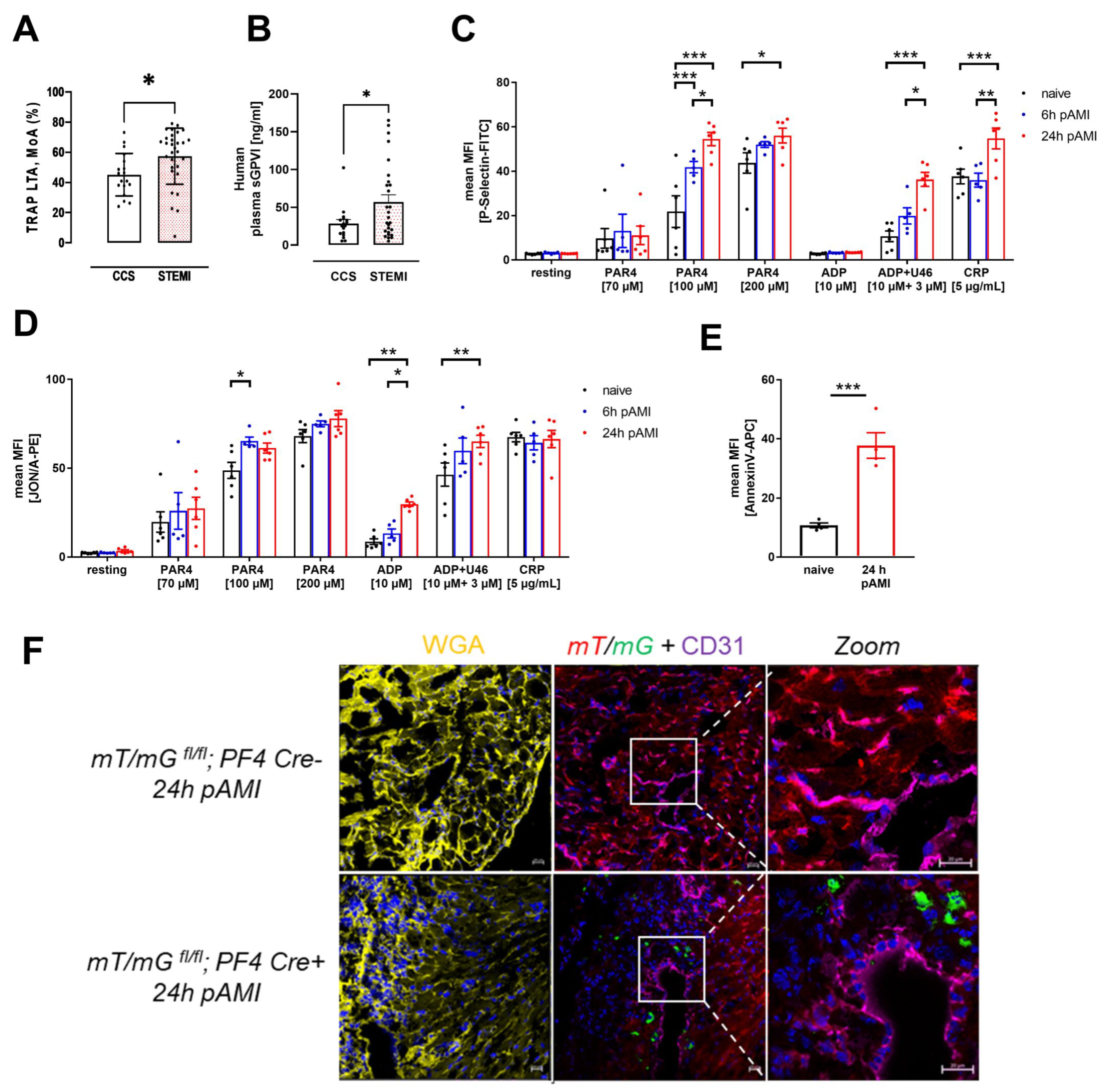
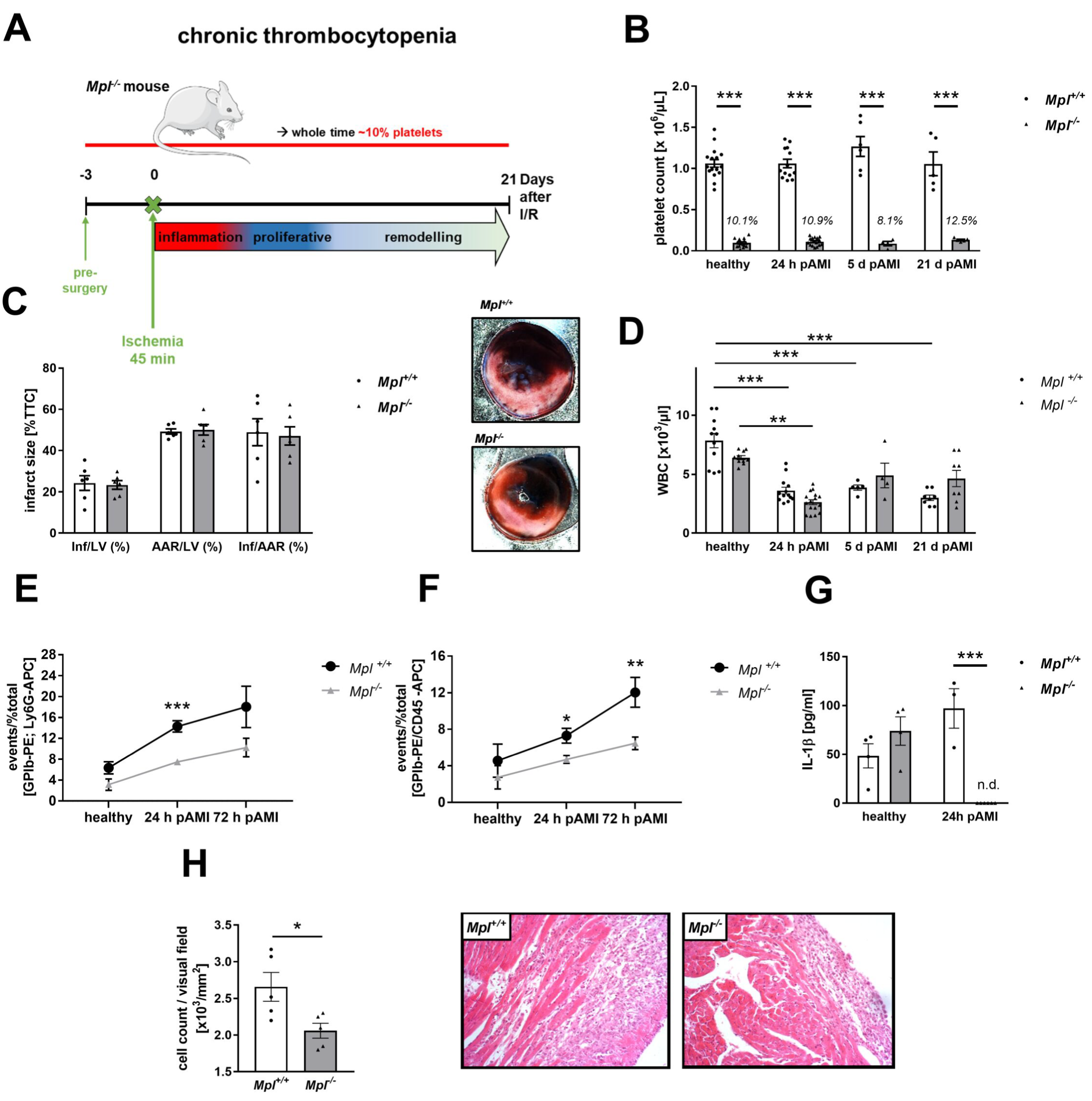
3.1. Enhanced Platelet Reactivity in Patients and Experimental Mice after Myocardial Infarction Causes Platelet Invasion
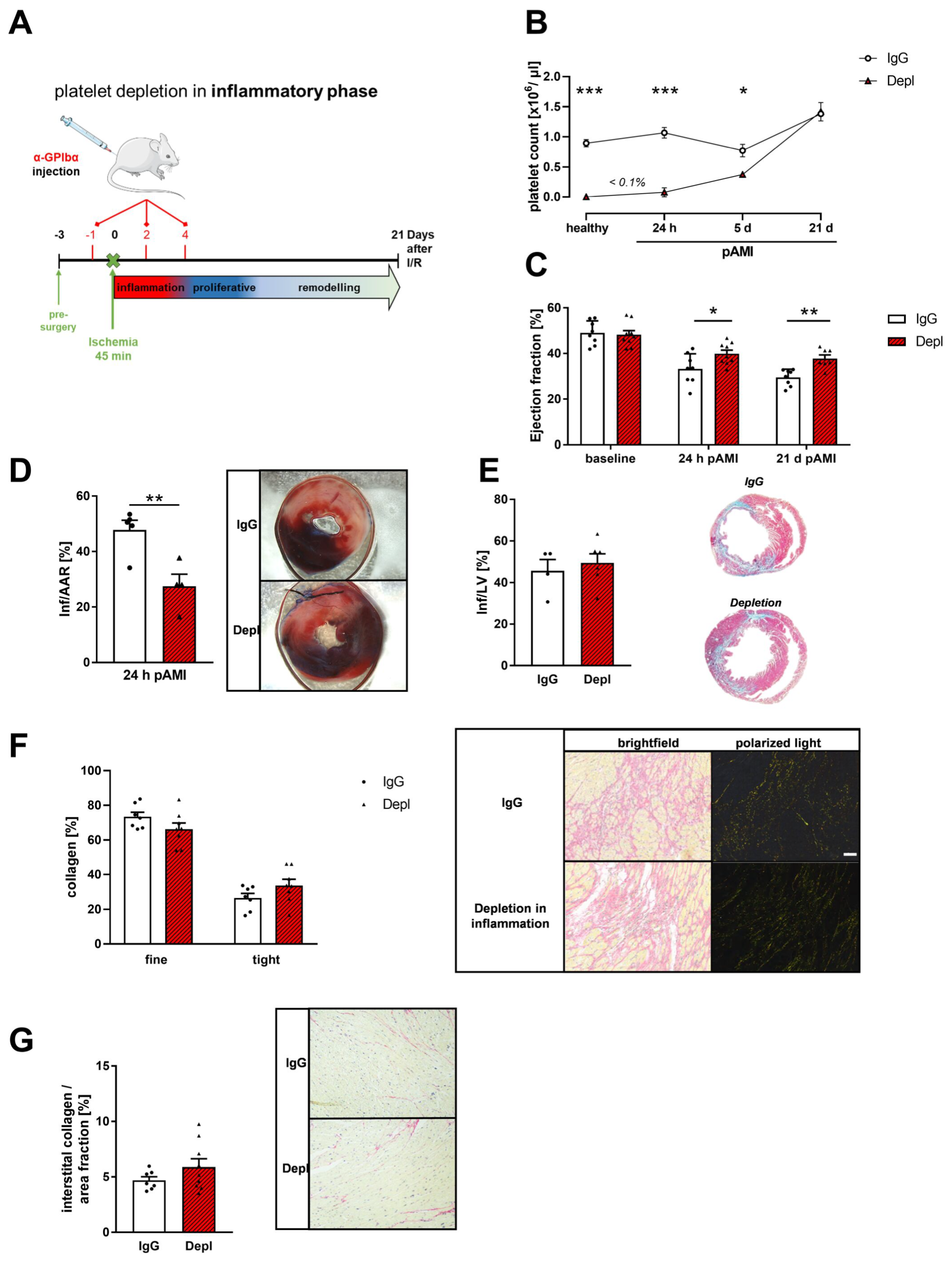
3.2. Platelet Depletion in the Early Phase after AMI Reduces Infarct Size and Improves Cardiac Function
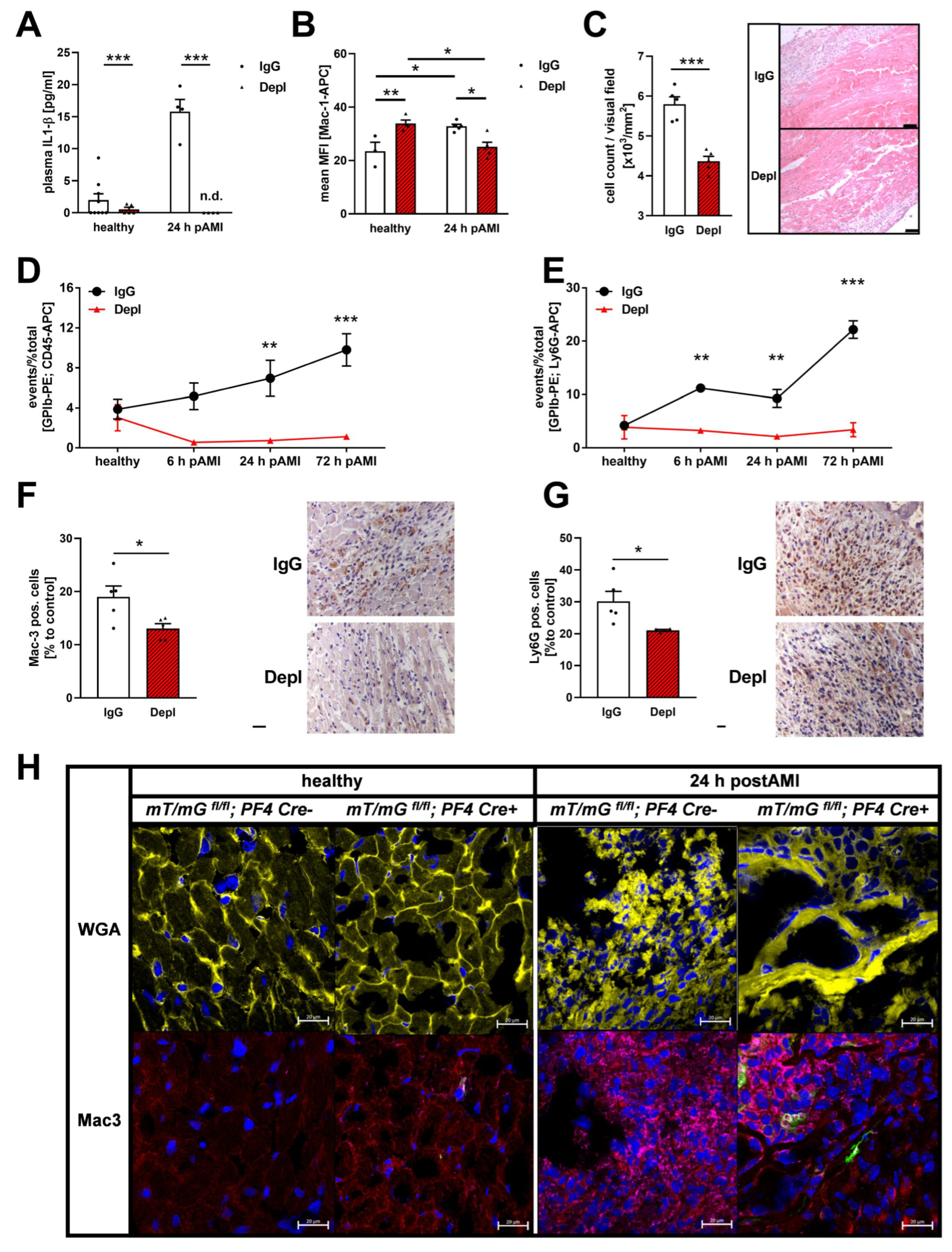
3.3. Reduced Inflammatory Responses in the Acute Phase after MI in Platelet Depleted Mice
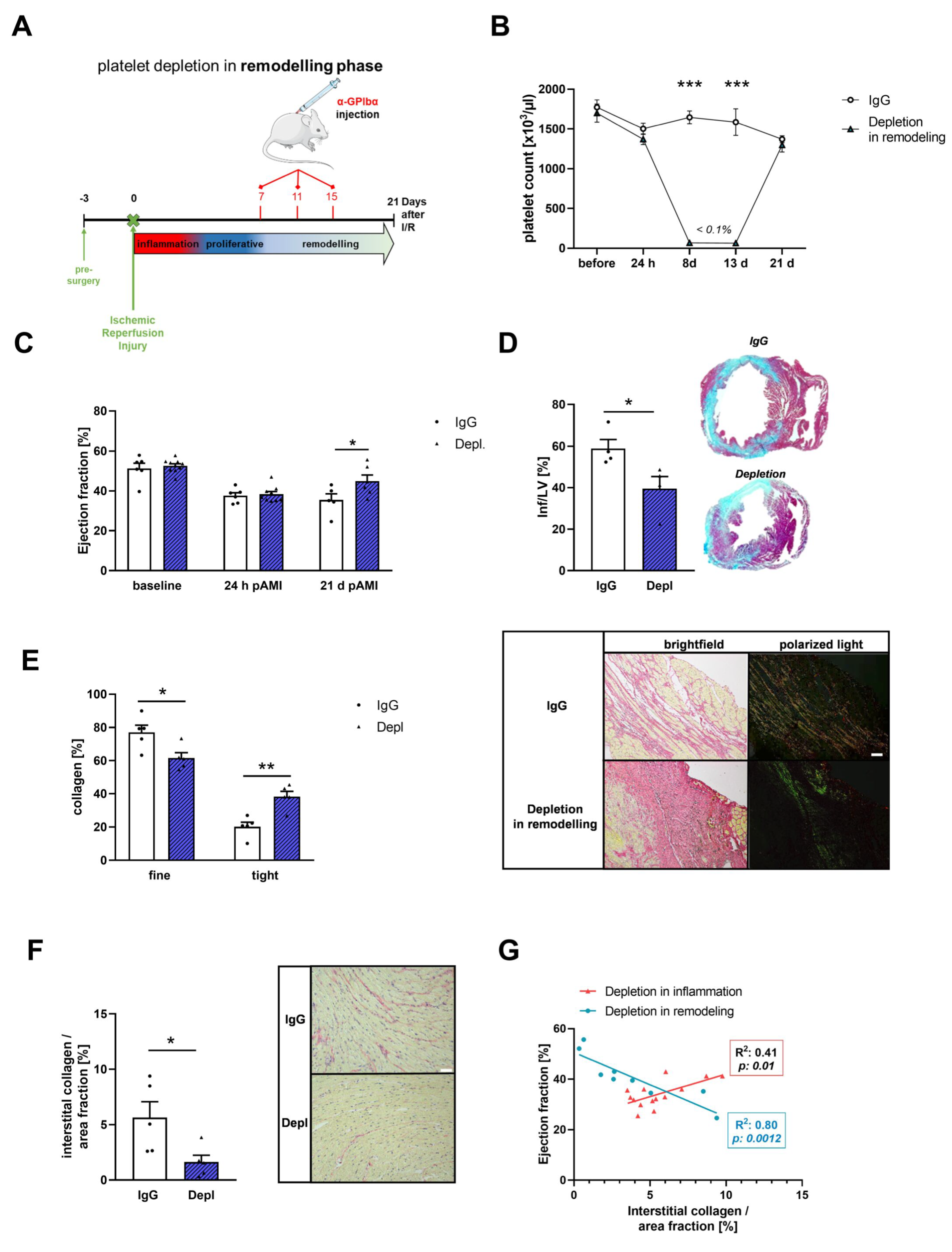
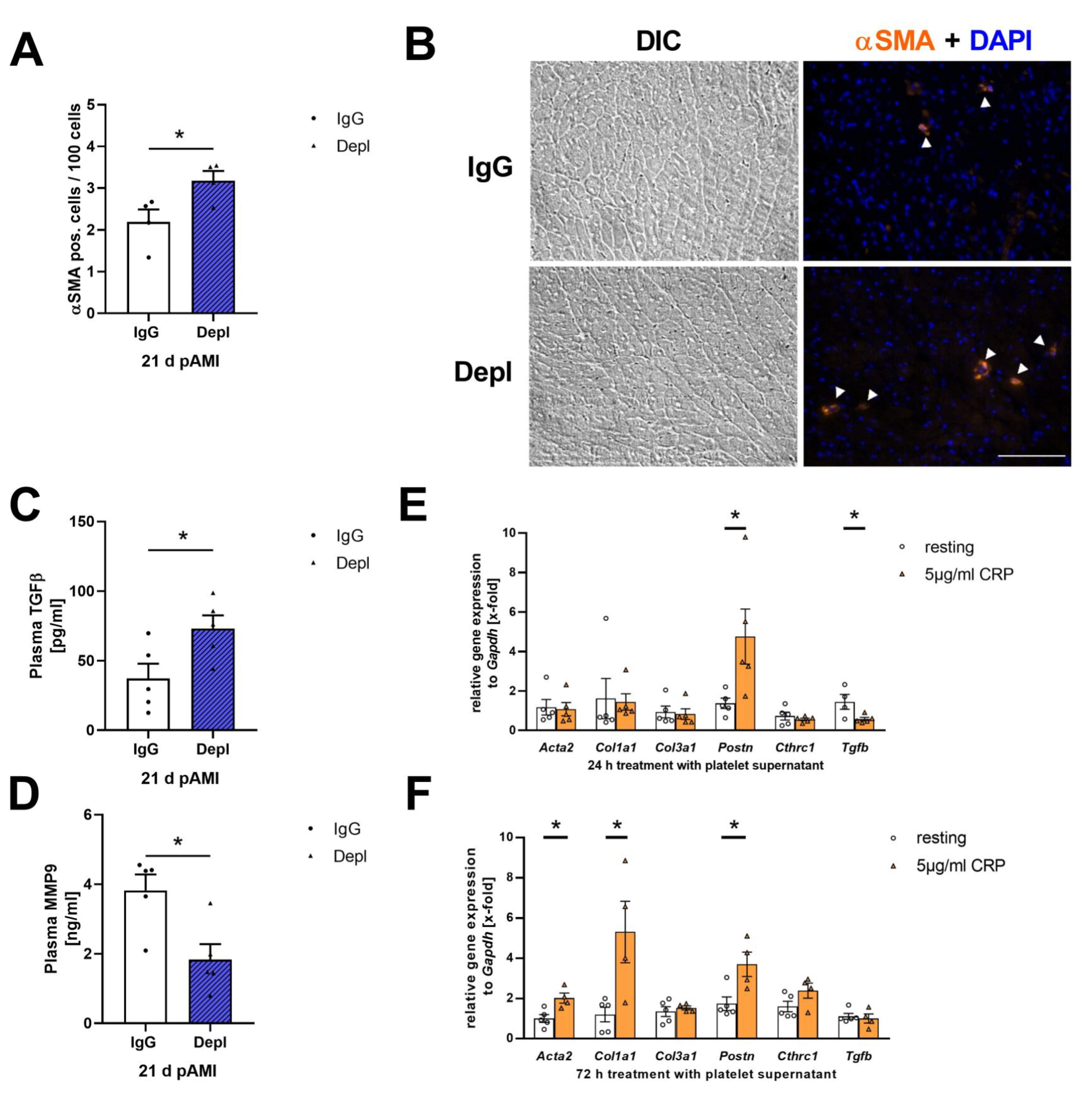
3.4. Platelets Modulate Cardiac Remodeling and Scar Formation after AMI
3.5. Platelet Activation Affects Cardiac Fibroblast Transformation in the Remodeling Phase after AMI
3.6. Reduced Platelet-Mediated Inflammation in Mice with Chronic Thrombocytopenia
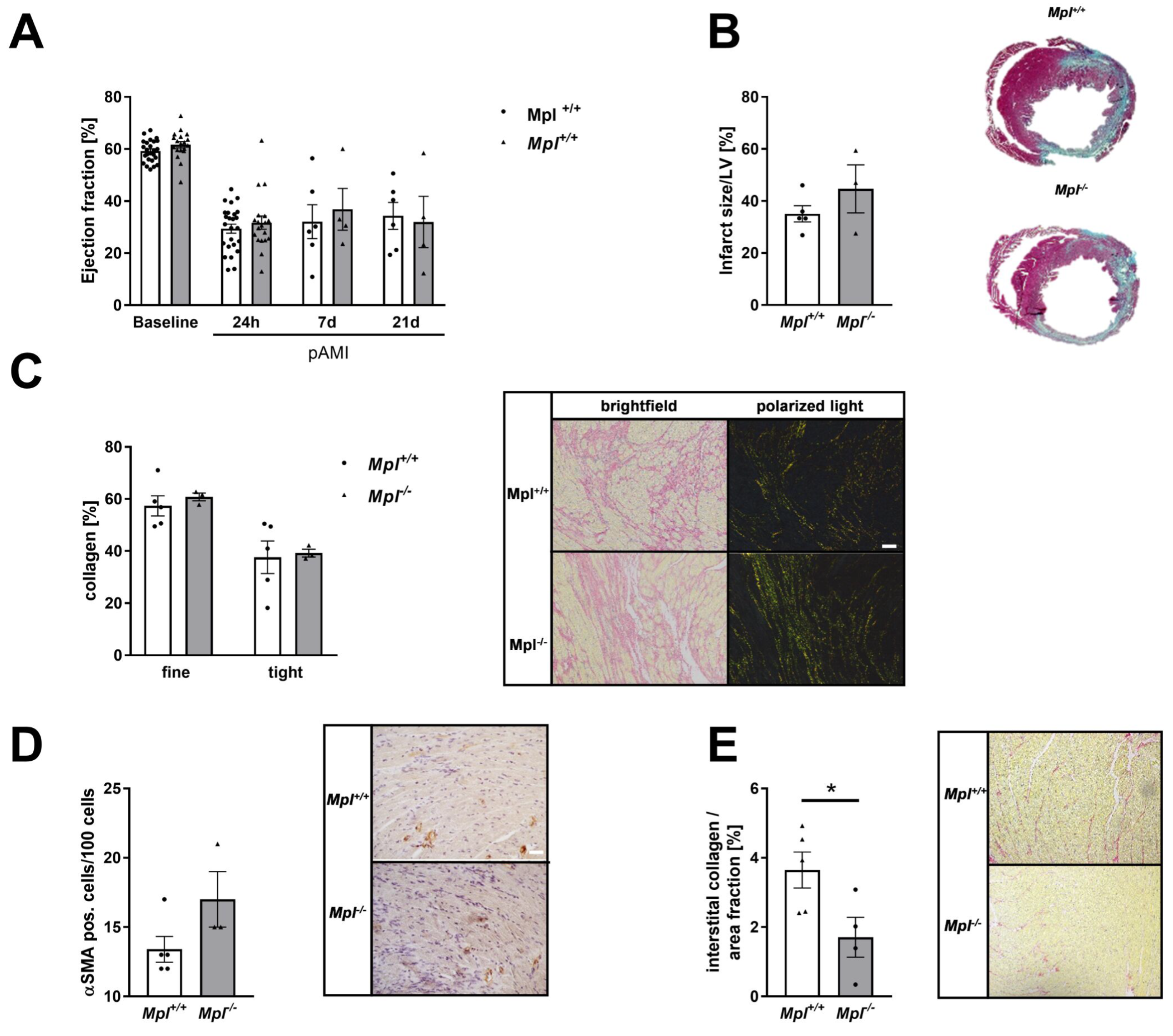
3.7. Unaltered Collagen Composition and Left Ventricular Function in Mice with Chronic Thrombocytopenia
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schanze, N.; Bode, C.; Duerschmied, D. Platelet Contributions to Myocardial Ischemia/Reperfusion Injury. Front. Immunol. 2019, 10, 1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, R.W.; Sidney, S.; Chandra, M.; Sorel, M.; Selby, J.V.; Go, A.S. Population trends in the incidence and outcomes of acute myocardial infarction. N. Engl. J. Med. 2010, 362, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, B.; James, S.; Agewall, S.; Antunes, M.J.; Bucciarelli-Ducci, C.; Bueno, H.; Caforio, A.L.P.; Crea, F.; Goudevenos, J.A.; Halvorsen, S.; et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2018, 39, 119–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndrepepa, G.; Alger, P.; Kufner, S.; Mehilli, J.; Schömig, A.; Kastrati, A. ST-segment resolution after primary percutaneous coronary intervention in patients with acute ST-segment elevation myocardial infarction. Cardiol. J. 2012, 19, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.M.; Jiang, L.X.; Chen, Y.P.; Xie, J.X.; Pan, H.C.; Peto, R.; Collins, R.; Liu, L.S.; COMMIT (ClOpidogrel and Metoprolol in Myocardial Infarction Trial) Collaborative Group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: Randomised placebo-controlled trial. Lancet 2005, 366, 1607–1621. [Google Scholar] [CrossRef]
- Chen, Z.M.; Pan, H.C.; Chen, Y.P.; Peto, R.; Collins, R.; Jiang, L.X.; Xie, J.X.; Liu, L.S. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: Randomised placebo-controlled trial. Lancet 2005, 366, 1622–1632. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, X.M.; Fang, L.; Jennings, N.L.; Su, Y.; Q, X.; Samson, A.L.; Kiriazis, H.; Wang, X.F.; Shan, L.; et al. Novel role of platelets in mediating inflammatory responses and ventricular rupture or remodeling following myocardial infarction. Arter. Thromb. Vasc. Biol. 2011, 31, 834–841. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Geraghty, O.C.; Mehta, Z.; Rothwell, P.M. Age-specific risks, severity, time course, and outcome of bleeding on long-term antiplatelet treatment after vascular events: A population-based cohort study. Lancet 2017, 390, 490–499. [Google Scholar] [CrossRef] [Green Version]
- Lebas, H.; Yahiaoui, K.; Martos, R.; Boulaftali, Y. Platelets Are at the Nexus of Vascular Diseases. Front. Cardiovasc. Med. 2019, 6, 132. [Google Scholar] [CrossRef]
- Fruchter, O.; Blich, M.; Jacob, G. Fatal acute myocardial infarction during severe thrombocytopenia in a patient with idiopathic thrombocytopenic purpura. Am. J. Med. Sci. 2002, 323, 279–280. [Google Scholar] [CrossRef]
- Rubinfeld, G.D.; Smilowitz, N.R.; Berger, J.S.; Newman, J.D. Association of Thrombocytopenia, Revascularization, and In-Hospital Outcomes in Patients with Acute Myocardial Infarction. Am. J. Med. 2019, 132, 942–948.e5. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.; Movahed, M.R.; Hashemzadeh, M.; Hashemzadeh, M. The presence of idiopathic thrombocytopenic purpura correlates with lower rate of acute ST-elevation myocardial infarction. Future Cardiol. 2021, 17, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Pachel, C.; Mathes, D.; Arias-Loza, A.-P.; Heitzmann, W.; Nordbeck, P.; Deppermann, C.; Lorenz, V.; Hofmann, U.; Nieswandt, B.; Frantz, S. Inhibition of Platelet GPVI Protects Against Myocardial Ischemia—Reperfusion Injury. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 629–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönberger, T.; Ziegler, M.; Borst, O.; Konrad, I.; Nieswandt, B.; Massberg, S.; Ochmann, C.; Jürgens, T.; Seizer, P.; Langer, H.; et al. The dimeric platelet collagen receptor GPVI-Fc reduces platelet adhesion to activated endothelium and preserves myocardial function after transient ischemia in mice. Am. J. Physiol. Cell Physiol. 2012, 303, C757–C766. [Google Scholar] [CrossRef]
- Xu, Y.; Huo, Y.; Toufektsian, M.C.; Ramos, S.I.; Ma, Y.; Tejani, A.D.; French, B.A.; Yang, Z. Activated platelets contribute importantly to myocardial reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H692–H699. [Google Scholar] [CrossRef] [PubMed]
- Kolpakov, M.A.; Rafiq, K.; Guo, X.; Hooshdaran, B.; Wang, T.; Vlasenko, L.; Bashkirova, Y.V.; Zhang, X.; Chen, X.; Iftikhar, S.; et al. Protease-activated receptor 4 deficiency offers cardioprotection after acute ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2016, 90, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Gawaz, M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc. Res. 2004, 61, 498–511. [Google Scholar] [CrossRef]
- Wagner, D.D.; Burger, P.C. Platelets in inflammation and thrombosis. Arter. Thromb. Vasc. Biol. 2003, 23, 2131–2137. [Google Scholar] [CrossRef]
- von Hundelshausen, P.; Weber, C. Platelets as immune cells: Bridging inflammation and cardiovascular disease. Circ. Res. 2007, 100, 27–40. [Google Scholar] [CrossRef]
- Walsh, T.G.; Poole, A.W. Do platelets promote cardiac recovery after myocardial infarction: Roles beyond occlusive ischemic damage. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H1043–H1048. [Google Scholar] [CrossRef]
- Koehler, F.; Winkler, S.; Schieber, M.; Sechtem, U.; Stangl, K.; Böhm, M.; Boll, H.; Baumann, G.; Honold, M.; Koehler, K.; et al. Impact of remote telemedical management on mortality and hospitalizations in ambulatory patients with chronic heart failure: The telemedical interventional monitoring in heart failure study. Circulation 2011, 123, 1873–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hałucha, K.; Rak-Pasikowska, A.; Bil-Lula, I. Protective Role of Platelets in Myocardial Infarction and Ischemia/Reperfusion Injury. Cardiol. Res. Pract. 2021, 2021, 5545416. [Google Scholar] [CrossRef] [PubMed]
- Gawaz, M.; Vogel, S. Platelets in tissue repair: Control of apoptosis and interactions with regenerative cells. Blood 2013, 122, 2550–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stellos, K.; Langer, H.; Daub, K.; Schoenberger, T.; Gauss, A.; Geisler, T.; Bigalke, B.; Mueller, I.; Schumm, M.; Schaefer, I.; et al. Platelet-derived stromal cell-derived factor-1 regulates adhesion and promotes differentiation of human CD34+ cells to endothelial progenitor cells. Circulation 2008, 117, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Alexander, W.S.; Roberts, A.W.; Nicola, N.A.; Li, R.; Metcalf, D. Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-Mpl. Blood 1996, 87, 2162–2170. [Google Scholar] [CrossRef] [Green Version]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef]
- Tiedt, R.; Schomber, T.; Hao-Shen, H.; Skoda, R.C. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood 2007, 109, 1503–1506. [Google Scholar] [CrossRef] [Green Version]
- Gorressen, S.; Stern, M.; van de Sandt, A.M.; Cortese-Krott, M.M.; Ohlig, J.; Rassaf, T.; Gödecke, A.; Fischer, J.W.; Heusch, G.; Merx, M.W.; et al. Circulating NOS3 modulates left ventricular remodeling following reperfused myocardial infarction. PLoS ONE 2015, 10, e0120961. [Google Scholar] [CrossRef]
- Gorski, D.J.; Petz, A.; Reichert, C.; Twarock, S.; Grandoch, M.; Fischer, J.W. Cardiac fibroblast activation and hyaluronan synthesis in response to hyperglycemia and diet-induced insulin resistance. Sci. Rep. 2019, 9, 1827. [Google Scholar] [CrossRef] [Green Version]
- Donner, L.; Fälker, K.; Gremer, L.; Klinker, S.; Pagani, G.; Ljungberg, L.U.; Lothmann, K.; Rizzi, F.; Schaller, M.; Gohlke, H.; et al. Platelets contribute to amyloid-β aggregation in cerebral vessels through integrin αIIbβ3-induced outside-in signaling and clusterin release. Sci. Signal 2016, 9, ra52. [Google Scholar] [CrossRef]
- Petzold, T.; Thienel, M.; Dannenberg, L.; Mourikis, P.; Helten, C.; Ayhan, A.; M’Pembele, R.; Achilles, A.; Trojovky, K.; Konsek, D.; et al. Rivaroxaban Reduces Arterial Thrombosis by Inhibition of FXa-Driven Platelet Activation via Protease Activated Receptor-1. Circ. Res. 2020, 126, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Krüger, I.; Reusswig, F.; Krott, K.J.; Lersch, C.F.; Spelleken, M.; Elvers, M. Genetic Labeling of Cells Allows Identification and Tracking of Transgenic Platelets in Mice. Int. J. Mol. Sci. 2021, 22, 3710. [Google Scholar] [CrossRef] [PubMed]
- Emde, B.; Heinen, A.; Gödecke, A.; Bottermann, K. Wheat germ agglutinin staining as a suitable method for detection and quantification of fibrosis in cardiac tissue after myocardial infarction. Eur. J. Histochem. 2014, 58, 2448. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, K.E.; Quinn, K.P.; Tang, K.M.; Georgakoudi, I.; Black, L.D., 3rd. Extracellular matrix remodeling following myocardial infarction influences the therapeutic potential of mesenchymal stem cells. Stem Cell Res. Ther. 2014, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Junqueira, L.C.; Bignolas, G.; Brentani, R.R. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem. J. 1979, 11, 447–455. [Google Scholar] [CrossRef]
- Ong, S.B.; Hernández-Reséndiz, S.; Crespo-Avilan, G.E.; Mukhametshina, R.T.; Kwek, X.Y.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol. Ther. 2018, 186, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Cronin, K.; Crane, J.S. Biochemistry, Collagen Synthesis. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef]
- Lighthouse, J.K.; Small, E.M. Transcriptional control of cardiac fibroblast plasticity. J. Mol. Cell. Cardiol. 2016, 91, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.; Wang, W.; Qu, J.; Croft, L.; Degen, J.L.; Coller, B.S.; Ahamed, J. Platelet TGF-β1 contributions to plasma TGF-β1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood 2012, 119, 1064–1074. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.H.; Chang, Y.; Reed, N.I.; Sheppard, D. α-Smooth muscle actin is an inconsistent marker of fibroblasts responsible for force-dependent TGFβ activation or collagen production across multiple models of organ fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L824–L836. [Google Scholar] [CrossRef]
- Tousoulis, D.; Paroutoglou, I.P.; Papageorgiou, N.; Charakida, M.; Stefanadis, C. Recent therapeutic approaches to platelet activation in coronary artery disease. Pharmacol. Ther. 2010, 127, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Kayikçioglu, M.; Can, L.; Mete-Erdem, N.; Kültürsay, H.; Payzin, S.; Kokuludag, A.; Türkoglu, C. Soluble P-selectin and the success of thrombolysis in acute myocardial infarction. Int. J. Cardiol. 2001, 79, 223–229. [Google Scholar] [CrossRef]
- Weyrich, A.S.; Ma, X.Y.; Lefer, D.J.; Albertine, K.H.; Lefer, A.M. In vivo neutralization of P-selectin protects feline heart and endothelium in myocardial ischemia and reperfusion injury. J. Clin. Investig. 1993, 91, 2620–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabet, M.; Garcia-Dorado, D.; Inserte, J.; Barrabés, J.A.; Lidón, R.M.; Soriano, B.; Azevedo, M.; Padilla, F.; Agulló, L.; Ruiz-Meana, M.; et al. Platelets activated by transient coronary occlusion exacerbate ischemia-reperfusion injury in rat hearts. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1134–H1141. [Google Scholar] [CrossRef] [Green Version]
- Maynard, D.M.; Heijnen, H.F.; Horne, M.K.; White, J.G.; Gahl, W.A. Proteomic analysis of platelet alpha-granules using mass spectrometry. J. Thromb. Haemost. 2007, 5, 1945–1955. [Google Scholar] [CrossRef]
- Michelson, A.D.; Barnard, M.R.; Krueger, L.A.; Valeri, C.R.; Furman, M.I. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: Studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation 2001, 104, 1533–1537. [Google Scholar] [CrossRef] [Green Version]
- Jugdutt, B.I. Ventricular remodeling after infarction and the extracellular collagen matrix: When is enough enough? Circulation 2003, 108, 1395–1403. [Google Scholar] [CrossRef] [Green Version]
- Yabanoglu, S.; Akkiki, M.; Seguelas, M.H.; Mialet-Perez, J.; Parini, A.; Pizzinat, N. Platelet derived serotonin drives the activation of rat cardiac fibroblasts by 5-HT2A receptors. J. Mol. Cell. Cardiol. 2009, 46, 518–525. [Google Scholar] [CrossRef]
- Shinde, A.V.; Humeres, C.; Frangogiannis, N.G. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 298–309. [Google Scholar] [CrossRef]
- Uchinaka, A.; Yoshida, M.; Tanaka, K.; Hamada, Y.; Mori, S.; Maeno, Y.; Miyagawa, S.; Sawa, Y.; Nagata, K.; Yamamoto, H.; et al. Overexpression of collagen type III in injured myocardium prevents cardiac systolic dysfunction by changing the balance of collagen distribution. J. Thorac. Cardiovasc. Surg. 2018, 156, 217–226.E3. [Google Scholar] [CrossRef]
- Wei, S.; Chow, L.T.; Shum, I.O.; Qin, L.; Sanderson, J.E. Left and right ventricular collagen type I/III ratios and remodeling post-myocardial infarction. J. Card. Fail. 1999, 5, 117–126. [Google Scholar] [CrossRef]
- Xie, Y.; Chen, J.; Han, P.; Yang, P.; Hou, J.; Kang, Y.J. Immunohistochemical detection of differentially localized up-regulation of lysyl oxidase and down-regulation of matrix metalloproteinase-1 in rhesus monkey model of chronic myocardial infarction. Exp. Biol. Med. 2012, 237, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Auerbach, H.E. New-onset acute thrombocytopenia in hospitalized patients: Pathophysiology and diagnostic approach. J. Community Hosp. Intern. Med. Perspect. 2017, 7, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Chehab, O.; Abdallah, N.; Kanj, A.; Pahuja, M.; Adegbala, O.; Morsi, R.Z.; Mishra, T.; Afonso, L.; Abidov, A. Impact of immune thrombocytopenic purpura on clinical outcomes in patients with acute myocardial infarction. Clin. Cardiol. 2020, 43, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.J.; Shanbhag, A.; Chiang, C.C.; Girardo, M.E.; Seri, A.R.; Khalid, M.U.; Rayfield, C.; O’Shea, M.P.; Fatunde, O.; Fortuin, F.D. Baseline thrombocytopenia in acute coronary syndrome: The lower, the worse. Int. J. Cardiol. 2021, 332, 1–7. [Google Scholar] [CrossRef]
- Klier, M.; Gorressen, S.; Urbahn, M.-A.; Barbosa, D.; Ouwens, M.; Fischer, J.W.; Elvers, M. Enzymatic Activity Is Not Required for Phospholipase D Mediated TNF-α Regulation and Myocardial Healing. Front. Physiol. 2018, 9, 1698. [Google Scholar] [CrossRef] [PubMed]
- Klose, A.M.; Klier, M.; Gorressen, S.; Elvers, M. Enhanced Integrin Activation of PLD2-Deficient Platelets Accelerates Inflammation after Myocardial Infarction. Int. J. Mol. Sci. 2020, 21, 3210. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reusswig, F.; Polzin, A.; Klier, M.; Dille, M.A.; Ayhan, A.; Benkhoff, M.; Lersch, C.; Prinz, A.; Gorressen, S.; Fischer, J.W.; et al. Only Acute but Not Chronic Thrombocytopenia Protects Mice against Left Ventricular Dysfunction after Acute Myocardial Infarction. Cells 2022, 11, 3500. https://doi.org/10.3390/cells11213500
Reusswig F, Polzin A, Klier M, Dille MA, Ayhan A, Benkhoff M, Lersch C, Prinz A, Gorressen S, Fischer JW, et al. Only Acute but Not Chronic Thrombocytopenia Protects Mice against Left Ventricular Dysfunction after Acute Myocardial Infarction. Cells. 2022; 11(21):3500. https://doi.org/10.3390/cells11213500
Chicago/Turabian StyleReusswig, Friedrich, Amin Polzin, Meike Klier, Matthias Achim Dille, Aysel Ayhan, Marcel Benkhoff, Celina Lersch, Anika Prinz, Simone Gorressen, Jens Walter Fischer, and et al. 2022. "Only Acute but Not Chronic Thrombocytopenia Protects Mice against Left Ventricular Dysfunction after Acute Myocardial Infarction" Cells 11, no. 21: 3500. https://doi.org/10.3390/cells11213500
APA StyleReusswig, F., Polzin, A., Klier, M., Dille, M. A., Ayhan, A., Benkhoff, M., Lersch, C., Prinz, A., Gorressen, S., Fischer, J. W., Kelm, M., & Elvers, M. (2022). Only Acute but Not Chronic Thrombocytopenia Protects Mice against Left Ventricular Dysfunction after Acute Myocardial Infarction. Cells, 11(21), 3500. https://doi.org/10.3390/cells11213500